


Design, Synthesis, In Silico Studies and In Vitro Evaluation of New Indole- and/or Donepezil-like Hybrids as Multitarget-Directed Agents for Alzheimer’s Disease

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
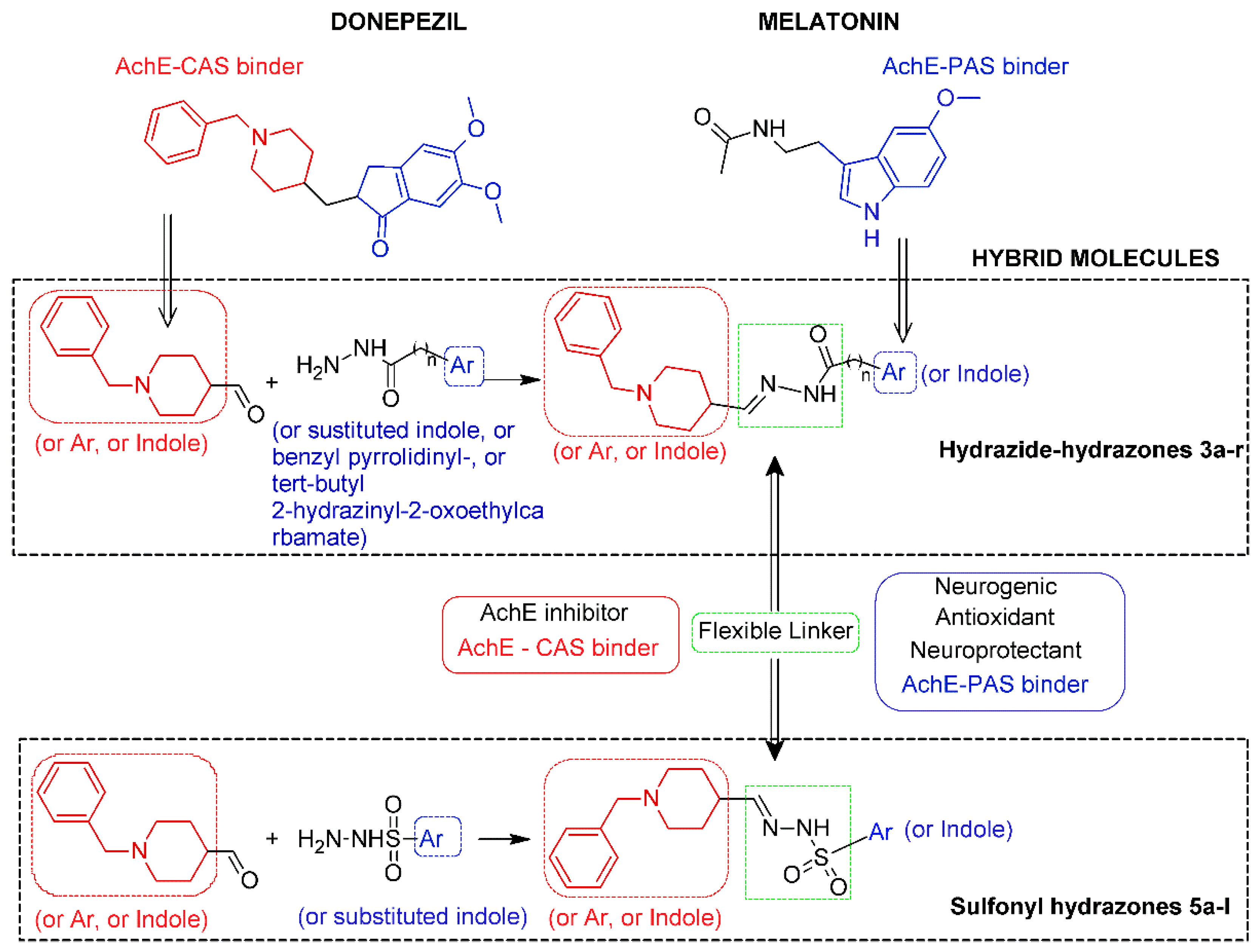
2.1. Chemistry and Design Strategy of the Multifunctional Donepezil–Melatonin Hybrids
2.2. In Vitro AChE and BChE Inhibition Assays
2.3. Cytotoxicity of the Compounds
2.4. Determination of Antioxidant Activity
2.5. DPPH Radical Scavenging Activity
2.6. ABTS Radical Scavenging Assay
2.7. Ferric Reducing Antioxidant Power (FRAP)
2.8. Determination of Antioxidant Activity in Linoleic Acid System by the FTC Method
