


De Novo Drug Design Using Transformer-Based Machine Translation and Reinforcement Learning of an Adaptive Monte Carlo Tree Search


Abstract
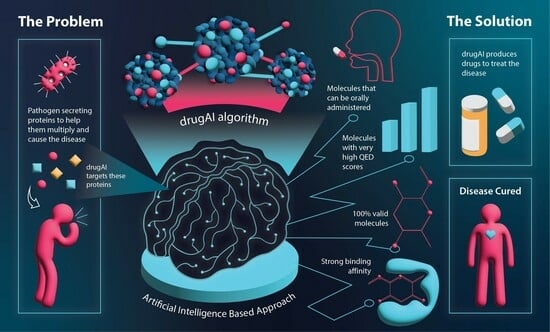
:
1. Introduction
2. Results
2.1. Effectiveness of DrugAI
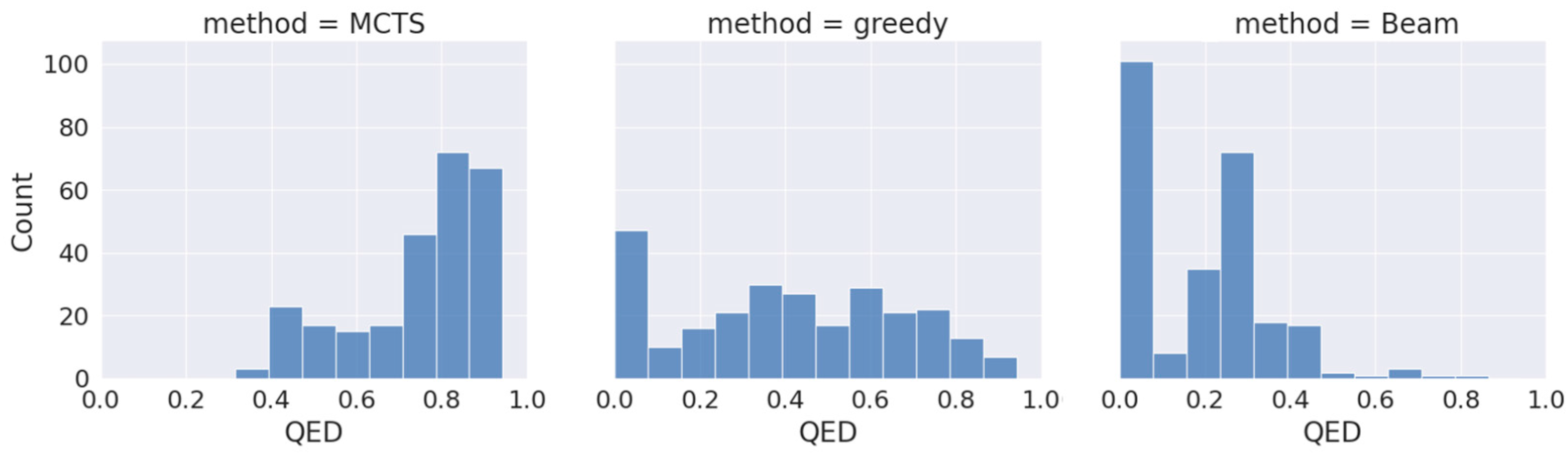
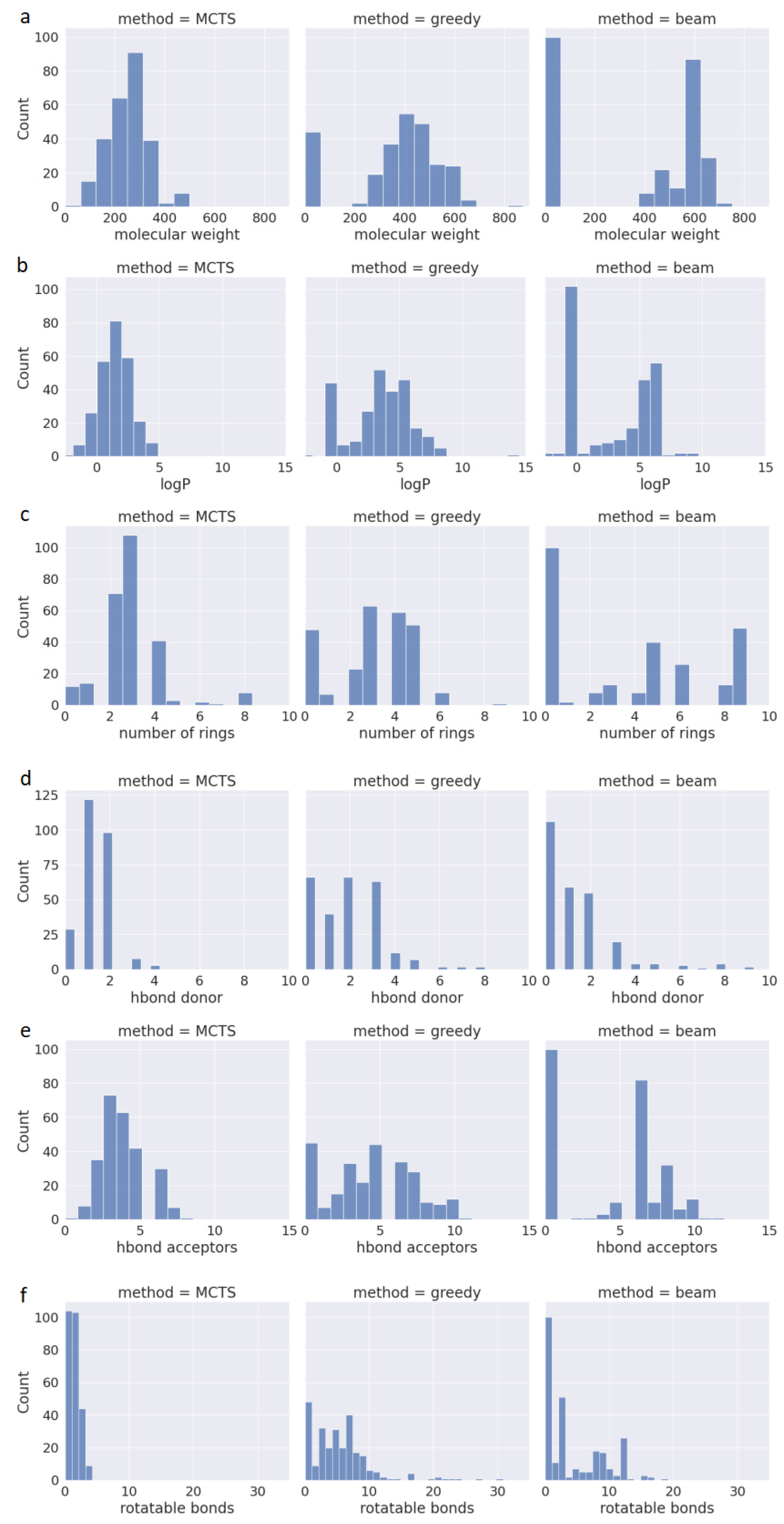
2.2. Physicochemical Properties of the Generated Molecules
2.3. Demonstrating the Flexibility of DrugAI and Comparing It to Traditional Virtual Screening Approaches
3. Discussion
4. Materials and Methods
4.1. Data
- The field “Target Source Organism According to Curator or DataSource” equals “Homo sapiens”;
- The record has an IC50 value less than 100 nm; if the IC50 is missing, then Kd is less than 100 nm; if both are missing, then EC50 is less than 100 nm;
- The record has SMILES representation.
