
Molecular and Structural Aspects of Clinically Relevant Mutations of SARS-CoV-2 RNA-Dependent RNA Polymerase in Remdesivir-Treated Patients
 , ,
, ,  , ,
, , 
 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Patients’ Characteristics
2.2. RdRp Mutational Profiles and Response to Remdesivir
2.3. Molecular Modeling
2.3.1. Molecular Docking and Thermodynamics Analysis
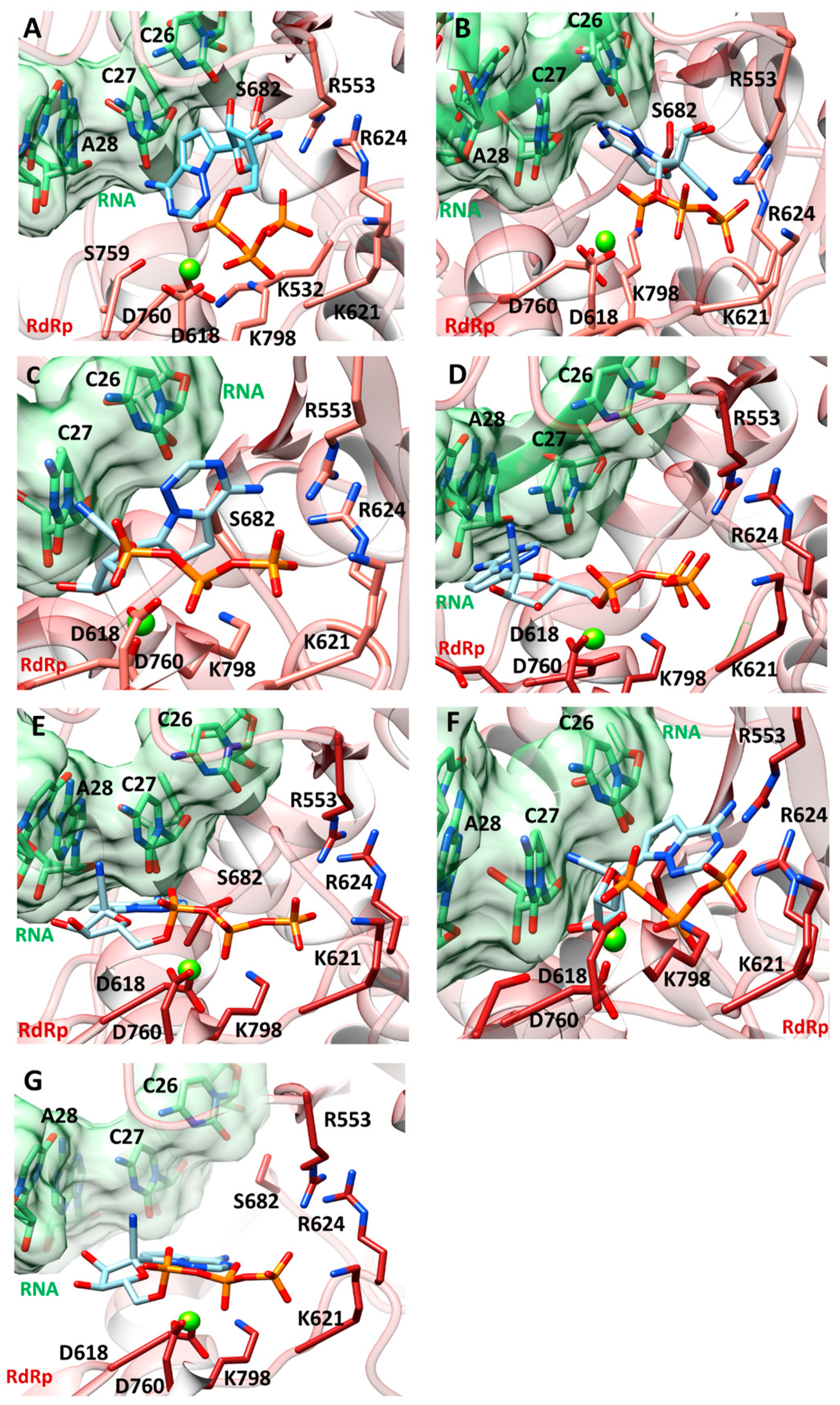
2.3.2. Structural Effects on the Global Motions of the 3M2 and 5M Mutants
2.3.3. Representative Pharmacophore Models (RPM) of Remdesivir Extracted during MDs
3. Discussion
4. Materials and Methods
4.1. Study Population and Mutational Analysis of RdRp
4.2. Evaluation of the Impact of RdRp Mutations on the Response to Remdesivir
4.3. SARS-CoV-2 Polymerase Model Preparation
4.4. Preparation of the SARS-CoV-2 Polymerase Mutant Models
4.5. Molecular Docking and Thermodynamics Analysis
4.6. Molecular Dynamics Simulations (MDs)
4.7. Principal Component Analysis (PCA)
4.8. Pharmacophore Model Generation along the MDs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Helmy, Y.A.; Fawzy, M.; Elaswad, A.; Sobieh, A.; Kenney, S.P.; Shehata, A.A. The COVID-19 pandemic: A comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. J. Clin. Med. 2020, 9, 1225. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Chen, S.; Chen, Z. Advances in COVID-19: The virus, the pathogenesis, and evidence based control and therapeutic strategies. Front. Med. 2020, 14, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.-A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Imbert, I.; Snijder, E.J.; Dimitrova, M.; Guillemot, J.-C.; Lécine, P.; Canard, B. The SARS-Coronavirus PLnc domain of nsp3 as a replication/transcription scaffolding protein. Virus Res. 2008, 133, 136–148. [Google Scholar] [CrossRef]
- Zardecki, C.; Dutta, S.; Goodsell, D.S.; Lowe, R.; Voigt, M.; Burley, S.K. Educational resources supporting molecular explorations through biology and medicine. Protein Sci. 2022, 31, 129–140. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [Green Version]
- McDonald, S.M. RNA synthetic mechanisms employed by diverse families of RNA viruses. Wiley Interdiscip. Rev. RNA 2013, 4, 351–367. [Google Scholar] [CrossRef]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci.USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020, 182, 417–428.e13. [Google Scholar] [CrossRef]
- Das, A.; Gerlits, O.; Parks, J.M.; Langan, P.; Kovalevsky, A.; Heller, W.T. Protein kinase A catalytic subunit primed for action: Time-lapse crystallography of michaelis complex formation. Structure 2015, 23, 2331–2340. [Google Scholar] [CrossRef] [Green Version]