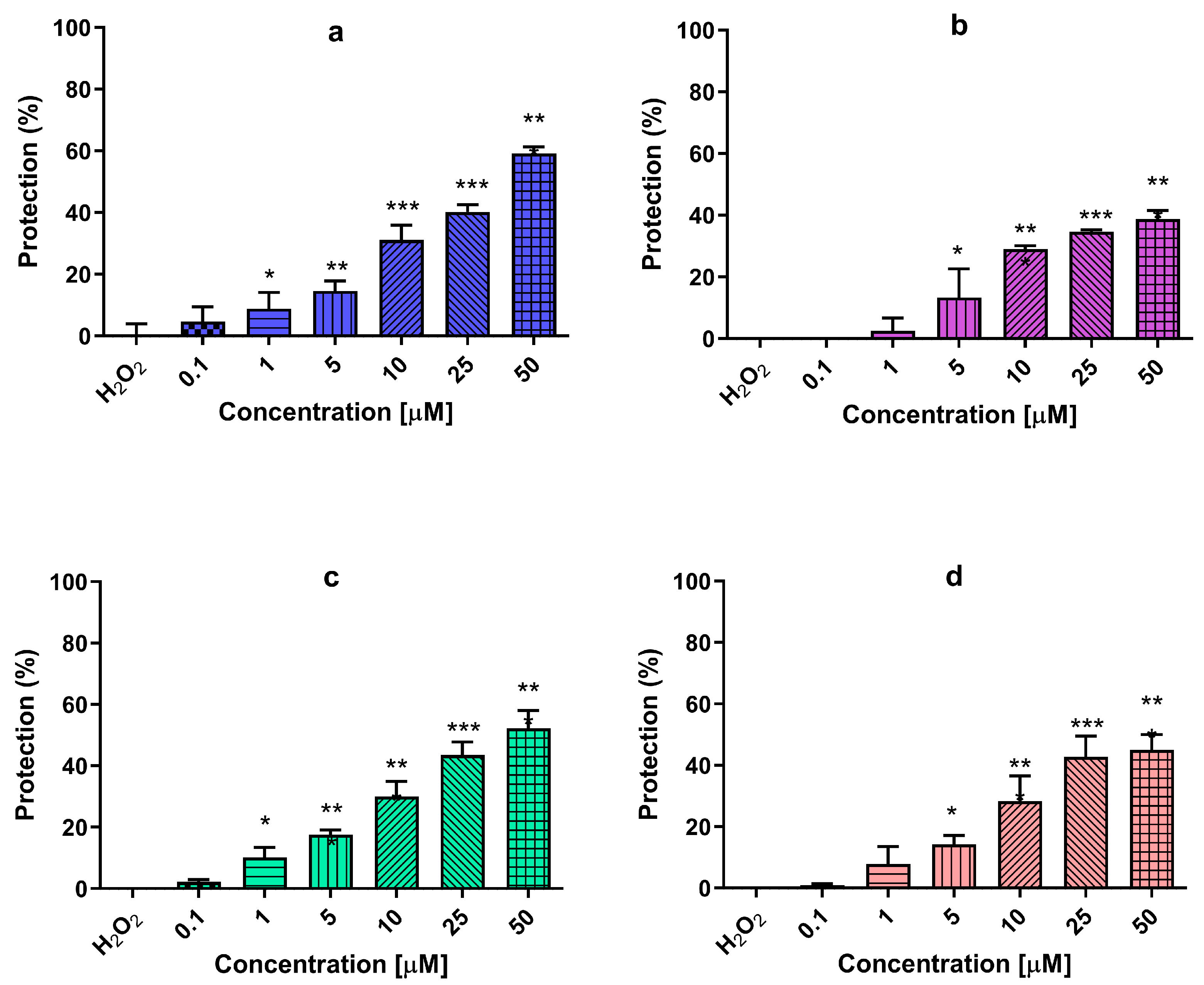
2.9. Neuroprotection against Oxidative Stress
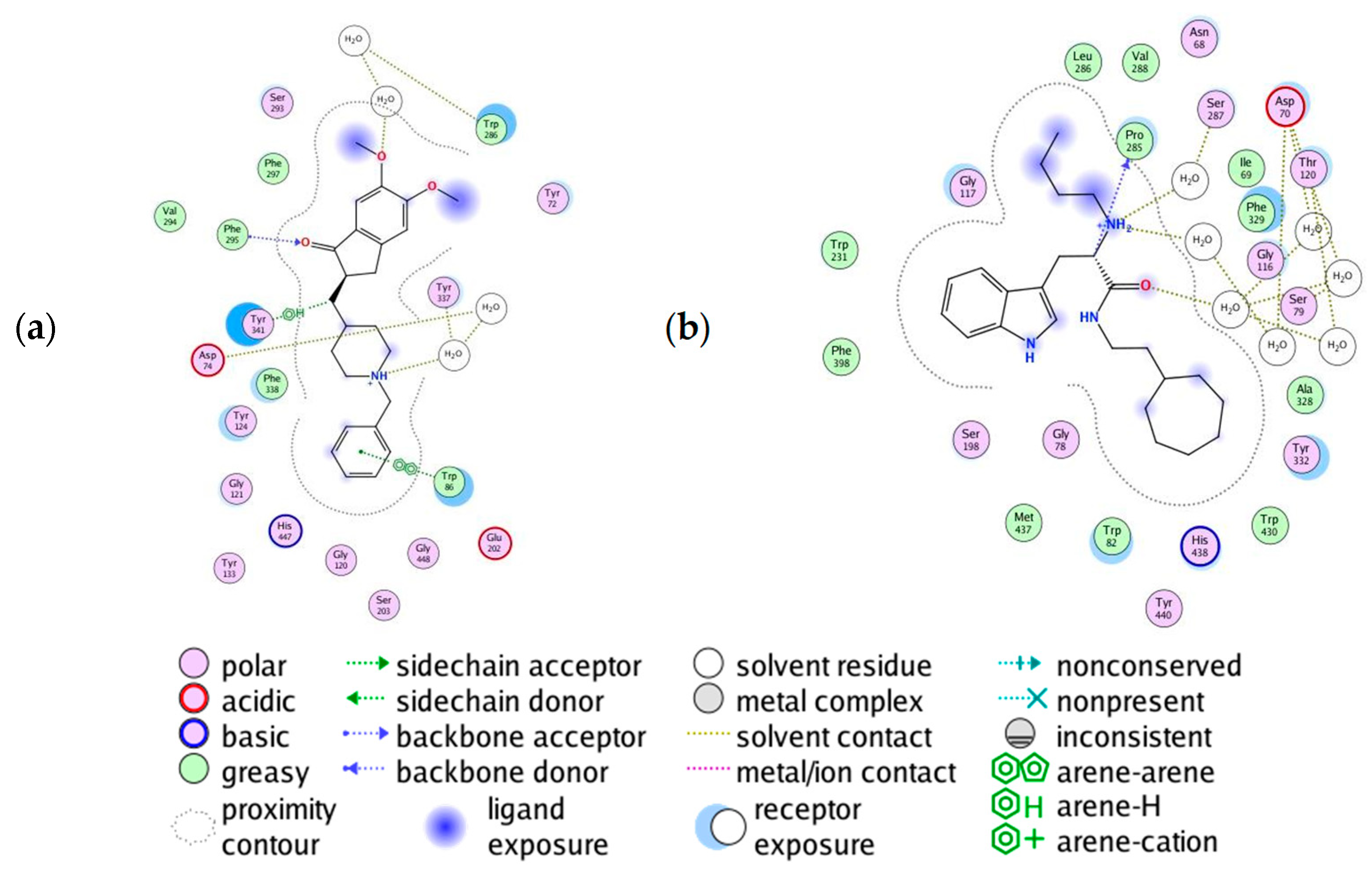
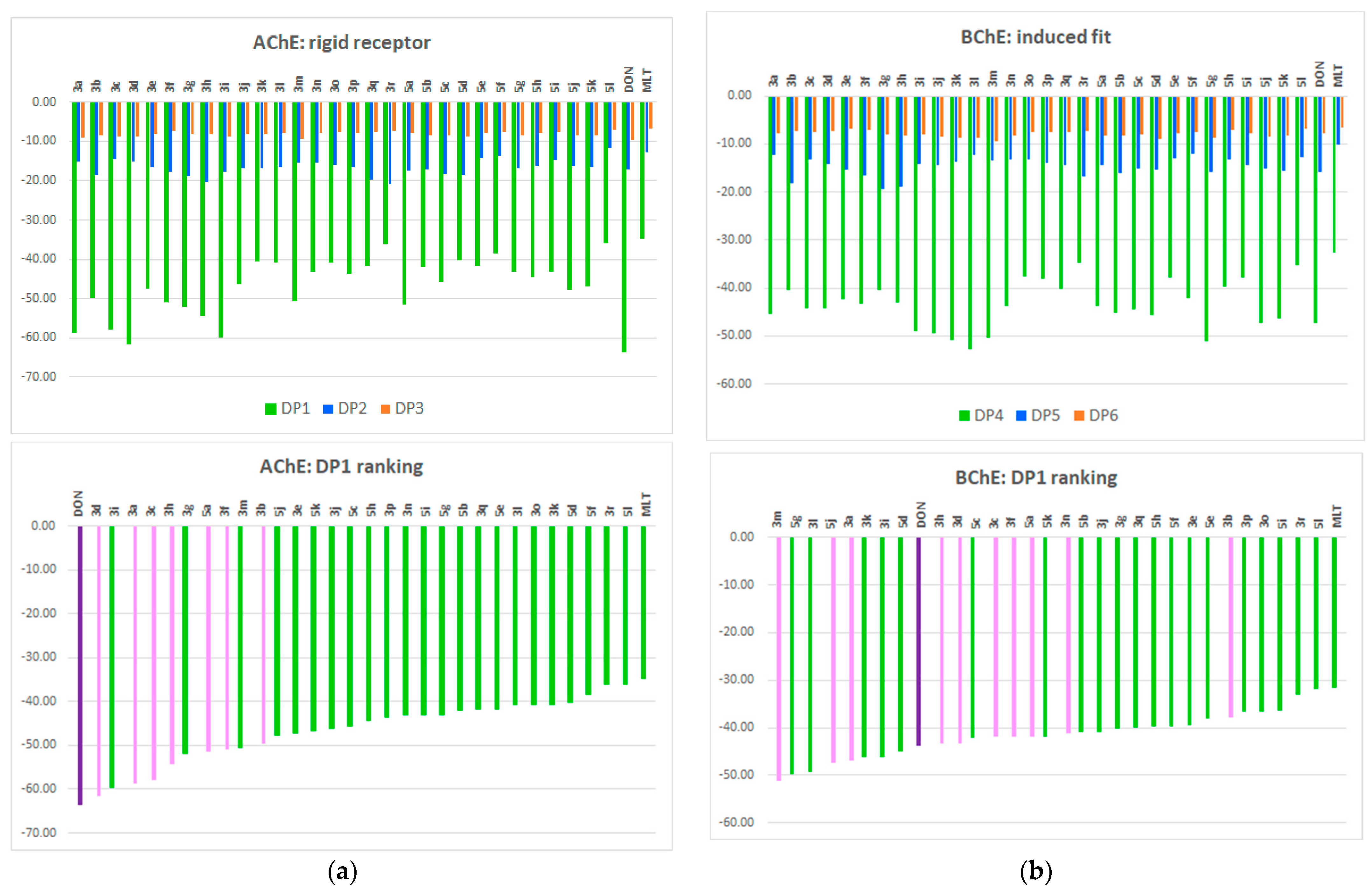
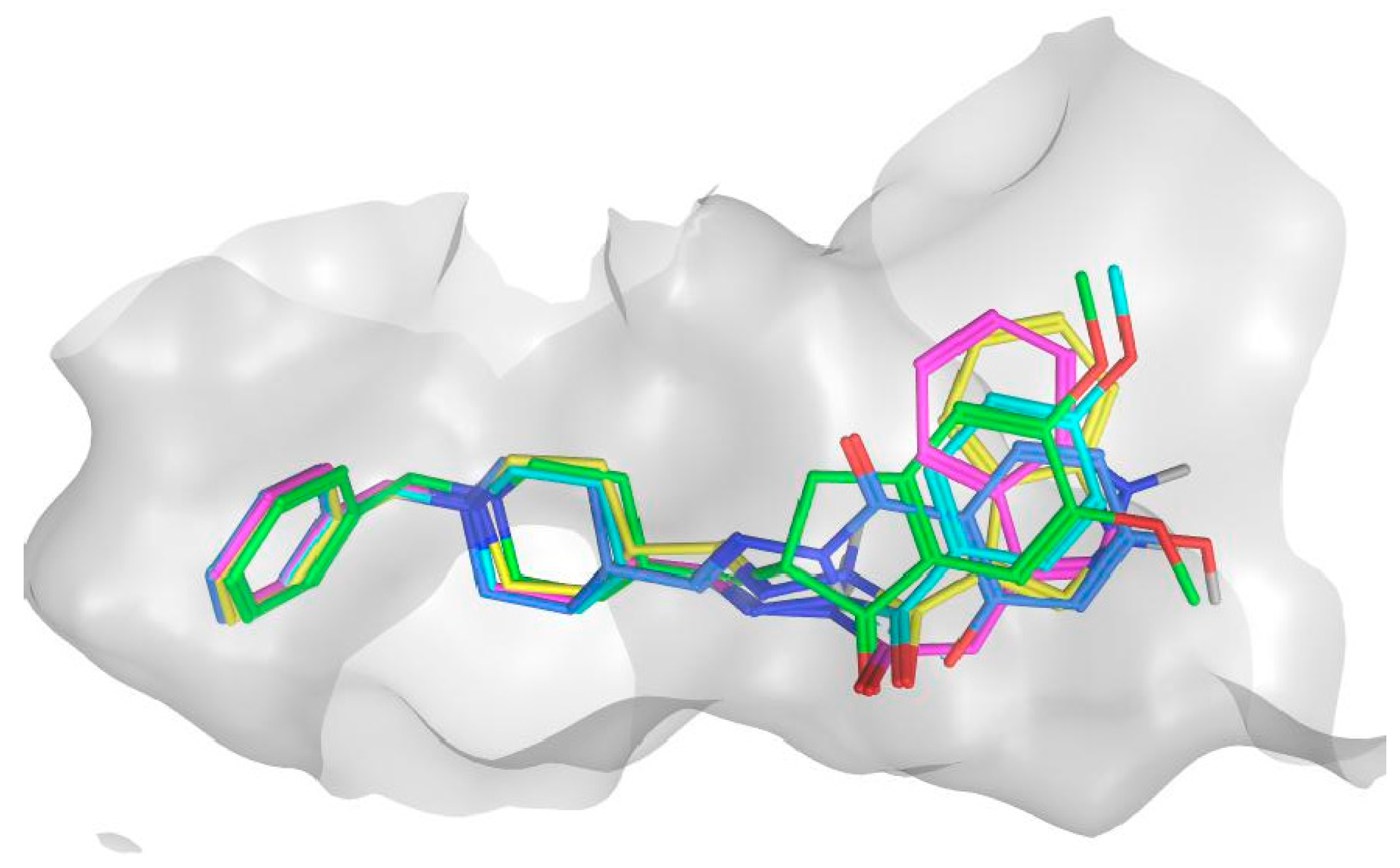
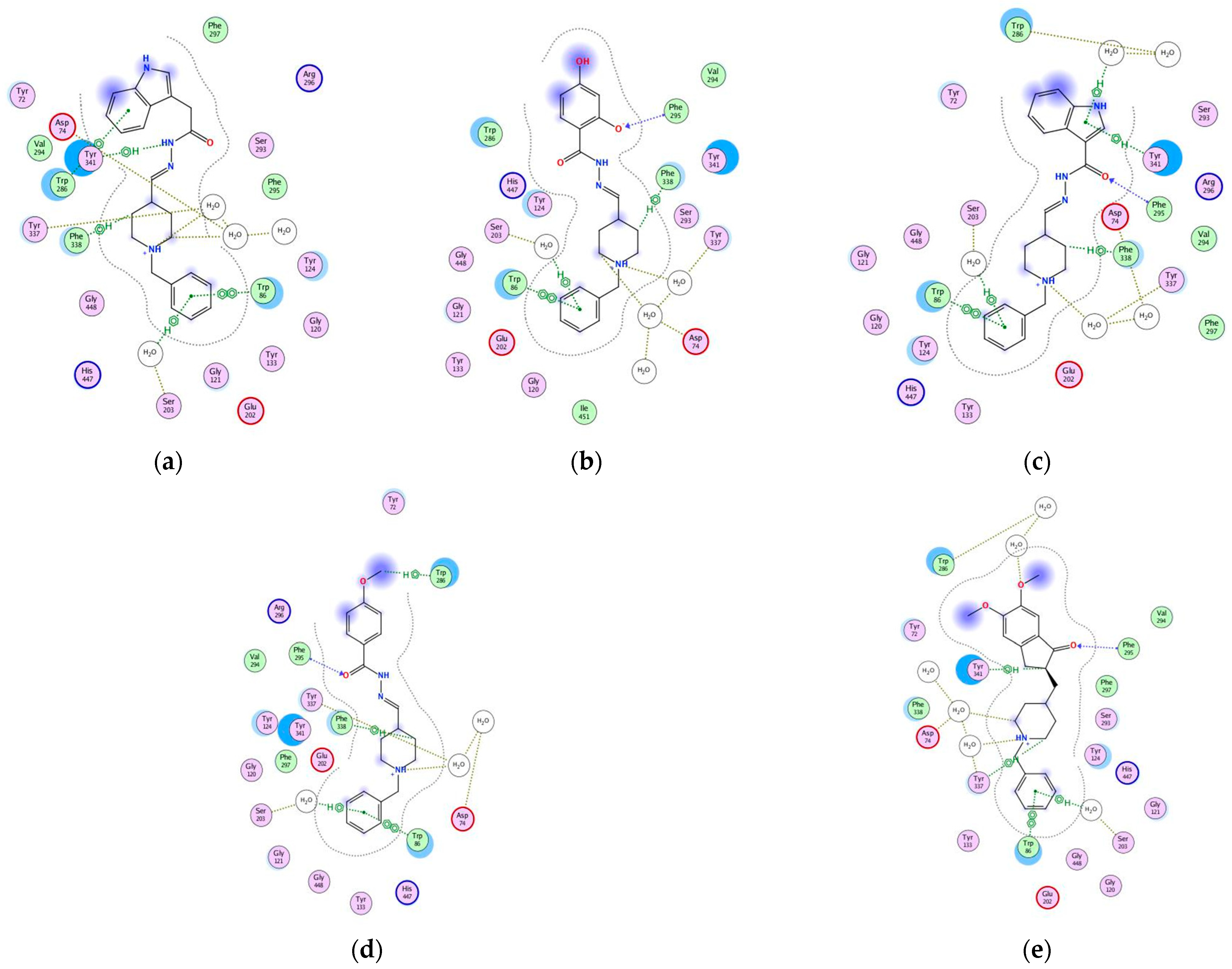
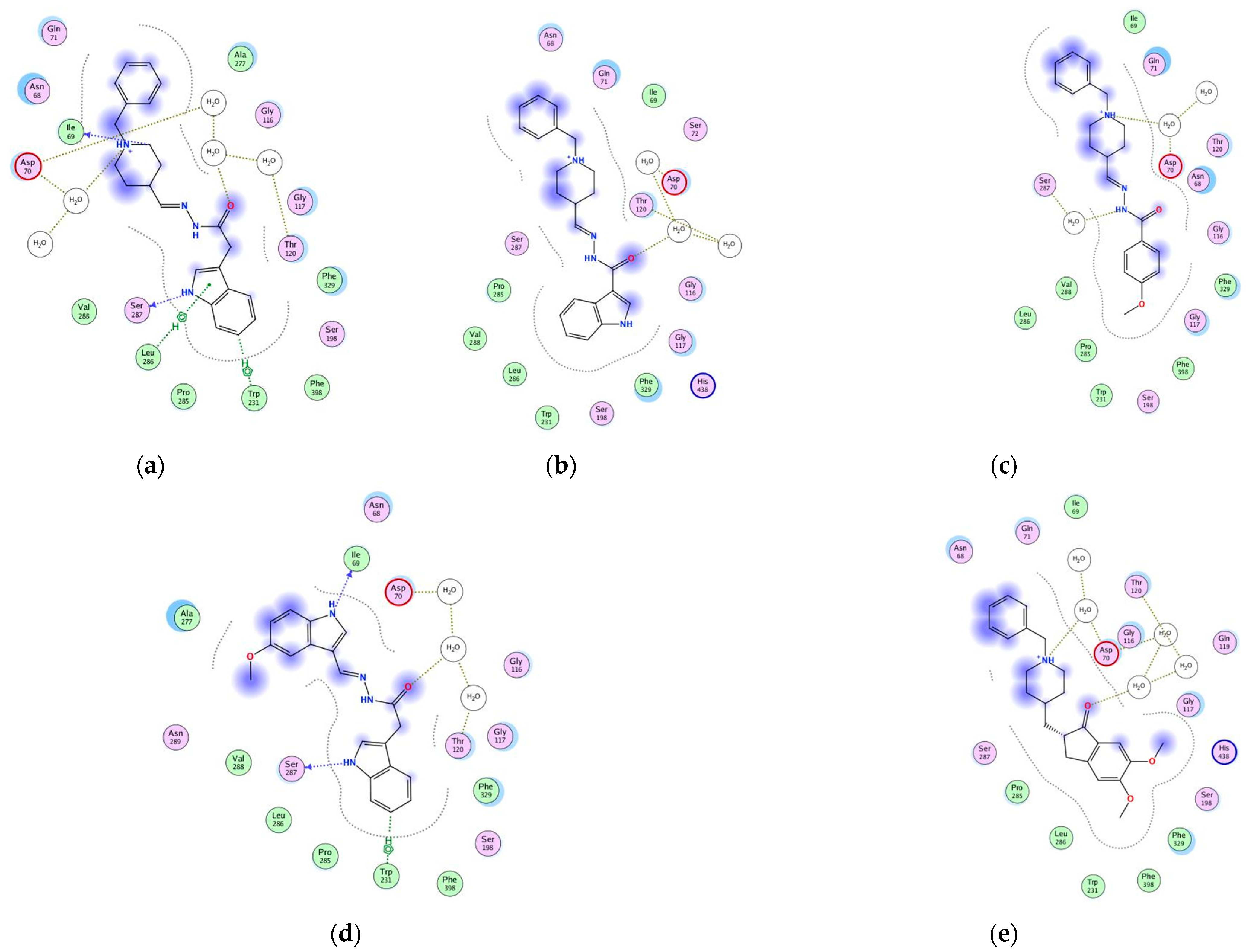
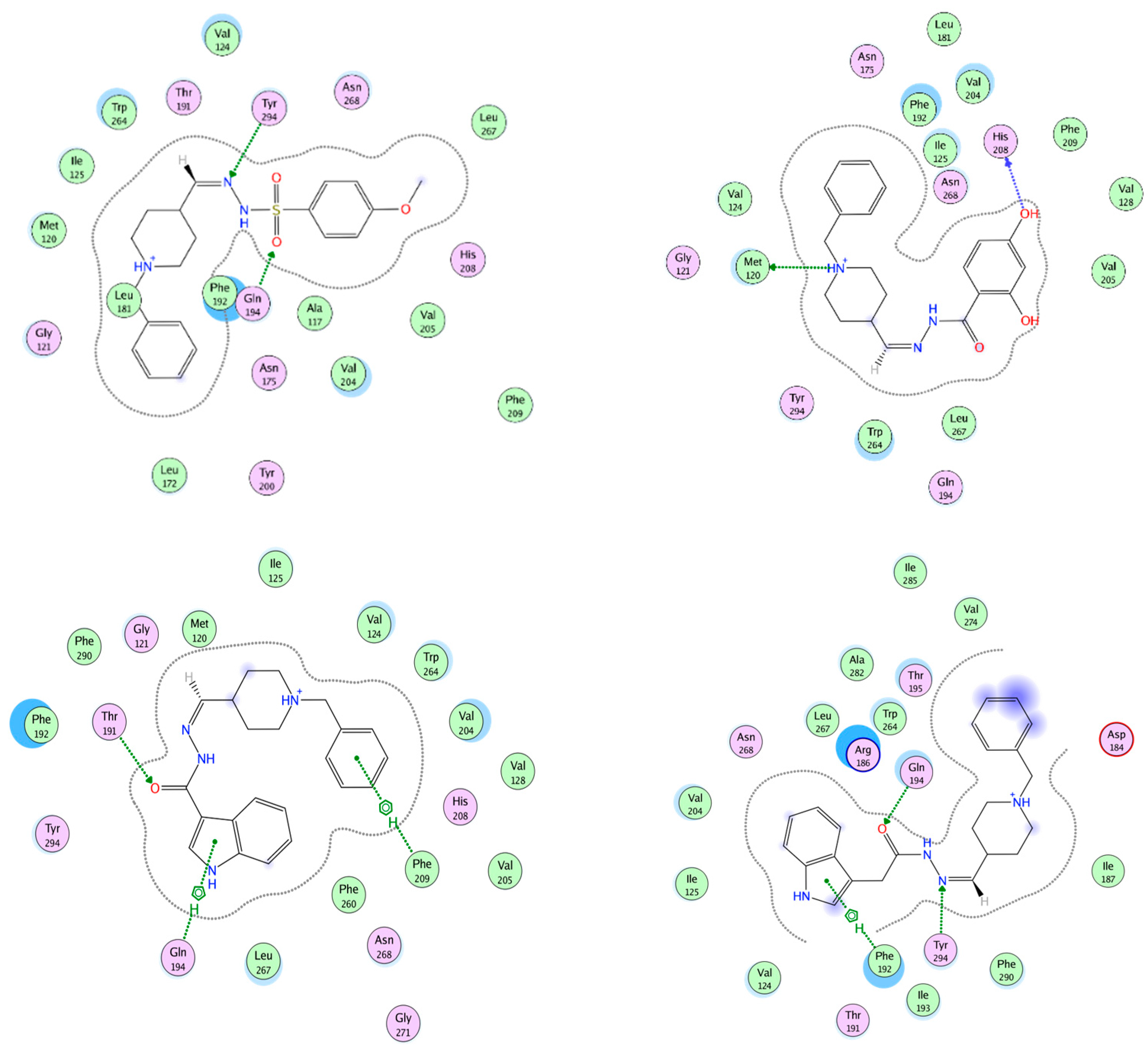
2.10. Molecular Docking of Human AChE and of Human BChE
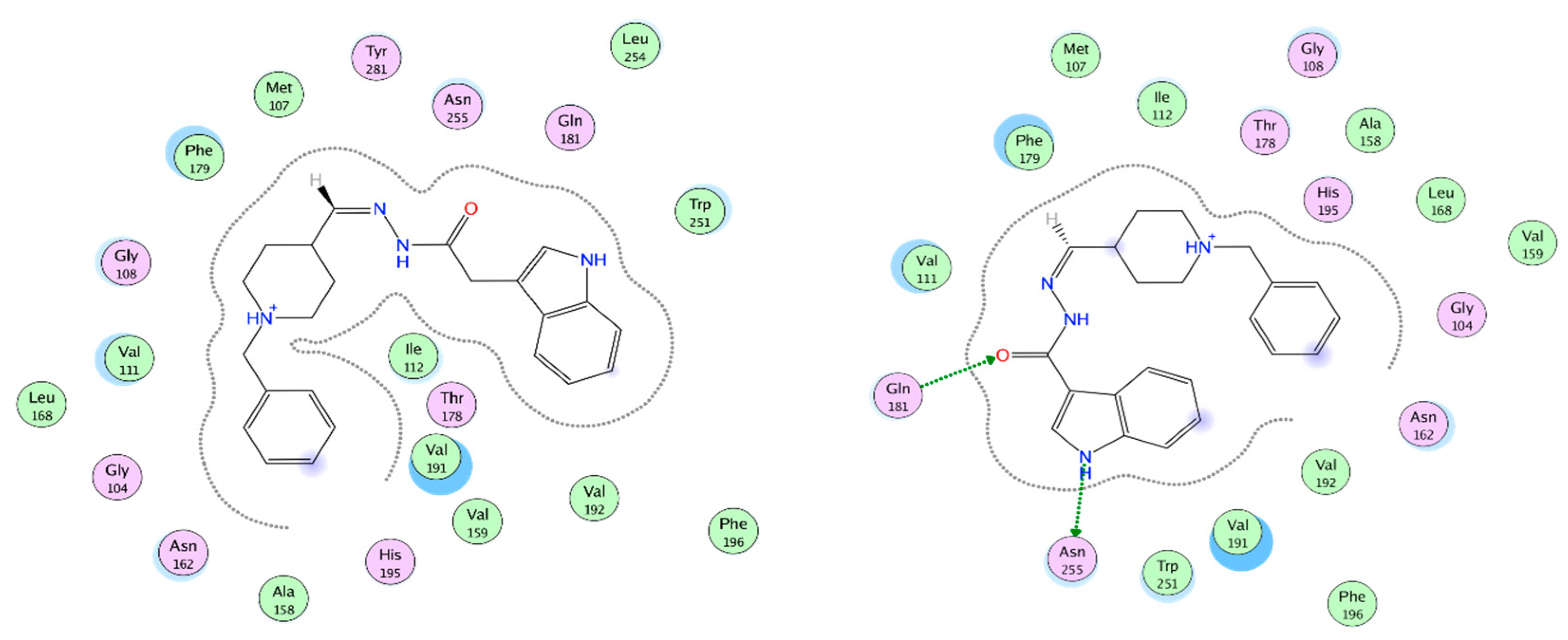
2.11. Docking of Ligand Set in MT1 and MT2 Receptors
2.12. MT1 Receptor Docking Results
2.13. MT2 Receptor Docking Results
2.14. In Silico ADME/Tox Study
2.15. Drug-Likeness Properties