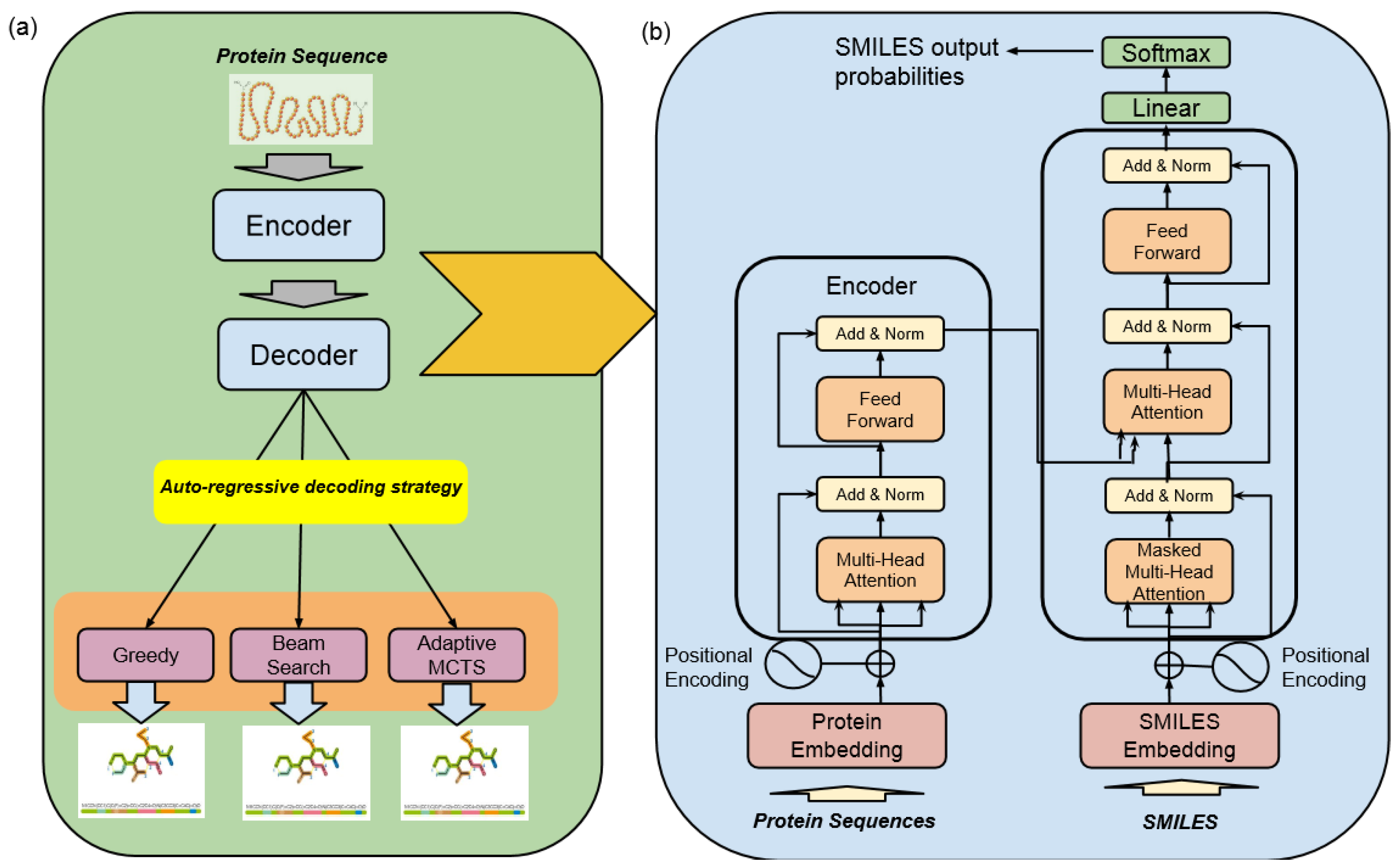
4.2. High-Level Architecture of DrugAI
- Encoder–Decoder TransformerAt its core, this architecture employs a transformer model, which comprises an encoder and a decoder. The encoder takes input data in the form of protein sequences and transforms them into latent representations. Subsequently, the decoder utilizes these representations to systematically generate molecular sequences. The transformer model used in this study was trained with six layers of transformer blocks, each having a size of 512, a learning rate of 0.0001, and eight attention heads. The training process employed the Adam Optimizer with a batch size of five and a total of 25 epochs.
- Monte Carlo Tree Search (MCTS)The MCTS is a heuristic search algorithm used in conjunction with the transformer model. It facilitates the exploration of the vast and complex chemical space by considering different molecular modifications iteratively. The MCTS simulates the potential outcomes of these modifications, allowing for efficient decision making.
4.3. Encoder–Decoder Transformer
- Transformers excel at modeling sequential data. We see molecule generation as a machine translation task that needs to follow a sequence-to-sequence model (seq-to-seq).
- Transformers are parallelizable, and this makes it efficient to parallelize the training and inference steps against Graphics Processing Units (GPUs) and Tensor Processing Units (TPUs).
