
The Potential Therapeutic Application of Simvastatin for Brain Complications and Mechanisms of Action

Abstract
:1. Introduction
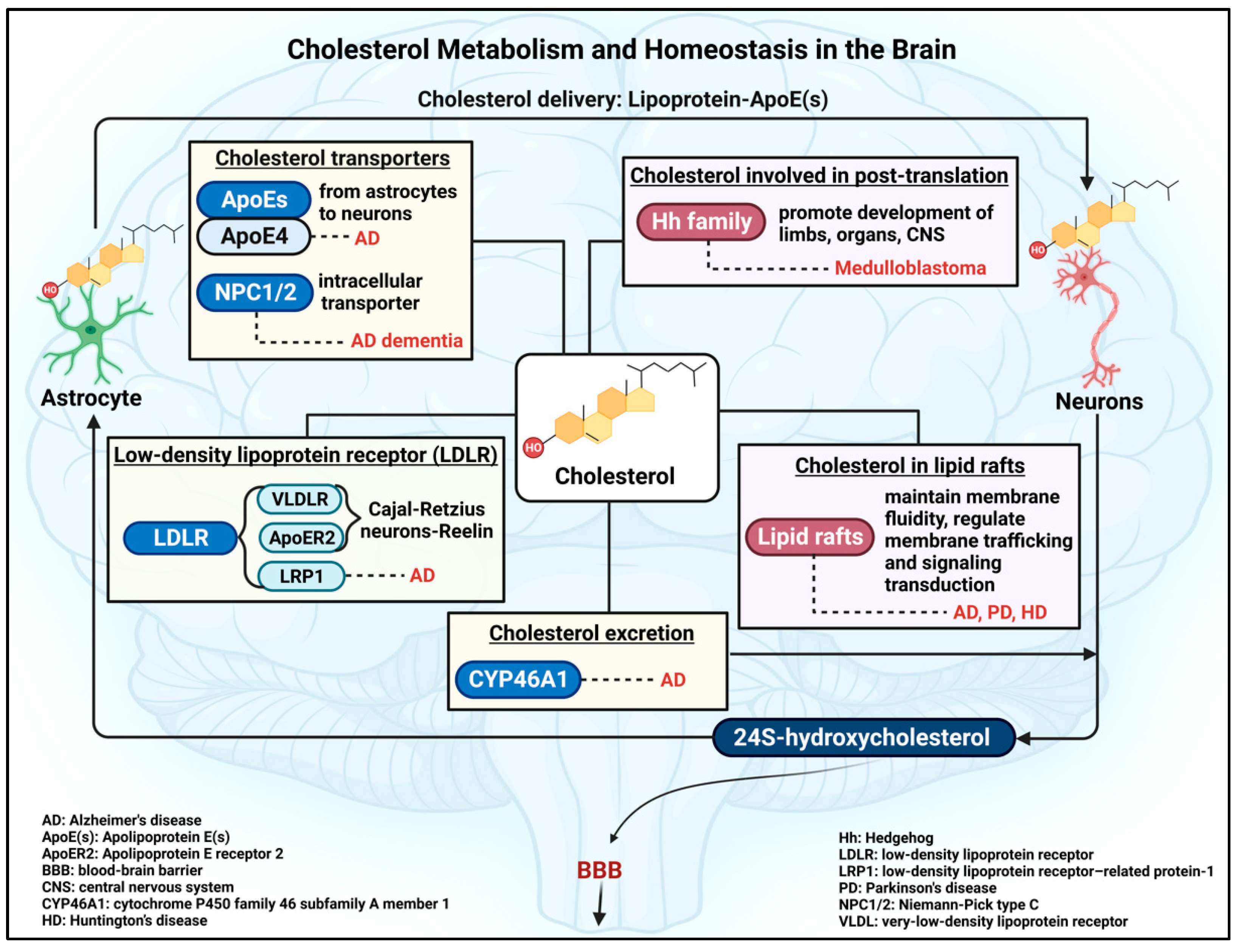
2. Cholesterol Metabolism and Homeostasis in the Human Body
2.1. Peripheral Tissues: Protective Mechanisms for Balancing Cholesterol