2.16. In Silico Prediction of BBB Permeability
2.17. Predicted Toxicity
2.18. BBB Permeability by In Vitro PAMPA Test
3. Materials and Methods
3.1. Chemistry
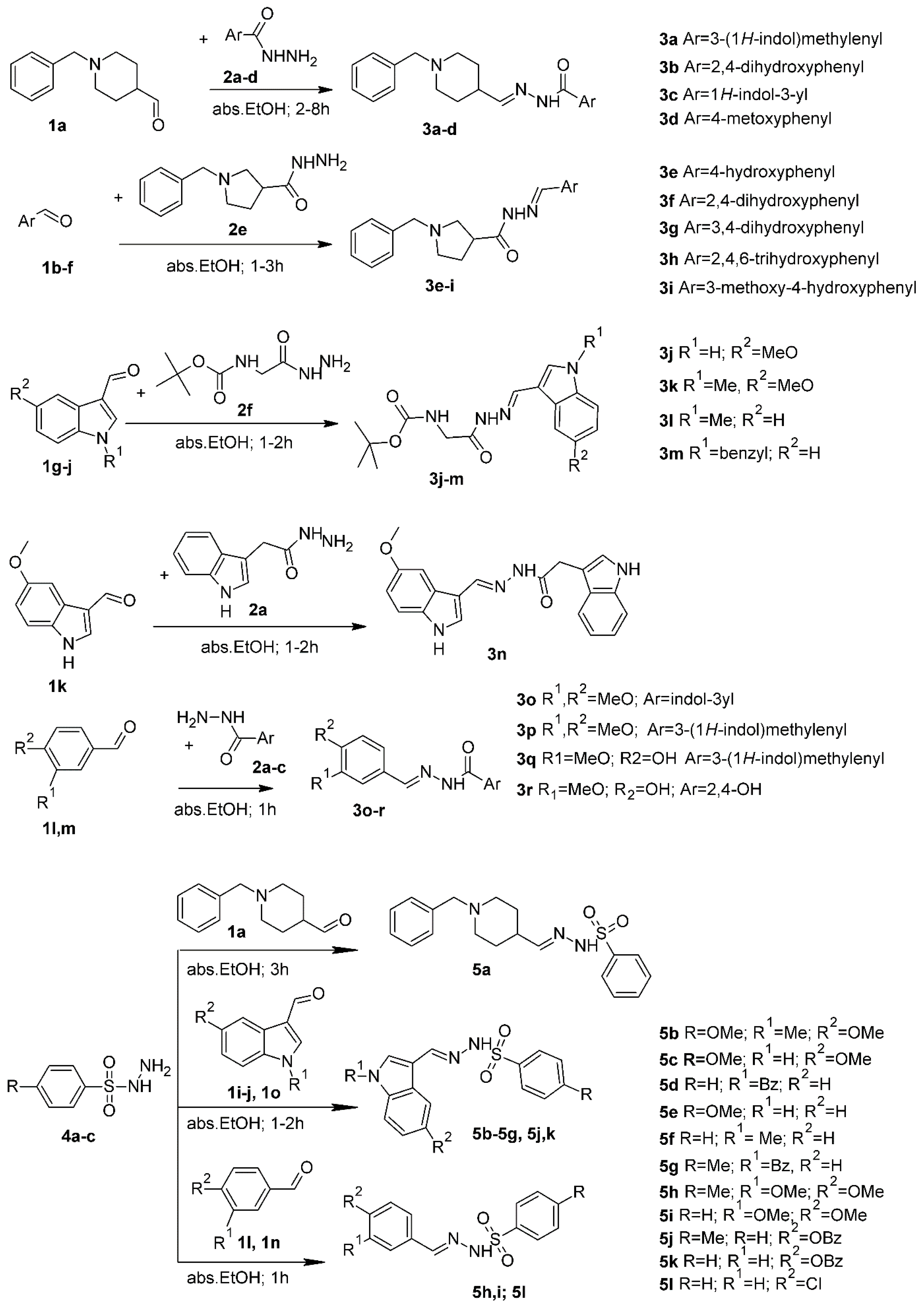
3.2. General Procedure for the Synthesis of Compounds 3a–r
3.3. General Procedure for the Synthesis of Compounds 5a–5l
3.4. Biological Evaluation
Assessment of AChE and BChE Inhibitory Activity
3.5. Cytotoxicity of the Compounds
3.5.1. Cell Lines and Culture Conditions
3.5.2. MTT Assay
3.6. Determination of Antioxidant Activity
3.6.1. DPPH Radical Scavenging Activity
3.6.2. ABTS Radical Scavenging Assay
3.6.3. Ferric Reducing/Antioxidant Power (FRAP)
3.6.4. Determination of Antioxidant Activity in Linoleic Acid System by the FTC Method
3.7. Model of H2O2-Induced Oxidative Stress in SH-SY5Y Cell Line
3.7.1. Model
3.7.2. Statistical Analysis
3.8. In Silico Studies
3.8.1. Molecular Docking of Human AChE and of Human BChE
- (1)
- AChE, complexed with 1-Benzyl-4-[(5,6-dimethoxy-1-indanon-2-yl)methyl]piperidine (E20) and co-factor 2-acetamido-2-deoxy-beta-D-glucopyranose (NAG), retrieved from Protein Data Bank (http://www.rcsb.org/) with PDB ID 4EY7;
- (2)
- BChE, complexed with butyl-[(2~(S))-1-(2-cycloheptylethylamino)-3-(1~(H)-indol-3-yl)-1-oxidanylidene-propan-2-yl]azanium (HUN) and NAD again (PDB ID 6QAA).
