
Mesenchymal Stem Cell-Derived Exosomes Ameliorate Doxorubicin-Induced Cardiotoxicity

Abstract
:1. Introduction
2. Results
2.1. MSC-Exo Confirmation and Characterization
2.2. Effect of MSC-Exos on Cell Viability of DOX-Treated H9c2 Cells
2.3. MSC-Exos Diminishes Pyroptotic HMGB1 in an In Vitro DIC Model
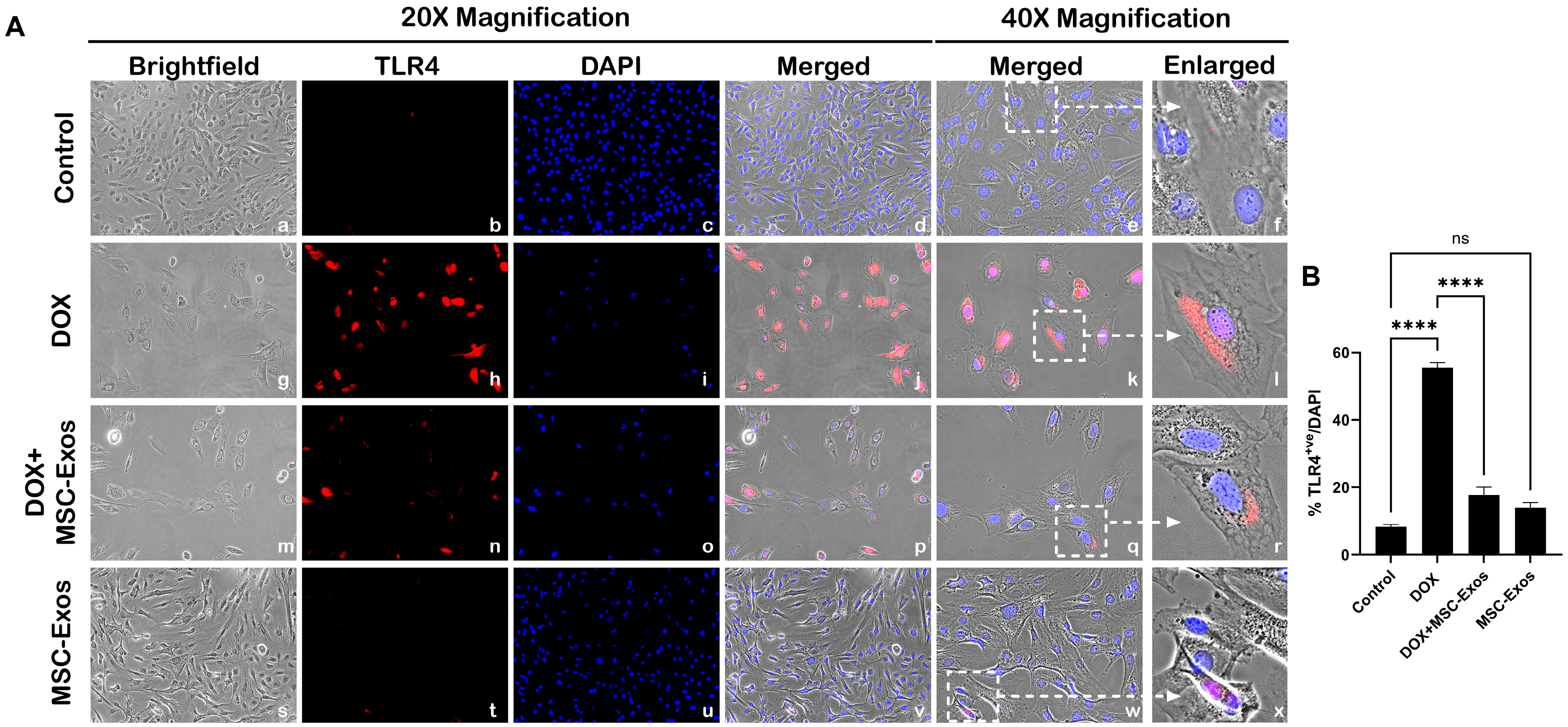
2.4. MSC-Exos Reduces TLR4 Expression in an In Vitro DIC Model
2.5. MSC-Exos Decreases NLRP3 Inflammasome Formation in an In Vitro DIC Model
2.6. MSC-Exos Mitigates Pyroptotic Cascade Markers Caspase-1, IL-1β, and IL-18 in an In Vitro DIC Model
2.7. MSC-Exos Attenuates Pyroptotic Executioner GSDMD in an In Vitro DIC Model
3. Discussion
4. Materials and Methods
4.1. Isolation of Mesenchymal Stem Cell-Derived Exosomes
4.2. MSC Exosome Confirmation and Characterization
4.3. H9c2 Cell Culture
4.4. MTT Assay
4.5. Immunocytochemistry (ICC) Staining
4.6. Western Blot
4.7. RT-PCR
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Renu, K.; Abilash, V.G.; PB, T.P.; Arunachalam, S. Molecular Mechanism of Doxorubicin-Induced Cardiomyopathy—An Update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-Induced Cardiotoxicity: An Update on the Molecular Mechanism and Novel Therapeutic Strategies for Effective Management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Pokhrel, A.R.; Darsandhari, S.; Parajuli, P.; Sohng, J.K.; Pandey, R.P. Engineering Streptomyces Peucetius for Doxorubicin and Daunorubicin Biosynthesis. In Pharmaceuticals from Microbes: The Bioengineering Perspective; Arora, D., Sharma, C., Jaglan, S., Lichtfouse, E., Eds.; Environmental Chemistry for a Sustainable World; Springer International Publishing: Cham, Switzerland, 2019; pp. 191–209. ISBN 978-3-030-01881-8. [Google Scholar]
- Sritharan, S.; Sivalingam, N. A Comprehensive Review on Time-Tested Anticancer Drug Doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef]
- Rivankar, S. An Overview of Doxorubicin Formulations in Cancer Therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated Cell Death Pathways in Doxorubicin-Induced Cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]
- Saleh, Y.; Abdelkarim, O.; Herzallah, K.; Abela, G.S. Anthracycline-Induced Cardiotoxicity: Mechanisms of Action, Incidence, Risk Factors, Prevention, and Treatment. Heart Fail. Rev. 2021, 26, 1159–1173. [Google Scholar] [CrossRef]
- Singla, D.K.; Johnson, T.A.; Tavakoli Dargani, Z. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells 2019, 8, 1224. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli Dargani, Z.; Singla, D.K. Embryonic Stem Cell-Derived Exosomes Inhibit Doxorubicin-Induced TLR4-NLRP3-Mediated Cell Death-Pyroptosis. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H460–H471. [Google Scholar] [CrossRef]