- Agostini, M.L.; Pruijssers, A.J.; Chappell, J.D.; Gribble, J.; Lu, X.; Andres, E.L.; Bluemling, G.R.; Lockwood, M.A.; Sheahan, T.P.; Sims, A.C.; et al. Small-molecule antiviral b-d-N4-hydroxycytidine Inhibits a proofreading-intact coronavirus with a high genetic barrier to resistance. J. Virol. 2019, 93, e01348-19. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broadspectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3’->5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, e00221-e18. [Google Scholar] [CrossRef] [Green Version]
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef]
- Stevens, L.J.; Pruijssers, A.J.; Lee, H.W.; Gordon, C.J.; Tchesnokov, E.P.; Gribble, J.; George, A.S.; Hughes, T.M.; Lu, X.; Li, J.; et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022, 14, eabo0718. [Google Scholar] [CrossRef]
- Bravo, J.P.K.; Dangerfield, T.L.; Taylor, D.W.; Johnson, K.A. Remdesivir is a delayed translocation inhibitor of SARS-CoV-2 replication. Mol. Cell 2021, 81, 1548–1552. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of Covid-19-preliminary report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- WHO Solidarity Trial Consortium. Repurposed antiviral drugs for Covid-19-interim WHO solidarity trial results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef]
- Rehman, S.U.; Rehman, S.U.; Yoo, H.H. COVID-19 challenges and its therapeutics. Biomed. Pharmacother. 2021, 142, 112015. [Google Scholar] [CrossRef] [PubMed]
- Julander, J.G.; Demarest, J.F.; Taylor, R.; Gowen, B.B.; Walling, D.M.; Mathis, A.; Babu, Y.S. An update on the progress of galidesivir (BCX4430), a broad-spectrum antiviral. Antivir. Res. 2021, 195, 105180. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, R. Structural Basis of the Potential Binding Mechanism of Remdesivir to SARS-CoV-2 RNA-Dependent RNA Polymerase. J. Phys. Chem. B 2020, 124, 6955–6962. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication–transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Slanina, H.; Madhugiri, R.; Bylapudi, G.; Schultheiß, K.; Karl, N.; Gulyaeva, A.; Gorbalenya, A.E.; Linne, U.; Ziebuhr, J. Coronavirus replication-transcription complex: Vital and selective NMPylation of a conserved site in nsp9 by the NiRAN-RdRp subunit. Proc. Natl. Acad. Sci. USA 2021, 118, e2022310118. [Google Scholar] [CrossRef] [PubMed]
- Byléhn, F.; Menéndez, C.A.; Perez-Lemus, G.R.; Alvarado, W.; de Pablo, J.J. Modeling the Binding Mechanism of Remdesivir, Favilavir, and Ribavirin to SARS-CoV-2 RNA-Dependent RNA Polymerase. ACS Cent. Sci. 2021, 7, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of Terms Used in Medicinal Chemistry. Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Maruca, A.; Ambrosio, F.A.; Lupia, A.; Romeo, I.; Rocca, R.; Moraca, F.; Talarico, C.; Bagetta, D.; Catalano, R.; Costa, G. Computer-based techniques for lead identification and optimization I: Basics. Phys. Sci. Rev. 2019, 4, 20180113. [Google Scholar] [CrossRef]