3.8.2. In Silico Docking Studies of MT1 and MT2 Receptors
3.8.3. ADME/Tox
3.8.4. In Silico Prediction of BBB Permeability
3.8.5. BBB Permeability by In Vitro PAMPA Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Multiregional workshop on the implementation of the global action plan on public health response to dementia. East. Mediterr. Health J. 2023, 29, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Wallace, L.; Kuhn, I.; Mytton, O.; Lafortune, L.; Wills, W.; Mukadam, N.; Brayne, C. Are Population-Level Approaches to Dementia Risk Reduction Under-Researched? A Rapid Review of the Dementia Prevention Literature. J. Prev. Alzheimer’s Dis. 2023, 1–8. [Google Scholar] [CrossRef]
- Sheppard, O.; Coleman, M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis; Exon Publications: Brisbane, QLD, Australia, 2020; pp. 1–21. [Google Scholar]
- Nasb, M.; Tao, W.; Chen, N. Alzheimer’s Disease Puzzle: Delving into Pathogenesis Hypotheses. Aging Dis. 2023, 15, 2. [Google Scholar]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Buccellato, F.R.; D’Anca, M.; Fenoglio, C.; Scarpini, E.; Galimberti, D. Role of oxidative damage in Alzheimer’s disease and neurodegeneration: From pathogenic mechanisms to biomarker discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef]
- Fernández-Bolaños, J.G.; López, Ó. Butyrylcholinesterase inhibitors as potential anti-Alzheimer’s agents: An updated patent review (2018–present). Expert Opin. Ther. Pat. 2022, 32, 913–932. [Google Scholar] [CrossRef]
- Meghana, G.; Gowda, D.; Chidambaram, S.B.; Osmani, R.A. Amyloid–β pathology in Alzheimer’s Disease: A Nano delivery Approach. Vib. Spectrosc. 2023, 126, 103510. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Inestrosa, N.C. Interactions of AChE with Aβ aggregates in Alzheimer’s brain: Therapeutic relevance of IDN 5706. Front. Mol. Neurosci. 2011, 4, 19. [Google Scholar] [CrossRef]
- Moss, D.E. Improving anti-neurodegenerative benefits of acetylcholinesterase inhibitors in Alzheimer’s disease: Are irreversible inhibitors the future? Int. J. Mol. Sci. 2020, 21, 3438. [Google Scholar] [CrossRef]
- Zeb, M.W.; Riaz, A.; Szigeti, K. Donepezil: A review of pharmacological characteristics and role in the management of Alzheimer disease. Clin. Med. Insights. Geriatr. 2017, 10, 1–14. [Google Scholar]
- Lee, C.-H.; Hung, S.-Y. Physiologic Functions and Therapeutic Applications of α7 Nicotinic Acetylcholine Receptor in Brain Disorders. Pharmaceutics 2022, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Jann, M.W. Rivastigmine, a new-generation cholinesterase inhibitor for the treatment of Alzheimer’s disease. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2000, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- de Freitas Silva, M.; Dias, K.S.; Gontijo, V.S.; Ortiz, C.J.C.; Viegas, C., Jr. Multi-target directed drugs as a modern approach for drug design towards Alzheimer’s disease: An update. Curr. Med. Chem. 2018, 25, 3491–3525. [Google Scholar] [CrossRef] [PubMed]
- Papagiouvannis, G.; Theodosis-Nobelos, P.; Kourounakis, P.N.; Rekka, E.A. Multi-target directed compounds with antioxidant and/or anti-inflammatory properties as potent agents for alzheimer’s disease. Med. Chem. 2021, 17, 1086–1103. [Google Scholar] [CrossRef]
- Grosjean, S.; Pachón-Angona, I.; Dawra, M.; Refouvelet, B.; Ismaili, L. Multicomponent reactions as a privileged tool for multitarget-directed ligand strategies in Alzheimer’s disease therapy. Future Med. Chem. 2022, 14, 1583–1606. [Google Scholar] [CrossRef]
- Ramalakshmi, N.; RS, R.; CN, N. Multitarget directed ligand approaches for Alzheimer’s disease: A Comprehensive Review. Mini Rev. Med. Chem. 2021, 21, 2361–2388. [Google Scholar] [CrossRef]
- Eissa, K.I.; Kamel, M.M.; Mohamed, L.W.; Kassab, A.E. Development of new Alzheimer’s disease drug candidates using donepezil as a key model. Arch. Der Pharm. 2023, 356, 2200398. [Google Scholar] [CrossRef]
- Gulcan, H.O.; Kosar, M. The hybrid compounds as multi-target ligands for the treatment of Alzheimer’s disease: Considerations on donepezil. Curr. Top. Med. Chem. 2022, 22, 395–407. [Google Scholar] [CrossRef]
- Luo, Z.; Sheng, J.; Sun, Y.; Lu, C.; Yan, J.; Liu, A.; Luo, H.-b.; Huang, L.; Li, X. Synthesis and evaluation of multi-target-directed ligands against Alzheimer’s disease based on the fusion of donepezil and ebselen. J. Med. Chem. 2013, 56, 9089–9099. [Google Scholar] [CrossRef]
- Pravin, N.; Jozwiak, K. Effects of linkers and substitutions on multitarget directed ligands for Alzheimer’s diseases: Emerging paradigms and strategies. Int. J. Mol. Sci. 2022, 23, 6085. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin regulates Aβ production/clearance balance and Aβ neurotoxicity: A potential therapeutic molecule for Alzheimer’s disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; Egea, J.; de Los Ríos, C.; Marco-Contelles, J.; Romero, A. Melatonin as a versatile molecule to design novel multitarget hybrids against neurodegeneration. Future Med. Chem. 2017, 9, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Singh, S.; Rizvi, S.I. Therapeutic potential of melatonin and its derivatives in aging and neurodegenerative diseases. Biogerontology 2023, 24, 183–206. [Google Scholar] [CrossRef]
- Brunner, P.; Sozer Topcular, N.; Jockers, R.; Ravid, R.; Angeloni, D.; Fraschini, F.; Eckert, A.; Muller Spahn, F.; Savaskan, E. Pineal and cortical melatonin receptors MT1 and MT2 are decreased in Alzheimer’s disease. Eur. J. Histochem. 2006, 50, 311–316. [Google Scholar] [PubMed]
- Halder, A.K.; Mitra, S.; Cordeiro, M.N.D. Designing multi-target drugs for the treatment of major depressive disorder. Expert Opin. Drug Discov. 2023, 18, 643–658. [Google Scholar] [CrossRef]
- Anastassova, N.; Stefanova, D.; Hristova-Avakumova, N.; Georgieva, I.; Kondeva-Burdina, M.; Rangelov, M.; Todorova, N.; Tzoneva, R.; Yancheva, D. New Indole-3-Propionic Acid and 5-Methoxy-Indole Carboxylic Acid Derived Hydrazone Hybrids as Multifunctional Neuroprotectors. Antioxidants 2023, 12, 977. [Google Scholar] [CrossRef]
- Aslanhan, Ö.; Kalay, E.; Tokalı, F.S.; Can, Z.; Şahin, E. Design, synthesis, antioxidant and anticholinesterase activities of novel isonicotinic hydrazide-hydrazone derivatives. J. Mol. Struct. 2023, 1279, 135037. [Google Scholar] [CrossRef]
- Bozbey, İ.; Özdemir, Z.; Uslu, H.; Özçelik, A.B.; Şenol, F.S.; Orhan, İ.E.; Uysal, M. A series of new hydrazone derivatives: Synthesis, molecular docking and anticholinesterase activity studies. Mini Rev. Med. Chem. 2020, 20, 1042–1060. [Google Scholar] [CrossRef]
- Demurtas, M.; Baldisserotto, A.; Lampronti, I.; Moi, D.; Balboni, G.; Pacifico, S.; Vertuani, S.; Manfredini, S.; Onnis, V. Indole derivatives as multifunctional drugs: Synthesis and evaluation of antioxidant, photoprotective and antiproliferative activity of indole hydrazones. Bioorg. Chem. 2019, 85, 568–576. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Ivanova, N.; Nenchovska, Z.; Tzoneva, R.; Stoyanova, T.; Uzunova, V.; Surcheva, S.; Tzonev, A.; Angelova, V.T.; Andreeva-Gateva, P. Evaluation of neurobiological and antioxidant effects of novel melatonin analogs in mice. Saudi Pharm. J. 2020, 28, 1566–1579. [Google Scholar] [CrossRef] [PubMed]
- Angelova, V.T.; Pencheva, T.; Vassilev, N.; Simeonova, R.; Momekov, G.; Valcheva, V. New indole and indazole derivatives as potential antimycobacterial agents. Med. Chem. Res. 2019, 28, 485–497. [Google Scholar] [CrossRef]
- Martins, F.; Santos, S.; Ventura, C.; Elvas-Leitão, R.; Santos, L.; Vitorino, S.; Reis, M.; Miranda, V.; Correia, H.F.; Aires-de-Sousa, J. Design, synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Eur. J. Med. Chem. 2014, 81, 119–138. [Google Scholar] [CrossRef]