- Kong, C.-Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.-P.; Teng, T.; Yan, L.; Tang, Q.-Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Ye, B.; Shi, X.; Xu, J.; Dai, S.; Xu, J.; Fan, X.; Han, B.; Han, J. Gasdermin D Mediates Doxorubicin-Induced Cardiomyocyte Pyroptosis and Cardiotoxicity via Directly Binding to Doxorubicin and Changes in Mitochondrial Damage. Transl. Res. 2022, 248, 36–50. [Google Scholar] [CrossRef]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Apoptosis, Pyroptosis, and Necroptosis—Oh My! The Many Ways a Cell Can Die. J. Mol. Biol. 2022, 434, 167378. [Google Scholar] [CrossRef] [PubMed]
- Sumneang, N.; Tanajak, P.; Oo, T.T. Toll-like Receptor 4 Inflammatory Perspective on Doxorubicin-Induced Cardiotoxicity. Molecules 2023, 28, 4294. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.; Zhang, G.; Zhang, Y.; Qian, W.; Rui, T. Role of HMGB1 in Doxorubicin-Induced Myocardial Apoptosis and Its Regulation Pathway. Basic. Res. Cardiol. 2012, 107, 267. [Google Scholar] [CrossRef] [PubMed]
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S. Gasdermin D in Pyroptosis. Acta Pharm. Sin. B 2021, 11, 2768–2782. [Google Scholar] [CrossRef]
- Ezquer, F.; Gutiérrez, J.; Ezquer, M.; Caglevic, C.; Salgado, H.C.; Calligaris, S.D. Mesenchymal Stem Cell Therapy for Doxorubicin Cardiomyopathy: Hopes and Fears. Stem Cell Res. Ther. 2015, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Gopalarethinam, J.; Nair, A.P.; Iyer, M.; Vellingiri, B.; Subramaniam, M.D. Advantages of Mesenchymal Stem Cell over the Other Stem Cells. Acta Histochem. 2023, 125, 152041. [Google Scholar] [CrossRef]
- Riggs, J.W.; Barrilleaux, B.L.; Varlakhanova, N.; Bush, K.M.; Chan, V.; Knoepfler, P.S. Induced Pluripotency and Oncogenic Transformation Are Related Processes. Stem Cells Dev. 2013, 22, 37–50. [Google Scholar] [CrossRef]
- Bagno, L.; Hatzistergos, K.E.; Balkan, W.; Hare, J.M. Mesenchymal Stem Cell-Based Therapy for Cardiovascular Disease: Progress and Challenges. Mol. Ther. 2018, 26, 1610–1623. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, Y.; Li, H.-J. Advances in Mesenchymal Stem Cell Exosomes: A Review. Stem Cell Res. Ther. 2021, 12, 71. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The Exosome Journey: From Biogenesis to Uptake and Intracellular Signalling. Cell Commun. Signal 2021, 19, 47. [Google Scholar] [CrossRef]