- Lupia, A.; Moraca, F.; Bagetta, D.; Maruca, A.; Ambrosio, F.A.; Rocca, R.; Catalano, R.; Romeo, I.; Talarico, C.; Ortuso, F. Computer-based techniques for lead identification and optimization II: Advanced search methods. Phys. Sci. Rev. 2019, 5, 20180114. [Google Scholar] [CrossRef]
- Moraca, F.; Marzano, S.; D’Amico, F.; Lupia, A.; Di Fonzo, S.; Vertecchi, E.; Salvati, E.; Di Porzio, A.; Catalanotti, B.; Randazzo, A.; et al. Ligand-based drug repurposing strategy identified SARS-CoV-2 RNA G-quadruplex binders. Chem. Commun. 2022, 58, 11913–11916. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound Ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, X.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from Sars-CoV-2 by Remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Ilmjärv, S.; Abdul, F.; Acosta-Gutiérrez, S.; Estarellas, C.; Galdadas, I.; Casimir, M.; Alessandrini, M.; Gervasio, F.L.; Krause, K.H. Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant. Sci. Rep. 2021, 11, 13705. [Google Scholar] [CrossRef]
- Salpini, R.; Alkhatib, M.; Costa, G.; Piermatteo, L.; Ambrosio, F.A.; Di Maio, V.C.; Scutari, R.; Duca, L.; Berno, G.; Fabeni, L.; et al. Key genetic elements, single and in clusters, underlying geographically dependent SARS-CoV-2 genetic adaptation and their impact on binding affinity for drugs and immune control. J. Antimicrob. Chemother. 2021, 76, 396–412. [Google Scholar] [CrossRef]
- Santos Bravo, M.; Alonso, R.; Soria, D.; Sánchez Palomino, S.; Sanzo Machuca, Á.; Rodríguez, C.; Alcamí, J.; Díez-Fuertes, F.; Simarro Redon, À.; Hurtado, J.C.; et al. Genetic Study of SARS-CoV-2 Non Structural Protein 12 in COVID-19 Patients Non Responders to Remdesivir. Microbiol. Spectr. 2022, 10, e0244822. [Google Scholar] [CrossRef] [PubMed]
- Genome Detective Virus Tool. Available online: https://www.genomedetective.com/app/typingtool/virus/. (accessed on 31 May 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Verbist, B.M.P.; Thys, K.; Reumers, J.; Wetzels, Y.; Van der Borght, K.; Talloen, W.; Aerssens, J.; Clement, L.; Thas, O. VirVarSeq: A low-frequency virus variant detection pipeline for Illumina sequencing using adaptive base-calling accuracy filtering. Bioinformatics 2015, 31, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Quasitools. Available online: https://phac-nml.github.io/quasitools/. (accessed on 31 May 2023).