- Oliveira, P.F.; Guidetti, B.; Chamayou, A.; André-Barrès, C.; Madacki, J.; Korduláková, J.; Mori, G.; Orena, B.S.; Chiarelli, L.R.; Pasca, M.R. Mechanochemical synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Molecules 2017, 22, 1457. [Google Scholar] [CrossRef] [PubMed]
- López, S.; Bastida, J.; Viladomat, F.; Codina, C. Acetylcholinesterase inhibitory activity of some Amaryllidaceae alkaloids and Narcissus extracts. Life Sci. 2002, 71, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Liu, D.; Li, J.; Feng, Y.; Lu, Y.; Yin, G.; Li, Z.; Shi, T.; Wang, Z. Carbamate-based N-Substituted tryptamine derivatives as novel pleiotropic molecules for Alzheimer’s disease. Bioorg. Chem. 2022, 125, 105844. [Google Scholar] [CrossRef]
- Al-Mamary, M.; Al-Habori, M.; Al-Zubairi, A.S. The in vitro antioxidant activity of different types of palm dates (Phoenix dactylifera) syrups. Arab. J. Chem. 2014, 7, 964–971. [Google Scholar] [CrossRef]
- Alam, M.K.; Rana, Z.H.; Islam, S.N.; Akhtaruzzaman, M. Total phenolic content and antioxidant activity of methanolic extract of selected wild leafy vegetables grown in Bangladesh: A cheapest source of antioxidants. Potravinarstvo 2019, 13, 287–293. [Google Scholar] [CrossRef]
- Dai, S.; Yu, C.; Liang, M.; Cheng, H.; Li, W.; Lai, F.; Ma, L.; Liu, X. Oxidation characteristics and thermal stability of Butylated hydroxytoluene. Arab. J. Chem. 2023, 16, 104932. [Google Scholar] [CrossRef]
- Hoffmann, L.F.; Martins, A.; Majolo, F.; Contini, V.; Laufer, S.; Goettert, M.I. Neural regeneration research model to be explored: SH-SY5Y human neuroblastoma cells. Neural Regen. Res. 2023, 18, 1265. [Google Scholar] [PubMed]
- Lopez-Suarez, L.; Al Awabdh, S.; Coumoul, X.; Chauvet, C. The SH-SY5Y human neuroblastoma cell line, a relevant in vitro cell model for investigating neurotoxicology in human: Focus on organic pollutants. Neurotoxicology 2022, 92, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Z.-M.; Li, X.-M.; Li, F.; Wu, J.-J.; Kong, L.-Y.; Wang, X.-B. Synthesis and evaluation of multi-target-directed ligands for the treatment of Alzheimer’s disease based on the fusion of donepezil and melatonin. Bioorg. Med. Chem. 2016, 24, 4324–4338. [Google Scholar] [CrossRef] [PubMed]
- Alov, P.; Stoimenov, H.; Lessigiarska, I.; Pencheva, T.; Tzvetkov, N.T.; Pajeva, I.; Tsakovska, I. In Silico Identification of Multi-Target Ligands as Promising Hit Compounds for Neurodegenerative Diseases Drug Development. Int. J. Mol. Sci. 2022, 23, 13650. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Elsevier: Amsterdam, The Netherlands, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Wang, Q.; Lu, Q.; Guo, Q.; Teng, M.; Gong, Q.; Li, X.; Du, Y.; Liu, Z.; Tao, Y. Structural basis of the ligand binding and signaling mechanism of melatonin receptors. Nat. Commun. 2022, 13, 454. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.H.; Miyauchi, H.; Inoue, A.; Raimondi, F.; Tsujimoto, H.; Kusakizako, T.; Shihoya, W.; Yamashita, K.; Suno, R.; Nomura, N. Cryo-EM structure of the human MT1–Gi signaling complex. Nat. Struct. Mol. Biol. 2021, 28, 694–701. [Google Scholar] [CrossRef]
- Clement, N.; Renault, N.; Guillaume, J.L.; Cecon, E.; Journé, A.S.; Laurent, X.; Tadagaki, K.; Cogé, F.; Gohier, A.; Delagrange, P. Importance of the second extracellular loop for melatonin MT1 receptor function and absence of melatonin binding in GPR50. Br. J. Pharmacol. 2018, 175, 3281–3297. [Google Scholar] [CrossRef]
- Stauch, B.; Johansson, L.C.; McCorvy, J.D.; Patel, N.; Han, G.W.; Huang, X.-P.; Gati, C.; Batyuk, A.; Slocum, S.T.; Ishchenko, A. Structural basis of ligand recognition at the human MT1 melatonin receptor. Nature 2019, 569, 284–288. [Google Scholar] [CrossRef]
- Herrera-Arozamena, C.; Estrada-Valencia, M.; Perez, C.; Lagartera, L.; Morales-Garcia, J.A.; Perez-Castillo, A.; Franco-Gonzalez, J.F.; Michalska, P.; Duarte, P.; Leon, R. Tuning melatonin receptor subtype selectivity in oxadiazolone-based analogues: Discovery of QR2 ligands and NRF2 activators with neurogenic properties. Eur. J. Med. Chem. 2020, 190, 112090. [Google Scholar] [CrossRef]
- Boutin, J.A.; Witt-Enderby, P.A.; Sotriffer, C.; Zlotos, D.P. Melatonin receptor ligands: A pharmaco-chemical perspective. J. Pineal Res. 2020, 69, e12672. [Google Scholar] [CrossRef] [PubMed]
- Pala, D.; Lodola, A.; Bedini, A.; Spadoni, G.; Rivara, S. Homology models of melatonin receptors: Challenges and recent advances. Int. J. Mol. Sci. 2013, 14, 8093–8121. [Google Scholar] [CrossRef] [PubMed]
- Farce, A.; Chugunov, A.O.; Logé, C.; Sabaouni, A.; Yous, S.; Dilly, S.; Renault, N.; Vergoten, G.; Efremov, R.G.; Lesieur, D. Homology modeling of MT1 and MT2 receptors. Eur. J. Med. Chem. 2008, 43, 1926–1944. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, J.; Nikolic, K.; Dobričić, V.; Agbaba, D. Prediction of blood–brain barrier permeation of α-adrenergic and imidazoline receptor ligands using PAMPA technique and quantitative-structure permeability relationship analysis. Eur. J. Pharm. Sci. 2015, 68, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Artursson, P.; Avdeef, A.; Ecker, G.F.; Faller, B.; Fischer, H.; Houston, J.B.; Kansy, M.; Kerns, E.H.; Krämer, S.D. Evidence-based approach to assess passive diffusion and carrier-mediated drug transport. Drug Discov. Today 2012, 17, 905–912. [Google Scholar] [CrossRef]
- Reis, M.; Sinko, B.; HR Serra, C. Parallel artificial membrane permeability assay (PAMPA)-Is it better than Caco-2 for human passive permeability prediction? Mini Rev. Med. Chem. 2010, 10, 1071–1076. [Google Scholar] [CrossRef]
- van de Waterbeemd, H.; Camenisch, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef]
- Didziapetris, R.; Japertas, P.; Avdeef, A.; Petrauskas, A. Classification analysis of P-glycoprotein substrate specificity. J. Drug Target. 2003, 11, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A’rule of three’for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Karabeliov, V.R.; Kondeva-Burdina, M.S.; Vassilev, N.G.; Elena, K.; Angelova, V.T. Neuroprotective evaluation of novel substituted 1, 3, 4-oxadiazole and aroylhydrazone derivatives. Bioorg. Med. Chem. Lett. 2022, 59, 128516. [Google Scholar] [CrossRef] [PubMed]
- Angelova, V.T.; Tatarova, T.; Mihaylova, R.; Vassilev, N.; Petrov, B.; Zhivkova, Z.; Doytchinova, I. Novel Arylsulfonylhydrazones as Breast Anticancer Agents Discovered by Quantitative Structure-Activity Relationships. Molecules 2023, 28, 2058. [Google Scholar] [CrossRef] [PubMed]
- Angelova, V.T.; Pencheva, T.; Vassilev, N.; K-Yovkova, E.; Mihaylova, R.; Petrov, B.; Valcheva, V. Development of new antimycobacterial sulfonyl hydrazones and 4-methyl-1, 2, 3-thiadiazole-based hydrazone derivatives. Antibiotics 2022, 11, 562. [Google Scholar] [CrossRef]
- Ellman, G.; Coutney Jr, K.V. Anders and RM Featherstone. Biochem. Pharmacol. 1961, 7, 85–95. [Google Scholar]
- Blois, M.S. Antioxidant determinations by the use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Grochowski, D.M.; Uysal, S.; Aktumsek, A.; Granica, S.; Zengin, G.; Ceylan, R.; Locatelli, M.; Tomczyk, M. In vitro enzyme inhibitory properties, antioxidant activities, and phytochemical profile of Potentilla thuringiaca. Phytochem. Lett. 2017, 20, 365–372. [Google Scholar] [CrossRef]
- Arnao, M.B.; Cano, A.; Acosta, M. The hydrophilic and lipophilic contribution to total antioxidant activity. Food Chem. 2001, 73, 239–244. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Jaworek, A.K.; Szepietowski, J.C.; Hałubiec, P.; Wojas-Pelc, A.; Jaworek, J. Melatonin as an antioxidant and immunomodulator in atopic dermatitis—A new look on an old story: A Review. Antioxidants 2021, 10, 1179. [Google Scholar] [CrossRef] [PubMed]