- Transformers can capture distant or long-range contexts and dependencies in the data between distant positions in the input or output sequences. Thus, longer connections can be learned, which makes it ideal for learning and capturing amino acid sequences whose residues can be hundreds or even thousands in length.
- Transformers make no assumptions about the temporal/spatial relationships across the data.
4.4. SMILES Decoding Strategies
4.4.1. Greedy Search
4.4.2. Beam Search
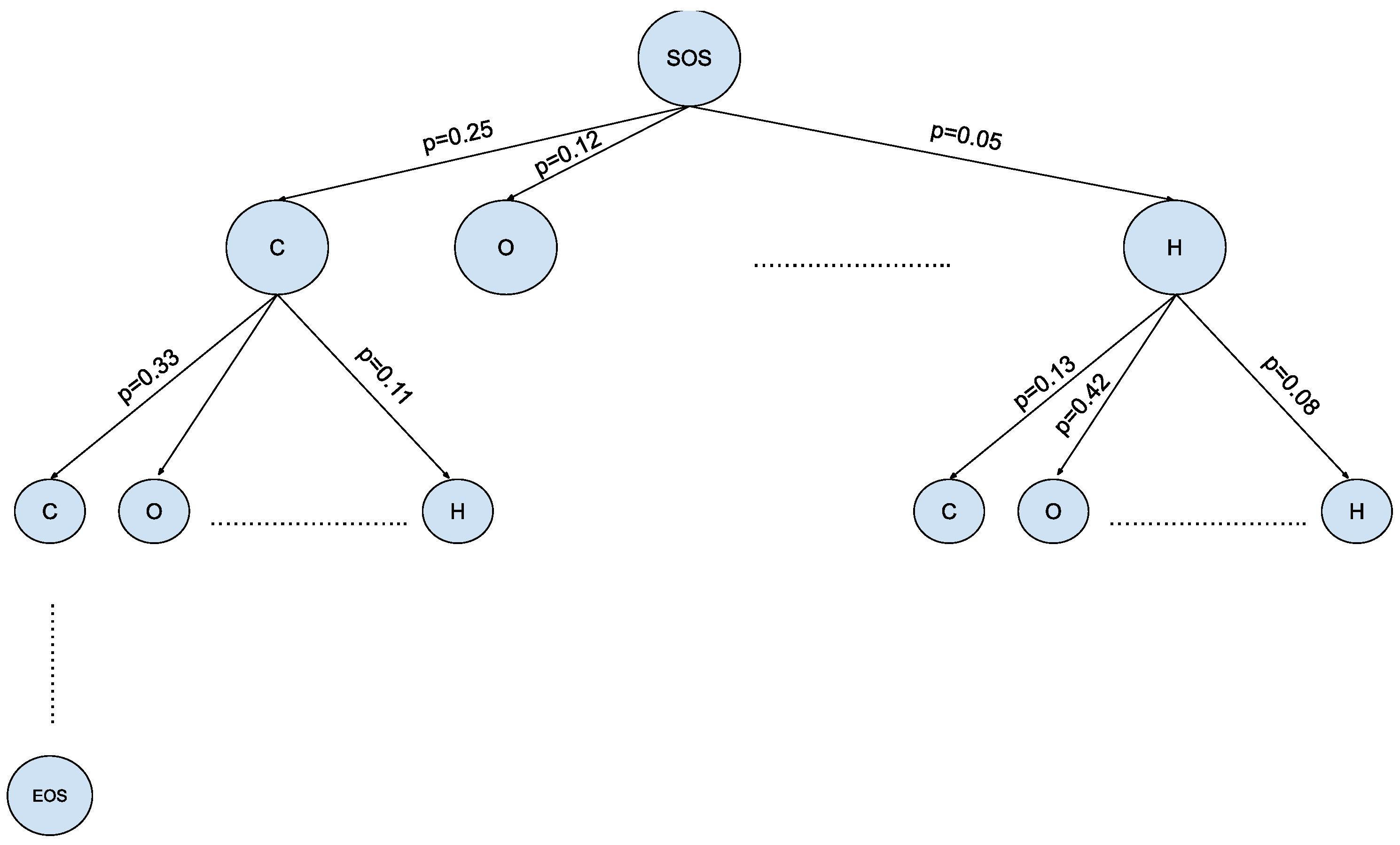
4.4.3. Monte Carlo Tree Search
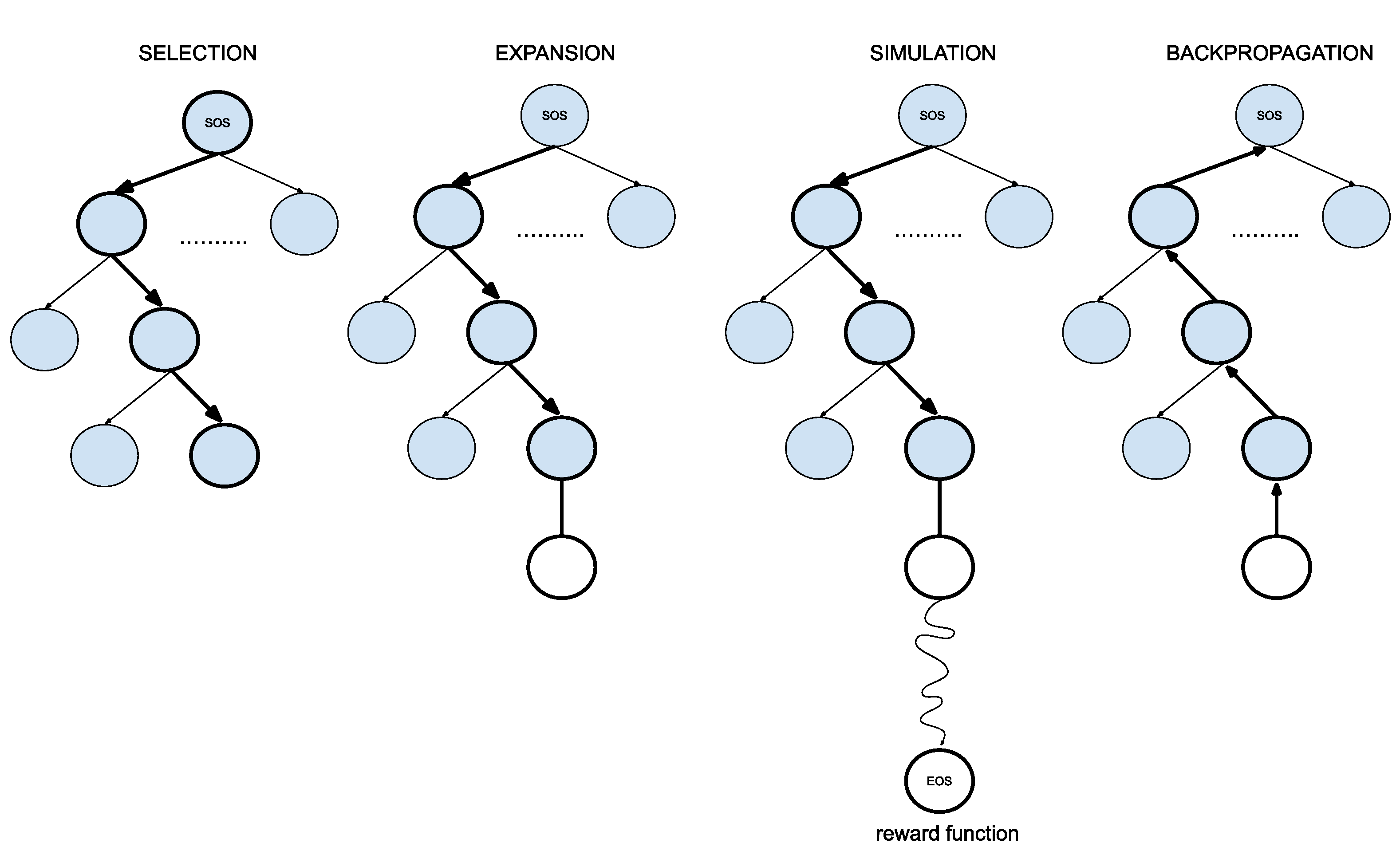
- 1.