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Shannon, A.; Fattorini, V.; Sama, B.; Selisko, B.; Feracci, M.; Falcou, C.; Gauffre, P.; Kazzi, P.E.; Delpal, A.; Decroly, E.; et al. A dual mechanism of action of AT-527 against Sars-CoV-2 Polymerase. Nat. Commun. 2022, 13, 621. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 45. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction, Proteins, 2004, 55, 351-67. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2021-4: Maestro; Schrödinger, LLC.: New York, NY, USA, 2021.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-4: MacroModel; Schrödinger, LLC.: New York, NY, USA, 2021.
- Lindahl, E.; Abraham, M.; Hess, B.; van der Spoel, D. GROMACS 2020.6 Source Code; Zenodo. 2020. Available online: https://zenodo.org/record/4576060 (accessed on 4 March 2021).
- Wang, J.M.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. et al. Gaussian16, Revision C.01. Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: https://gaussian.com/citation/ (accessed on 6 August 2023).
- Träg, J.; Zahn, D. Improved GAFF2 parameters for fluorinated alkanes and mixed hydro- and fluorocarbons. J. Mol. Model. 2019, 25, 39. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1996, 18, 1463–1472. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rdrp Model | G-Score | ΔGbind |
|---|---|---|
| WT | −8.63 | −122.70 |
| 1M | −8.74 | −34.50 |
| 2M | −8.58 | −119.05 |
| 3M1 | −8.72 | −129.41 |
| 3M2 | −8.81 | −84.78 |
| 4M | −8.44 | −125.56 |
| 5M | −8.76 | −96.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gratteri, C.; Ambrosio, F.A.; Lupia, A.; Moraca, F.; Catalanotti, B.; Costa, G.; Bellocchi, M.; Carioti, L.; Salpini, R.; Ceccherini-Silberstein, F.; et al. Molecular and Structural Aspects of Clinically Relevant Mutations of SARS-CoV-2 RNA-Dependent RNA Polymerase in Remdesivir-Treated Patients. Pharmaceuticals 2023, 16, 1143. https://doi.org/10.3390/ph16081143
Gratteri C, Ambrosio FA, Lupia A, Moraca F, Catalanotti B, Costa G, Bellocchi M, Carioti L, Salpini R, Ceccherini-Silberstein F, et al. Molecular and Structural Aspects of Clinically Relevant Mutations of SARS-CoV-2 RNA-Dependent RNA Polymerase in Remdesivir-Treated Patients. Pharmaceuticals. 2023; 16(8):1143. https://doi.org/10.3390/ph16081143
Chicago/Turabian StyleGratteri, Carmen, Francesca Alessandra Ambrosio, Antonio Lupia, Federica Moraca, Bruno Catalanotti, Giosuè Costa, Maria Bellocchi, Luca Carioti, Romina Salpini, Francesca Ceccherini-Silberstein, and et al. 2023. "Molecular and Structural Aspects of Clinically Relevant Mutations of SARS-CoV-2 RNA-Dependent RNA Polymerase in Remdesivir-Treated Patients" Pharmaceuticals 16, no. 8: 1143. https://doi.org/10.3390/ph16081143