- Un, H.; Ugan, R.A.; Kose, D.; Yayla, M.; Tastan, T.B.; Bayir, Y.; Halici, Z. A new approach to sepsis treatment by rasagiline: A molecular, biochemical and histopathological study. Mol. Biol. Rep. 2022, 49, 3875–3883. [Google Scholar] [CrossRef]
- Aktar, B.S.K.; Sıcak, Y.; Tatar, G.; Oruç-Emre, E.E. Synthesis, Antioxidant and Some Enzyme Inhibition Activities of New Sulfonyl Hydrazones and their Molecular Docking Simulations. Pharm. Chem. J. 2022, 56, 559–569. [Google Scholar] [CrossRef]
- Doytchinova, I.; Atanasova, M.; Valkova, I.; Stavrakov, G.; Philipova, I.; Zhivkova, Z.; Zheleva-Dimitrova, D.; Konstantinov, S.; Dimitrov, I. Novel hits for acetylcholinesterase inhibition derived by docking-based screening on ZINC database. J. Enzym. Inhib. Med. Chem. 2018, 33, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood–brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Formula | Activity a [%] | AChE IC50 [μM] | Activity b [%] | BChE IC50 [μM] | SI c AChE | SI d BChE |
|---|---|---|---|---|---|---|---|
| 3a |  | 78.93 | 76.51 ± 3.04 | 80.43 | 56.50 ± 0.20 | - | 1.35 |
| 3b |  | 89.27 | 19.84 ± 0.95 | 75.94 | 262.40 ± 8.75 | 13.22 | - |
| 3c |  | 72.02 | 10.76 ± 1.66 | 76.95 | 26.32 ± 3.11 | 2.45 | - |
| 3d |  | 81.16 | 9.77 ± 0.76 | 72.88 | 197.27 ± 18.65 | 20.19 | |
| 3e |  | 3.84 | >1000 | 12.44 | >1000 | - | - |
| 3f |  | 64.07 | 549.73 ± 37.08 | 57.16 | 708.73 ± 41.32 | 1.28 | - |
| 3g |  | 2.74 | >1000 | 28.19 | >1000 | - | - |
| 3h |  | 73.31 | 351.37 ± 17.93 | 76.49 | 178.60 ± 11.54 | - | 1.96 |
| 3i |  | 20.64 | >1000 | 17.57 ± 2.45 | >1000 | - | - |
| 3j |  | na | >1000 | 17.39 | >1000 | - | - |
| 3k |  | 3.26 | >1000 | 30.28 | >1000 | - | - |
| 3l |  | na | >1000 | 39.09 | >1000 | - | - |
| 3m |  | 2.01 | >1000 | 59.40 | 326.90 ± 34.35 | - | 3.05 |
| 3n |  | 29.24 | >1000 | 73.65 | 21.12 ± 1.48 | - | 47.34 |
| 3o |  | na | >1000 | na | >1000 | - | - |
| 3p |  | na | >1000 | 23.96 | >1000 | - | - |
| 3q |  | 12.85 | >1000 | 13.90 | >1000 | - | - |
| 3r |  | na | >1000 | 16.89 | >1000 | - | - |
| Donepezil |  | 65.64 | 0.20 ± 0.04 | 69.14 | 1.83 ± 0.35 | 9.15 | - |
| Galanthamine |  | 85.22 | 1.48 ± 0.11 | 85.83 | 29.55 ± 0.96 | 16.5 | - |
| Melatonin |  | 1.24 | >1000 | 4.21 ± 0.88 | >1000 | - | - |
| Compd. | Formula | Activity a [%] | AChE IC50 [μM] | Activity b [%] | BChE IC50 [μM] | SI c AChE | SI d BChE |
|---|---|---|---|---|---|---|---|
| 5a |  | 76.99 | 162.83 ± 10.73 | 79.44 | 259.20 ± 4.90 | 1.81 | - |
| 5b |  | na | >1000 | na | >1000 | - | - |
| 5c |  | na | >1000 | 18.21 | >1000 | - | - |
| 5d |  | 41.72 | >1000 | na | >1000 | - | - |
| 5e |  | na | >1000 | na | >1000 | - | - |
| 5f |  | na | >1000 | na | >1000 | - | - |
| 5g |  | 2.18 | >1000 | 40.65 | >1000 | - | - |
| 5h |  | 10.53 | >1000 | 26.65 ± 5.11 | >1000 | - | - |
| 5i |  | 2.63 | >1000 | 7.67 ± 0.99 | >1000 | - | - |
| 5j |  | 13.17 | >1000 | 56.92 | 865.40 ± 159.28 | - | 1.15 |
| 5k |  | 35.41 | >1000 | 4.97 | >1000 | - | - |
| 5l |  | na | >1000 | na | >1000 | - | - |
| Donepezil |  | 65.64 | 0.20 ± 0.04 | 69.14 | 1.83 ± 0.35 | 9.15 | - |
| Galanthamine |  | 85.22 | 1.48 ± 0.11 | 85.83 | 29.55 ± 0.96 | 16.5 | - |
| Melatonin |  | 1.24 | >1000 | 4.21 ± 0.88 | >1000 | - | - |
| Compd | Neuro-2a IC50 μM | SH-SY5Y IC50 μM | CCL-1 IC50 μM |
|---|---|---|---|
| 3a | >300 | 129 ± 7.5 | 138.5 ± 9.8 |
| 3b | >300 | 138.4 | 153.5 ± 7.3 |
| 3c | >300 | 97.3 | 167.8 ± 10.4 |
| 3d | >300 | 122.3 ± 9.2 | 220 |
| 3e | >300 | 170.2 ± 10.6 | >300 |
| 3f | 12.5 ± 2.4 | 35.8 ± 5.5 | 150 ± 11.2 |
| 3g | 110.5 ± 9.1 | >300 | >300 |
| 3h | >300 | >300 | >300 |
| 3i | >300 | 178.1 ± 15.7 | >800 |
| 3j | >300 | 150.4 ± 12.3 | >300 |
| 3k | >300 | 130.8 ± 11.8 | >300 |
| 3l | >300 | 106.7 ± 8.9 | >300 |
| 3m | 108.7 ± 9.1 | 7.2 ± 1.8 | 65.4 ± 5.1 |
| 3n | 83.7 ± 8.3 | 107.8 ± 9.4 | 48.2 ± 6.4 |
| 3o | 125.8 ± 10.7 | 70.0 ± 7.2 | >300 |
| 3p | 115.5 ± 12.8 | 87.4 ± 7.7 | >300 |
| 3q | 254.5 ± 13.7 | 140.8 ± 8.8 | 170.2 ± 14.0 |
| 3r | >300 | 155.6 ± 11.1 | >300 |
| 5a | >300 | 142.0 ± 13.1 | 156.9 ± 8.5 |
| 5b | 30.5 ± 3.2 | 70.4 ± 4.9 | 38.1 ± 3.5 |
| 5c | 7.5 ± 2.4 | 48.3 ± 5.2 | 25.7 ± 2.9 |
| 5d | 117.4 ± 11.5 | 99.5 ± 7.8 | 140.3 ± 8.4 |
| 5e | 19.9 ± 1.7 | 83.3 ± 4.9 | 180 ± 9.6 |
| 5f | 134.9 ± 10.9 | 131.8 ± 10.3 | 167.4 ± 11.8 |
| 5g | 134.9 ± 10.9 | 92.1 ± 9.3 | 50.7 ± 3.6 |
| 5h | 135.5 ± 12.6 | 42.0 ± 5.2 | 159.6 |
| 5i | 109.2 ± 5.1 | 40.3 ± 6.1 | 147.3 |
| 5j | 65.4 ± 0.1 | 33.1 ± 0.1 | 41.2 ± 4.2 |
| 5k | 77.7 ± 6.9 | 134.7 ± 14.0 | 321.4 ± 16.2 |
| 5l | 117.4 ± 11.5 | 99.5 ± 7.8 | 131.4 ± 11.4 |
| Donepezil | 248.1 ± 15.4 | 79.3 ± 6.2 | 52.0 ± 5.3 |
| Galanthamine | >300 | >300 | >300 |
| Compd. | DPPH Activity | ABTS Activity | FRAP | |
|---|---|---|---|---|
| [%] | [%] | IC50 [mM] | μMTE | |
| 3a | 5.74 ± 0.39 | 27.44 ± 2.57 | 27.44 ± 2.57 | 15.42 ± 0.74 |
| 3b | 13.30 ± 0.83 | 93.22 ± 0.30 | 2.57 ± 0.10 | 16.63 ± 0.98 |
| 3c | 30.05 ± 0.55 | 36.69 ± 0.70 | >5 | 38.97 ± 3.58 |
| 3d | 40.80 ± 3.10 | ≥100 | 1.05 ± 0.04 | 15.87 ± 1.41 |
| 3i | 6.56 ± 0.78 | ≥100 | 2.37 ± 0.05 | 14.67 ± 1.14 |
| 3m | 32.06 ± 4.70 | 36.20 ± 1.40 | >5 | 19.06 ± 1.96 |
| 3n | 40.62 ± 0.83 | ≥100 | 0.77 ± 0.06 | 14.30 ± 0.56 |
| 3q | 20.40 ± 0.83 | ≥100 | 2.38 ± 0.07 | 15.60 ± 0.90 |
| 3r | 45.17 ± 4.65 | 91.41 ± 0.47 | 2.50 ± 0.23 | 14.30 ± 0.56 |
| 5a | 6.56 ± 0.78 | 43.80 ± 0.23 | >5 | 17.48 ± 0.90 |
| 5g | 45.72 ± 1.38 | ≥100 | 2.08 ± 0.10 | 15.87 ± 1.13 |
| 5h | 16.94 ± 1.09 | ≥100 | 1.85 ± 0.03 | 20.37 ± 0.90 |
| 5i | 16.21 ± 3.01 | 65.29 ± 2.34 | 3.74 ± 0.05 | 14.67 ± 0.81 |
| 5j | 47.36 ± 2.75 | ≥100 | 2.18 ± 0.16 | 18.59 ± 2.28 |
| 5k | 47.18 ± 1.14 | ≥100 | 2.57 ± 0.13 | 22.52 ± 1.29 |
| Donepezil | na | na | >5 | 20.09 ± 0.16 |
| Melatonin | na | ≥100 | 0.50 ± 0.02 | 13.73 ± 0.84 |
| BHT | 7.78 ± 0.77 | 29.53 ± 1.65 | >5 | 15.79 ± 0.58 |
| Compd. | Absorbance | ||||
|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | |
| 3a | 0.87 ± 0.03 | 0.87 ± 0.08 | 0.72 ± 0.07 | 0.72 ± 0.05 | 0.71 ± 0.08 |
| 3b | 0.92 ± 0.06 | 0.93 ± 0.09 | 0.95 ± 0.05 | 0.96 ± 0.01 | 1.00 ± 0.02 |
| 3c | 0.89 ± 0.07 | 0.87 ± 0.02 | 0.85 ± 0.05 | 0.85 ± 0.07 | 0.74 ± 0.05 |
| 3d | 0.85 ± 0.02 | 0.86 ± 0.02 | 0.88 ± 0.05 | 0.85 ± 0.07 | 0.84 ± 0.09 |
| 3i | 0.80 ± 0.09 | 0.83 ± 0.04 | 0.08 ± 0.07 | 0.82 ± 0.03 | 0.84 ± 0.07 |
| 3m | 0.89 ± 0.09 | 0.89 ± 0.01 | 0.92 ± 0.05 | 0.95 ± 0.01 | 0.96 ± 0.08 |
| 3n | 0.85 ± 0.05 | 0.87 ± 0.07 | 0.89 ± 0.04 | 0.90 ± 0.01 | 0.92 ± 0.06 |
| 3q | 0.76 ± 0.03 | 0.77 ± 0.08 | 0.79 ± 0.03 | 0.84 ± 0.05 | 0.91 ± 0.09 |
| 3r | 0.80 ± 0.04 | 0.81 ± 0.01 | 0.82 ± 0.02 | 0.87 ± 0.09 | 0.86 ± 0.09 |
| 5a | 0.83 ± 0.02 | 0.88 ± 0.08 | 0.89 ± 0.04 | 1.00 ± 0.04 | 1.10 ± 0.05 |
| 5g | 0.82 ± 0.04 | 0.87 ± 0.09 | 0.88 ± 0.09 | 0.87 ± 0.05 | 0.88 ± 0.03 |
| 5h | 0.70 ± 0.09 | 0.82 ± 0.06 | 0.83 ± 0.05 | 0.83 ± 0.06 | 0.80 ± 0.05 |
| 5i | 0.81 ± 0.08 | 0.83 ± 0.01 | 0.83 ± 0.09 | 0.81 ± 0.02 | 0.83 ± 0.03 |
| 5j | 0.83 ± 0.01 | 0.83 ± 0.08 | 0.84 ± 0.02 | 0.83 ± 0.05 | 0.82 ± 0.02 |
| 5k | 0.80 ± 0.04 | 0.80 ± 0.09 | 0.83 ± 0.05 | 0.84 ± 0.08 | 0.87 ± 0.09 |
| Donepezil | 0.79 ± 0.08 | 0.82 ± 0.08 | 0.84 ± 0.04 | 1.19 ± 0.24 | 2.00 ± 0.18 |
| Melatonin | 0.75 ± 0.07 | 0.80 ± 0.09 | 0.89 ± 0.05 | 0.89 ± 0.02 | 0.94 ± 0.01 |
| BHT | 0.74 ± 0.07 | 0.77 ± 0.03 | 0.78 ± 0.06 | 0.80 ± 0.06 | 0.81 ± 0.09 |
| Control | 0.83 ± 0.08 | 0.84 ± 0.07 | 0.85 ± 0.03 | 0.92 ± 0.02 | 1.02 ± 0.2 |
| Compd. | MW | Heavy Atoms | Arom. Heavy Atoms | RB | HBA | HBD | TPSA | MR | WLOGP | ESOL Class | BBB Perm. | Pgp Subs | GIA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 374.48 | 28 | 15 | 7 | 3 | 2 | 60.49 | 117.59 | 3.19 | Moderately soluble | Yes | Yes | High |
| 3b | 353.41 | 26 | 12 | 6 | 5 | 3 | 85.16 | 105.2 | 2.19 | Soluble | No | Yes | High |