- SelectionThe MCTS traverses the SMILES tree structure from the root node using a strategy called the Upper Confidence Bound (UCB) to optimally select the subsequent nodes with the highest estimated value of UCB. Values derived by UCB balance the exploration-exploitation trade-off, and during the tree traversal, a node is selected based on some parameters that return the maximum value. The formula of UCB is described as follows:is total cumulative rewards.is the number of simulations for the node considered after -th move.is the total number of simulations after -th move.is the exploration parameter (default value set to 2).In summary, the first term of the equation will help determine when the MCTS should prioritize making the most of what it knows (exploitation) and the second term of the equation will determine when it should focus on trying out new options (exploration). This ensures that the algorithm is balanced between ensuring that it explores new possibilities and also exploiting known good choices.
- 2.
- ExpansionDuring the traversal of a SMILES tree as part of the selection process in the Monte Carlo Tree Search (MCTS), the child node that yields the highest value from the equation will be chosen for further exploration. If this selected node is also a leaf node and not a terminal node, the MCTS will proceed with the expansion process. This involves creating all possible children of this leaf node based on the SMILES vocabulary.
- 3.
- SimulationThe posterior distribution is derived by using the distribution supplied by the decoder as an informative prior (in contrast to using uniform distribution as a prior where a large proportion of the posterior samples are in the invalid form).
- 4.
- Back propagationWhen the terminal node is reached, the complete SMILES string can be finalized by concatenating all the traversed and simulated nodes from the root until the terminal and the reward is calculated by running the reward function based on the newly constructed SMILES string. Thus, the MCTS needs to update the traversed nodes with this new reward by performing a back-propagation process where it back-propagates from the selected leaf node as a result of step 2 all the way back up to the root node. During this process, the number of simulations stored in each node is also incremented.
- 1.
- Valid SMILESThis binary variable is a straightforward check that assesses the validity of the newly generated SMILES string resulting from the simulation. The check is carried out by executing a basic function provided by RDKit, a toolkit commonly used in cheminformatics and drug discovery [35].
- 2.
- Lipinski’s Rule of Five (Ro5)Another binary variable that checks to see if the newly constructed SMILES string passes all the 5 conditions set forth in the Ro5 [22].
- 3.
- Quantitive Estimation of Drug-likeness (QED)A floating-point variable that reflects the underlying distribution of molecular properties. This metric is intuitive, transparent, and straightforward to implement in many practical settings and allows compounds to be ranked by their relative merit. Medicinal chemists often consider a compound to exhibit characteristics and properties typically desired in drug candidates if the correlation coefficient of the QED value falls within the range of 0.5 to 0.6 [23].
- 4.
- Binding Affinity (kcal/mol)A floating-point variable that refers to the strength by which two molecules interact or bind. The smaller its value, the greater the affinity between two molecules. This binding affinity score is generated by AutoDock Vina [36], a commonly used open-source program for doing molecular docking.
5. Conclusions and Future Perspectives
- pChEMBL values, including pKi, pKd, pIC50, or pEC50 [37];
- ADMET-related properties, such as acute oral toxicity, Ames mutagenicity, and Caco-2 permeability;
- Adherence to Oprea’s rules of drug-likeness [38];
- Avoidance of functional groups with toxic, reactive, or otherwise undesirable moieties defined by the REOS (Rapid Elimination of Swill) rules [39].