- Kou, M.; Huang, L.; Yang, J.; Chiang, Z.; Chen, S.; Liu, J.; Guo, L.; Zhang, X.; Zhou, X.; Xu, X.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles for Immunomodulation and Regeneration: A next Generation Therapeutic Tool? Cell Death Dis. 2022, 13, 580. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Zhang, Y.; Zhang, S.; Wang, D.; Ye, H. Exosomes: Cell-Free Therapy for Cardiovascular Diseases. J. Cardiovasc. Trans. Res. 2020, 13, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Shan, A.; Wei, Z.; Xu, B. Intravenous Mesenchymal Stem Cell-Derived Exosomes Ameliorate Myocardial Inflammation in the Dilated Cardiomyopathy. Biochem. Biophys. Res. Commun. 2018, 503, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Chung, J.; Byun, Y.; Kim, K.H.; An, S.H.; Kwon, K. Mesenchymal Stem Cell-Derived Small Extracellular Vesicles Protect Cardiomyocytes from Doxorubicin-Induced Cardiomyopathy by Upregulating Survivin Expression via the miR-199a-3p-Akt-Sp1/P53 Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 7102. [Google Scholar] [CrossRef]
- Chen, L.-M.; Chai, J.C.; Liu, B.; Strutt, T.M.; McKinstry, K.K.; Chai, K.X. Prostasin Regulates PD-L1 Expression in Human Lung Cancer Cells. Biosci. Rep. 2021, 41, BSR20211370. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, S.-H.; Yoo, K.H.; Chung, M.-H.; Lee, J.W.; Son, K.H. Exogenous 8-Hydroxydeoxyguanosine Attenuates Doxorubicin-Induced Cardiotoxicity by Decreasing Pyroptosis in H9c2 Cardiomyocytes. BMC Mol. Cell Biol. 2022, 23, 55. [Google Scholar] [CrossRef] [PubMed]
- Syukri, A.; Budu; Hatta, M.; Amir, M.; Rohman, M.S.; Mappangara, I.; Kaelan, C.; Wahyuni, S.; Bukhari, A.; Junita, A.R.; et al. Doxorubicin Induced Immune Abnormalities and Inflammatory Responses via HMGB1, HIF1-α and VEGF Pathway in Progressive of Cardiovascular Damage. Ann. Med. Surg. 2022, 76, 103501. [Google Scholar] [CrossRef]
- Aluganti Narasimhulu, C.; Singla, D.K. Amelioration of Diabetes-Induced Inflammation Mediated Pyroptosis, Sarcopenia, and Adverse Muscle Remodelling by Bone Morphogenetic Protein-7. J. Cachexia Sarcopenia Muscle 2021, 12, 403–420. [Google Scholar] [CrossRef]
- Wang, K.; You, S.; Hu, H.; Li, X.; Yin, J.; Shi, Y.; Qi, L.; Li, P.; Zhao, Y.; Yan, S. Effect of TLR4/MyD88/NF-kB Axis in Paraventricular Nucleus on Ventricular Arrhythmias Induced by Sympathetic Hyperexcitation in Post-Myocardial Infarction Rats. J. Cell. Mol. Med. 2022, 26, 2959–2971. [Google Scholar] [CrossRef]