2.2. Brain Cholesterol: De Novo Cholesterol Synthesis and Homeostatic Mechanism
2.3. Insights into Factors Involved in the Regulation of Brain Cholesterol Homeostasis
3. Brain Cholesterol: Beyond the General Roles
3.1. Cholesterol as a Post-Translational Modification
3.2. Cholesterol as an Important Composition of Lipid Rafts in Brain Cells
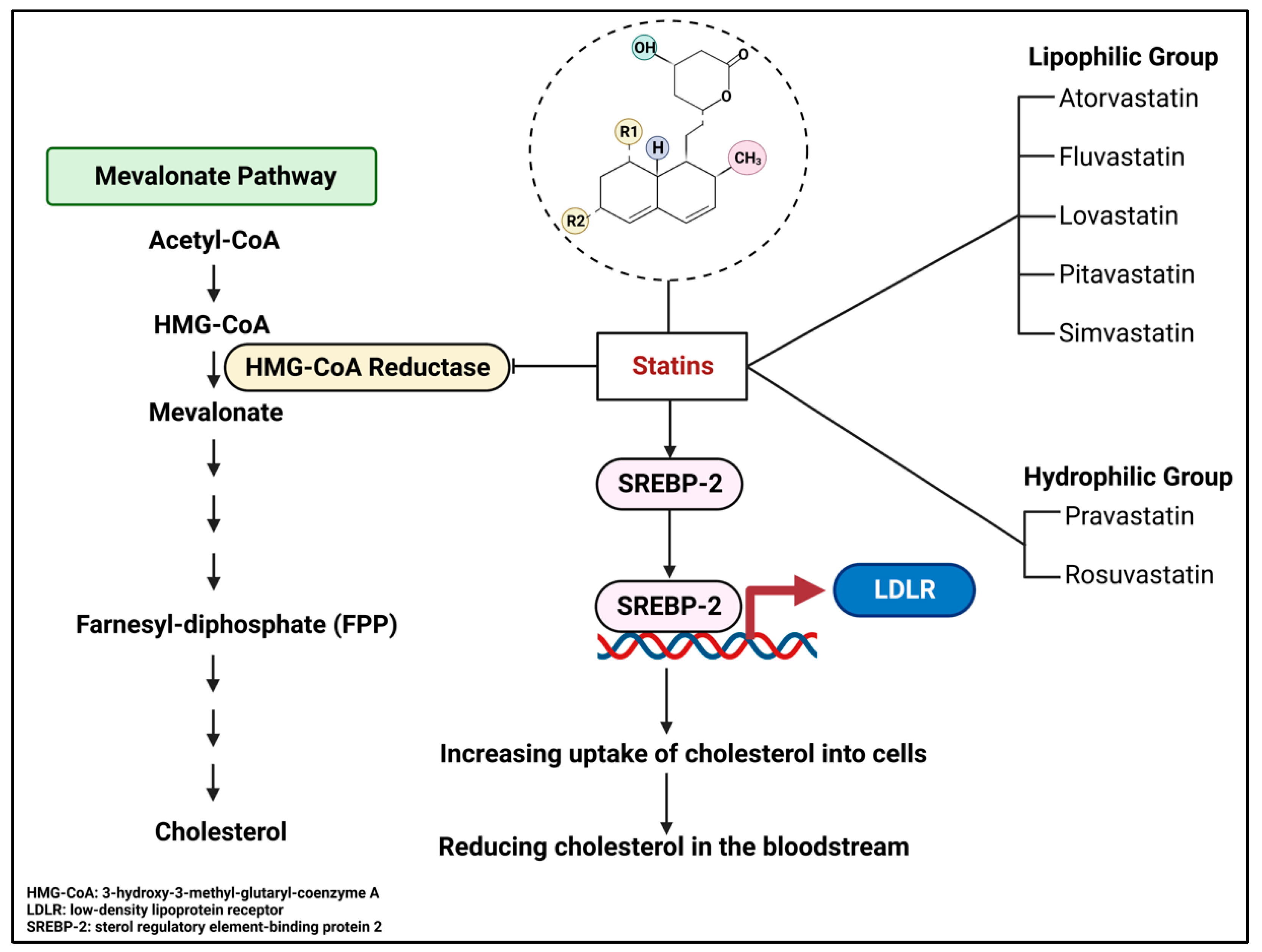