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walters, W.P. Virtual chemical libraries: Miniperspective. J. Med. Chem. 2018, 62, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Dreiman, G.H.; Bictash, M.; Fish, P.V.; Griffin, L.; Svensson, F. Changing the HTS paradigm: AI-driven iterative screening for hit finding. SLAS Discov. 2021, 26, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.R.; Fraga, C.A.; Dantas, R.F.; Silva, F.P., Jr. Filtering promiscuous compounds in early drug discovery: Is it a good idea? Drug Discov. Today 2016, 21, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.N.; Alam, A.; Hasnain, S.E. Protein promiscuity in drug discovery, drug-repurposing and antibiotic resistance. Biochimie 2020, 175, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S. Drug discovery: Playing dirty. Nature 2005, 437, 942. [Google Scholar] [CrossRef]
- Lin, X.; Li, X.; Lin, X. A review on applications of computational methods in drug screening and design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef]
- Sharma, V.; Wakode, S.; Kumar, H. Structure- and ligand-based drug design: Concepts, approaches, and challenges. In Chemoinformatics and Bioinformatics in the Pharmaceutical Sciences; Sharma, N., Ojha, H., Raghav, P.K., Goyal, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 27–53. [Google Scholar]
- Salo-Ahen, O.M.; Alanko, I.; Bhadane, R.; Bonvin, A.M.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Vanmeert, M. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 2020, 9, 71. [Google Scholar] [CrossRef]
- Cheng, Y.; Gong, Y.; Liu, Y.; Song, B.; Zou, Q. Molecular design in drug discovery: A comprehensive review of deep generative models. Brief. Bioinform. 2021, 22, bbab344. [Google Scholar] [CrossRef]
- Xie, W.; Wang, F.; Li, Y.; Lai, L.; Pei, J. Advances and challenges in de novo drug design using three-dimensional deep generative models. J. Chem. Inf. Model. 2022, 62, 2269–2279. [Google Scholar] [CrossRef]
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P.; Neelakantan, A.; Amodei, D. Language models are few-shot learners. In Proceedings of the Advances in Neural Information Processing Systems 33 (NeurIPS 2020), Virtual, 6–12 December 2020; pp. 1877–1901. [Google Scholar]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Chowdhary, K.R. Natural language processing. In Fundamentals of Artificial Intelligence; Springer: New Delhi, India, 2020; pp. 603–649. [Google Scholar]
- Kell, D.B.; Samanta, S.; Swainston, N. Deep learning and generative methods in cheminformatics and chemical biology: Navigating small molecule space intelligently. Biochem. J. 2020, 477, 4559–4580. [Google Scholar] [CrossRef] [PubMed]
- Sutskever, I.; Vinyals, O.; Le, Q.V. Sequence to sequence learning with neural networks. In Proceedings of the Advances in Neural Information Processing Systems 27 (NIPS 2014), Montreal, QC, Canada, 8–13 December 2014. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. In Proceedings of the Neural Information Processing Systems 30 (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Chickering, D.M. Optimal structure identification with greedy search. J. Mach. Learn. Res. 2002, 3, 507–554. [Google Scholar]
- Wiseman, S.; Rush, A.M. Sequence-to-sequence learning as beam-search optimization. arXiv 2016, arXiv:1606.02960. [Google Scholar]
- Leblond, R.; Alayrac, J.B.; Sifre, L.; Pislar, M.; Lespiau, J.B.; Antonoglou, I.; Simonyan, K.; Vinyals, O. Machine translation decoding beyond beam search. arXiv 2021, arXiv:2104.05336. [Google Scholar]
- Chaffin, A.; Claveau, V.; Kijak, E. PPL-MCTS: Constrained textual generation through discriminator-guided MCTS decoding. arXiv 2021, arXiv:2109.13582. [Google Scholar]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: Benchmarking models for de novo molecular design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Ang, D.; Kendall, R.; Atamian, H.S. Virtual and In Vitro Screening of Natural Products Identifies Indole and Benzene Derivatives as Inhibitors of SARS-CoV-2 Main Protease (Mpro). Biology 2023, 12, 519. [Google Scholar] [CrossRef] [PubMed]