- Moretti, I.F.; Lerario, A.M.; Trombetta-Lima, M.; Sola, P.R.; da Silva Soares, R.; Oba-Shinjo, S.M.; Marie, S.K.N. Late P65 Nuclear Translocation in Glioblastoma Cells Indicates Non-Canonical TLR4 Signaling and Activation of DNA Repair Genes. Sci. Rep. 2021, 11, 1333. [Google Scholar] [CrossRef]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Sheibani, M.; Azizi, Y.; Shayan, M.; Nezamoleslami, S.; Eslami, F.; Farjoo, M.H.; Dehpour, A.R. Doxorubicin-Induced Cardiotoxicity: An Overview on Pre-Clinical Therapeutic Approaches. Cardiovasc. Toxicol. 2022, 22, 292–310. [Google Scholar] [CrossRef]
- Sun, S.-J.; Wei, R.; Li, F.; Liao, S.-Y.; Tse, H.-F. Mesenchymal Stromal Cell-Derived Exosomes in Cardiac Regeneration and Repair. Stem Cell Rep. 2021, 16, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aal, S.A.; AbdElrahman, M.; Reda, A.M.; Afify, H.; Ragab, G.M.; El-Gazar, A.A.; Ibrahim, S.S.A. Galangin Mitigates DOX-Induced Cognitive Impairment in Rats: Implication of NOX-1/Nrf-2/HMGB1/TLR4 and TNF-α/MAPKs/RIPK/MLKL/BDNF. Neurotoxicology 2022, 92, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A New Paradigm of Cell Death for Fighting against Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153. [Google Scholar] [CrossRef]
- Luther, K.M.; Haar, L.; McGuinness, M.; Wang, Y.; Lynch, T.L., IV; Phan, A.; Song, Y.; Shen, Z.; Gardner, G.; Kuffel, G.; et al. Exosomal miR-21a-5p Mediates Cardioprotection by Mesenchymal Stem Cells. J. Mol. Cell. Cardiol. 2018, 119, 125–137. [Google Scholar] [CrossRef]
- Bridge, T.; Wegmann, U.; Crack, J.C.; Orman, K.; Shaikh, S.A.; Farndon, W.; Martins, C.; Saalbach, G.; Sachdeva, A. Site-specific encoding of photoactivity and photoreactivity into antibody fragments. Nat. Chem. Biol. 2023, 19, 740–749. [Google Scholar] [CrossRef]
- Srivastava, A.; Singla, D.K. PTEN-AKT Pathway Attenuates Apoptosis and Adverse Remodeling in Ponatinib-Induced Skeletal Muscle Toxicity Following BMP-7 Treatment. Physiol. Rep. 2023, 11, e15629. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Table | Forward Primer | Reverse Primer |
|---|---|---|
| NLRP3 | 5′-GGTGACCTTGTGTGTGCTTG-3′ | 5′-ATGTCCTGAGCCATGGAAGC-3′ |
| GAPDH | 5′-GCCCACTAAAGGGCATCCTG-3′ | 5′-GAGTTGGGATGGGGACTCTCA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, S.A.; Singla, D.K. Mesenchymal Stem Cell-Derived Exosomes Ameliorate Doxorubicin-Induced Cardiotoxicity. Pharmaceuticals 2024, 17, 93. https://doi.org/10.3390/ph17010093
Ali SA, Singla DK. Mesenchymal Stem Cell-Derived Exosomes Ameliorate Doxorubicin-Induced Cardiotoxicity. Pharmaceuticals. 2024; 17(1):93. https://doi.org/10.3390/ph17010093
Chicago/Turabian StyleAli, Sawdah A., and Dinender K. Singla. 2024. "Mesenchymal Stem Cell-Derived Exosomes Ameliorate Doxorubicin-Induced Cardiotoxicity" Pharmaceuticals 17, no. 1: 93. https://doi.org/10.3390/ph17010093