| 3c | 360.45 | 27 | 15 | 6 | 3 | 2 | 60.49 | 113.01 | 3.26 | Moderately soluble | Yes | Yes | High |
| 3d | 351.44 | 26 | 12 | 7 | 4 | 1 | 53.93 | 107.64 | 2.79 | Soluble | Yes | No | High |
| 3e | 323.39 | 24 | 12 | 6 | 4 | 2 | 64.93 | 98.14 | 1.83 | Soluble | Yes | Yes | High |
| 3f | 339.39 | 25 | 12 | 6 | 5 | 3 | 85.16 | 100.17 | 1.54 | Soluble | No | Yes | High |
| 3g | 339.39 | 25 | 12 | 6 | 5 | 3 | 85.16 | 100.17 | 1.46 | Soluble | No | Yes | High |
| 3h | 355.39 | 26 | 12 | 6 | 6 | 4 | 105.39 | 102.19 | 1.24 | Soluble | No | Yes | High |
| 3i | 353.41 | 26 | 12 | 7 | 5 | 2 | 74.16 | 104.64 | 1.84 | Soluble | Yes | Yes | High |
| 3j | 346.38 | 25 | 9 | 9 | 5 | 3 | 104.81 | 94.61 | 2.15 | Soluble | No | Yes | High |
| 3k | 360.41 | 26 | 9 | 9 | 5 | 2 | 99.51 | 93.95 | 2.16 | Soluble | No | Yes | High |
| 3l | 330.38 | 24 | 9 | 8 | 4 | 2 | 84.72 | 93.02 | 2.15 | Soluble | No | No | High |
| 3m | 406.48 | 30 | 15 | 10 | 4 | 2 | 84.72 | 117.5 | 3.66 | Moderately soluble | No | No | High |
| 3n | 346.38 | 26 | 18 | 6 | 3 | 3 | 82.27 | 102.4 | 3.35 | Soluble | No | Yes | High |
| 3o | 323.35 | 24 | 15 | 6 | 4 | 2 | 75.71 | 92.45 | 2.95 | Soluble | Yes | No | High |
| 3p | 337.37 | 25 | 15 | 7 | 4 | 2 | 75.71 | 97.04 | 2.88 | Soluble | Yes | No | High |
| 3q | 323.35 | 24 | 15 | 6 | 4 | 3 | 86.71 | 92.57 | 2.57 | Soluble | No | No | High |
| 3r | 302.28 | 22 | 12 | 5 | 6 | 4 | 111.38 | 80.17 | 1.58 | Soluble | No | No | High |
| donepezil | 379.49 | 28 | 12 | 6 | 4 | 0 | 38.77 | 115.31 | 3.83 | Moderately soluble | Yes | Yes | High |
| melatonin | 232.28 | 17 | 9 | 5 | 2 | 2 | 54.12 | 67.18 | 1.86 | Soluble | Yes | No | High |
| Compd. | MW | Heavy Atoms | Arom. Heavy Atoms | RB | HBA | HBD | TPSA | MR | WLOGP | ESOL Class | BBB Perm. | Pgp Subs | GIA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5a | 357.47 | 25 | 12 | 6 | 4 | 1 | 70.15 | 104.05 | 3.41 | Soluble | Yes | No | High |
| 5b | 373.43 | 26 | 15 | 6 | 5 | 1 | 90.3 | 100.25 | 3.59 | Soluble | No | No | High |
| 5c | 359.4 | 25 | 15 | 6 | 5 | 2 | 101.16 | 95.35 | 3.53 | Soluble | No | No | High |
| 5d | 389.47 | 28 | 21 | 6 | 3 | 1 | 71.84 | 111.75 | 5.08 | Moderately soluble | No | No | High |
| 5e | 343.4 | 24 | 15 | 5 | 4 | 1 | 81.07 | 93.76 | 3.58 | Soluble | No | No | High |
| 5f | 313.37 | 22 | 15 | 4 | 3 | 1 | 71.84 | 87.27 | 3.57 | Soluble | Yes | No | High |
| 5g | 403.5 | 29 | 21 | 6 | 3 | 1 | 71.84 | 116.72 | 5.39 | Moderately soluble | No | No | High |
| 5h | 334.39 | 23 | 12 | 6 | 5 | 1 | 85.37 | 88.46 | 3.41 | Soluble | No | No | High |
| 5i | 320.36 | 22 | 12 | 6 | 5 | 1 | 85.37 | 83.49 | 3.1 | Soluble | No | No | High |
| 5j | 419.5 | 30 | 21 | 7 | 4 | 2 | 91.93 | 118.31 | 5.3 | Moderately soluble | No | No | High |
| 5k | 405.47 | 29 | 21 | 7 | 4 | 2 | 91.93 | 113.35 | 4.99 | Moderately soluble | No | No | High |
| 5l | 294.76 | 19 | 12 | 4 | 3 | 1 | 66.91 | 75.52 | 3.73 | Soluble | Yes | No | High |
| donepezil | 379.49 | 28 | 12 | 6 | 4 | 0 | 38.77 | 115.31 | 3.83 | Moderately soluble | Yes | Yes | High |
| melatonin | 232.28 | 17 | 9 | 5 | 2 | 2 | 54.12 | 67.18 | 1.86 | Soluble | Yes | No | High |
| Comp. | LD50 (mg/kg) (Pred) | Toxicity Class (Pred) | Octanol/ Water Partition coeff. (logP) | Organ Toxicity (Hepatotoxicity) | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pred | Prob | Pred | Prob | Pred | Prob | Pred | Prob | Pred | Prob | ||||
| 3a | 225 | 3 | 4.05 | I | 0.64 | A | 0.65 | I | 0.99 | I | 0.59 | I | 0.56 |
| 3b | 1000 | 4 | 3.05 | I | 0.63 | A | 0.57 | I | 0.86 | I | 0.57 | I | 0.67 |
| 3c | 370 | 4 | 4.12 | I | 0.6 | A | 0.63 | I | 0.99 | I | 0.54 | I | 0.56 |
| 3d | 2000 | 4 | 3.65 | I | 0.69 | A | 0.63 | I | 0.88 | I | 0.57 | I | 0.63 |
| 3e | 1962 | 4 | 2.69 | I | 0.62 | A | 0.62 | I | 0.98 | I | 0.55 | I | 0.66 |
| 3f | 1962 | 4 | 2.64 | I | 0.57 | A | 0.54 | I | 0.74 | I | 0.58 | I | 0.65 |
| 3g | 1962 | 4 | 2.4 | I | 0.59 | A | 0.56 | I | 0.71 | I | 0.59 | I | 0.6 |
| 3h | 1962 | 4 | 2.1 | I | 0.57 | A | 0.52 | I | 0.97 | I | 0.59 | I | 0.66 |
| 3i | 1250 | 4 | 2.7 | I | 0.59 | A | 0.57 | A | 0.79 | I | 0.59 | I | 0.6 |
| 3j | 1120 | 4 | 2.93 | I | 0.5 | I | 0.52 | A | 0.5 | I | 0.58 | I | 0.61 |
| 3k | 1120 | 4 | 2.94 | I | 0.53 | I | 0.54 | A | 0.64 | I | 0.57 | I | 0.55 |
| 3l | 498 | 4 | 2.93 | I | 0.53 | I | 0.53 | I | 0.96 | I | 0.59 | I | 0.58 |
| 3m | 498 | 4 | 4.45 | I | 0.52 | I | 0.54 | I | 0.98 | I | 0.58 | I | 0.55 |
| 3n | 900 | 4 | 3.74 | A | 0.58 | A | 0.61 | A | 0.81 | I | 0.6 | I | 0.69 |
| 3o | 4540 | 5 | 3.34 | A | 0.64 | A | 0.65 | A | 0.57 | A | 0.51 | I | 0.67 |
| 3p | 6000 | 6 | 3.27 | A | 0.57 | A | 0.67 | A | 0.5 | I | 0.54 | I | 0.64 |
| 3q | 6000 | 6 | 2.97 | A | 0.59 | A | 0.6 | A | 0.6 | I | 0.56 | I | 0.62 |
| 3r | 4920 | 5 | 1.97 | A | 0.6 | A | 0.63 | A | 0.94 | A | 0.5 | I | 0.74 |
| 5a | 1398 | 4 | 4.27 | I | 0.71 | A | 0.53 | I | 0.99 | I | 0.61 | I | 0.62 |
| 5b | 500 | 4 | 3.98 | A | 0.5 | A | 0.5 | I | 0.8 | I | 0.51 | I | 0.74 |
| 5c | 500 | 4 | 3.97 | I | 0.5 | A | 0.52 | I | 0.84 | I | 0.54 | I | 0.82 |
| 5d | 500 | 4 | 5.47 | I | 0.6 | A | 0.5 | I | 0.99 | I | 0.56 | I | 0.61 |
| 5e | 500 | 4 | 3.97 | I | 0.5 | I | 0.51 | I | 0.96 | I | 0.5 | I | 0.72 |
| 5f | 500 | 4 | 3.96 | I | 0.53 | A | 0.51 | I | 0.99 | I | 0.55 | I | 0.63 |
| 5g | 500 | 4 | 5.78 | I | 0.62 | A | 0.51 | I | 0.99 | I | 0.55 | I | 0.63 |
| 5h | 500 | 4 | 3.8 | I | 0.51 | A | 0.54 | I | 0.99 | I | 0.54 | I | 0.86 |
| 5i | 500 | 4 | 3.49 | I | 0.51 | A | 0.54 | I | 0.99 | I | 0.55 | I | 0.88 |
| 5j | 500 | 4 | 5.84 | I | 0.51 | I | 0.52 | I | 0.99 | I | 0.54 | I | 0.8 |
| 5k | 500 | 4 | 5.53 | I | 0.5 | I | 0.53 | I | 0.99 | I | 0.55 | I | 0.78 |
| 5l | 500 | 4 | 4.12 | I | 0.63 | I | 0.6 | I | 0.99 | I | 0.66 | I | 0.7 |
| donepezil | 505 | 4 | 4.3 | I | 0.98 | A | 0.5 | A | 0.95 | I | 0.53 | A | 0.63 |
| melatonin | 963 | 4 | 2.25 | I | 0.84 | I | 0.68 | I | 0.51 | I | 0.9 | I | 0.73 |
| Compd. | PAMPA BBB −logPe | logP | logD7.4 | pKa,MB | PSA, Å2 | FRB | HBD | HBA | Ro5 |
|---|---|---|---|---|---|---|---|---|---|
| 3a | 4.420 | 3.33 | 2.74 | 7.86 | 82.27 | 6 | 3 | 5 | 0 |
| 3b | 5.873 | 3.3 | 2.56 | 8.00 | 85.16 | 7 | 3 | 6 | 0 |
| 3c | 4.466 | 3.31 | 2.72 | 7.85 | 60.49 | 5 | 2 | 5 | 0 |
| 3m | 4.377 | 5.05 | 5.05 | - | 84.72 | 8 | 2 | 7 | 1 |
| 3n | 4.084 | 3.52 | 3.52 | 0.83 | 82.27 | 5 | 3 | 6 | 0 |
| 5a | 4.749 | 3.16 | 2.58 | 7.83 | 70.15 | 5 | 1 | 5 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelova, V.T.; Georgiev, B.; Pencheva, T.; Pajeva, I.; Rangelov, M.; Todorova, N.; Zheleva-Dimitrova, D.; Kalcheva-Yovkova, E.; Valkova, I.V.; Vassilev, N.; et al. Design, Synthesis, In Silico Studies and In Vitro Evaluation of New Indole- and/or Donepezil-like Hybrids as Multitarget-Directed Agents for Alzheimer’s Disease. Pharmaceuticals 2023, 16, 1194. https://doi.org/10.3390/ph16091194
Angelova VT, Georgiev B, Pencheva T, Pajeva I, Rangelov M, Todorova N, Zheleva-Dimitrova D, Kalcheva-Yovkova E, Valkova IV, Vassilev N, et al. Design, Synthesis, In Silico Studies and In Vitro Evaluation of New Indole- and/or Donepezil-like Hybrids as Multitarget-Directed Agents for Alzheimer’s Disease. Pharmaceuticals. 2023; 16(9):1194. https://doi.org/10.3390/ph16091194
Chicago/Turabian StyleAngelova, Violina T., Borislav Georgiev, Tania Pencheva, Ilza Pajeva, Miroslav Rangelov, Nadezhda Todorova, Dimitrina Zheleva-Dimitrova, Elena Kalcheva-Yovkova, Iva V. Valkova, Nikolay Vassilev, and et al. 2023. "Design, Synthesis, In Silico Studies and In Vitro Evaluation of New Indole- and/or Donepezil-like Hybrids as Multitarget-Directed Agents for Alzheimer’s Disease" Pharmaceuticals 16, no. 9: 1194. https://doi.org/10.3390/ph16091194