- Harshvardhan, G.M.; Gourisaria, M.K.; Pandey, M.; Rautaray, S.S. A comprehensive survey and analysis of generative models in machine learning. Comput. Sci. Rev. 2020, 38, 100285. [Google Scholar]
- Martinelli, D.D. Generative machine learning for de novo drug discovery: A systematic review. Comput. Biol. Med. 2022, 145, 105403. [Google Scholar] [CrossRef] [PubMed]
- Grechishnikova, D. Transformer neural network for protein-specific de novo drug generation as a machine translation problem. Sci. Rep. 2021, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Latif, S.; Cuayáhuitl, H.; Pervez, F.; Shamshad, F.; Ali, H.S.; Cambria, E. A survey on deep reinforcement learning for audio-based applications. Artif. Intell. Rev. 2023, 56, 2193–2240. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de novo drug design: From conventional to machine learning methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef]
- Parenti, M.D.; Rastelli, G. Advances and applications of binding affinity prediction methods in drug discovery. Biotechnol. Adv. 2012, 30, 244–250. [Google Scholar] [CrossRef]
- Silver, D.; Huang, A.; Maddison, C.J.; Guez, A.; Sifre, L.; van den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; et al. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484–489. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org (accessed on 20 September 2023).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Kruger, F.A.; Overington, J.P. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I. Property distribution of drug-related chemical databases. J. Comput. Aided Mol. Des. 2020, 14, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P.; Namchuk, M. Designing screens: How to make your hits a hit. Nat. Rev. Drug Discov. 2003, 2, 259–266. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Benchmark | DrugAI | Greedy | Beam (K = 2) |
|---|---|---|---|
| Validity | 1.00 a (1) | 0.83 b (0.82–0.84) | 0.62 c (0.61–0.63) |
| Uniqueness | 0.84 a (0.83–0.85) | 0.87 a (0.86–0.88) | 0.37 b (0.36–0.38) |
| Novelty | 1.00 a (1) | 1.00 a (1) | 0.47 b (0.46–0.48) |
| Mean QED | 0.73 a (72.95–73.05) | 0.41 b (0.41) | 0.18 c (0.18) |
| Generated Molecule (SMILES) | Validity | Adherence to RO5 | Binding Affinity (kcal/mol) | QED Score |
|---|---|---|---|---|
| O=C1c2cc(N3CCNCC3)ccc2C(OC2CCc3ccccc32)c2ccccc21 | 1 | 1 | −9.87 | 0.69 |
| O=C1c2cc(N3CCN(c4cnc5ncccc5c4)CC3)ccc2CCCc2ccccc21 | 1 | 1 | −9.59 | 0.46 |
| O=C1c2cc(N3CCNCC3)cc(-c3cccc(/C=C/C(=O)Nc4ccccc4)c3)c2C(=O)N1 | 1 | 1 | −9.56 | 0.41 |
| O=C1c2cc(N3CCNC(c4ncc(C(F)(F)F)cc4Cl)CC3)ccc2COC1O | 1 | 1 | −9.45 | 0.74 |
| O=C1c2cc(N3CCNCC3)ccc2OC(COc2cccc([N+](=O)[O-])c2)Cc2ccccc21 | 1 | 1 | −9.39 | 0.46 |
| O=C1c2cc(N3CCNCC3)nc-c3ccc(C(=O)c4cc(F)cc(F)c4)cc3c2CCC1=O | 1 | 1 | −9.39 | 0.56 |
| O=C1c2cc(N3CCNC4C3CC5CC(C4)OC5)ccc2C(=O)c2ccc(Cl)cc2N1 | 1 | 1 | −9.30 | 0.61 |
| O=C1c2cc(N3CCNCC3)nc(-c3cccc(C(F)(F)F)c3)c2CCc2ccccc21 | 1 | 1 | −9.24 | 0.64 |
| O=C1c2cc(N3CCNCC3)ccc2C(OC2Cc3ccccc3C2)=C2C(=O)CCC(O)C(F)(F)C21 | 1 | 1 | −9.22 | 0.68 |
| O=C1c2cc(N3CCNCC3)ccc2OC/C1=C(\O)c1cccc(-c2ccncc2)c1 | 1 | 1 | −9.21 | 0.50 |
| Average | 1 | 1 | −9.42 | 0.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ang, D.; Rakovski, C.; Atamian, H.S. De Novo Drug Design Using Transformer-Based Machine Translation and Reinforcement Learning of an Adaptive Monte Carlo Tree Search. Pharmaceuticals 2024, 17, 161. https://doi.org/10.3390/ph17020161
Ang D, Rakovski C, Atamian HS. De Novo Drug Design Using Transformer-Based Machine Translation and Reinforcement Learning of an Adaptive Monte Carlo Tree Search. Pharmaceuticals. 2024; 17(2):161. https://doi.org/10.3390/ph17020161
Chicago/Turabian StyleAng, Dony, Cyril Rakovski, and Hagop S. Atamian. 2024. "De Novo Drug Design Using Transformer-Based Machine Translation and Reinforcement Learning of an Adaptive Monte Carlo Tree Search" Pharmaceuticals 17, no. 2: 161. https://doi.org/10.3390/ph17020161