4. Simvastatin: A Member of the Statin Family of Cholesterol-Lowering Drugs
4.1. Overview of Statins

4.2. Historical Perspectives and Clinical Trials of Simvastatin
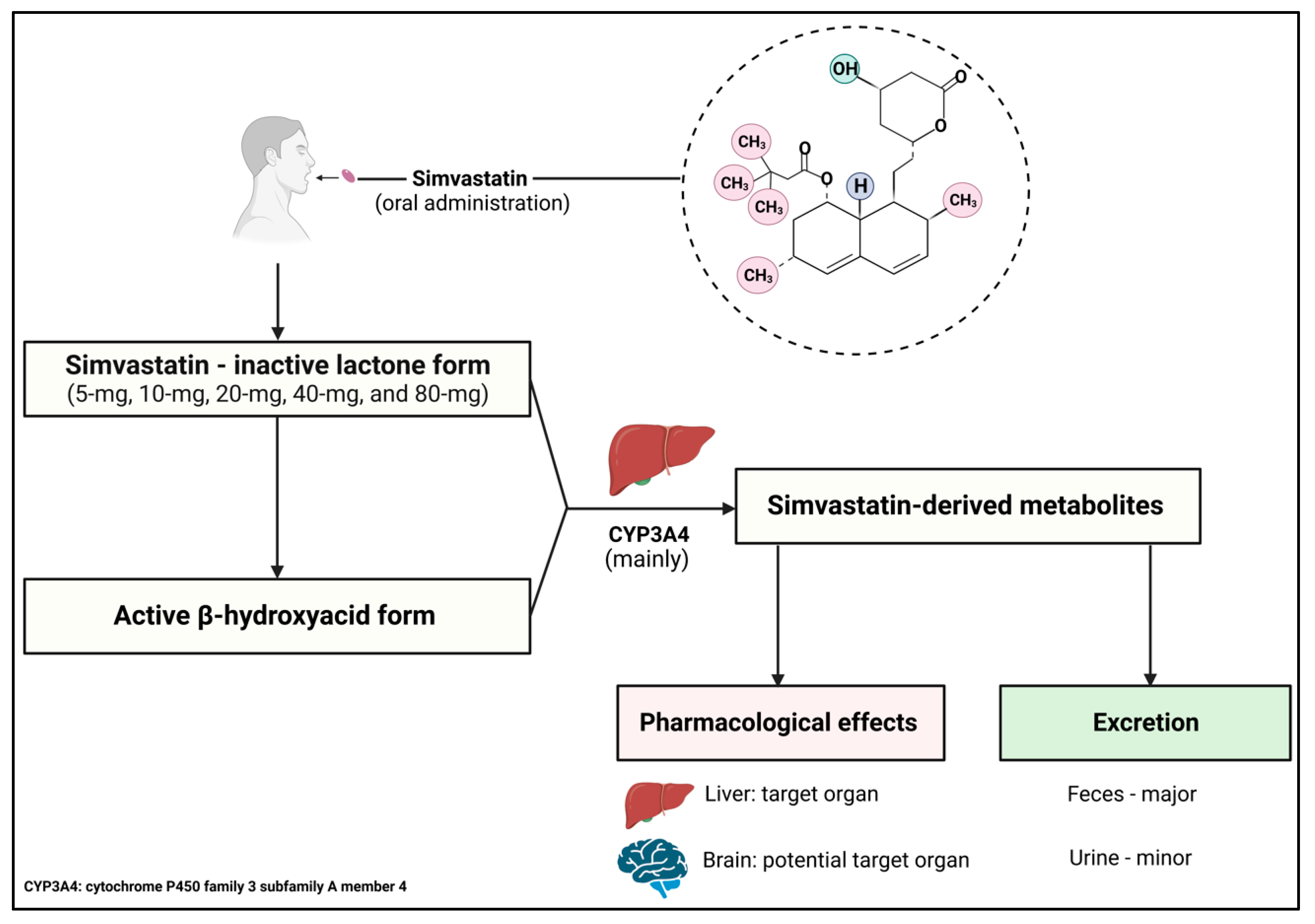
4.3. Mechanism of Action, Pharmacokinetics, and Pharmacodynamics

4.4. Drug Interaction of Simvastatin and Potential Side Effects
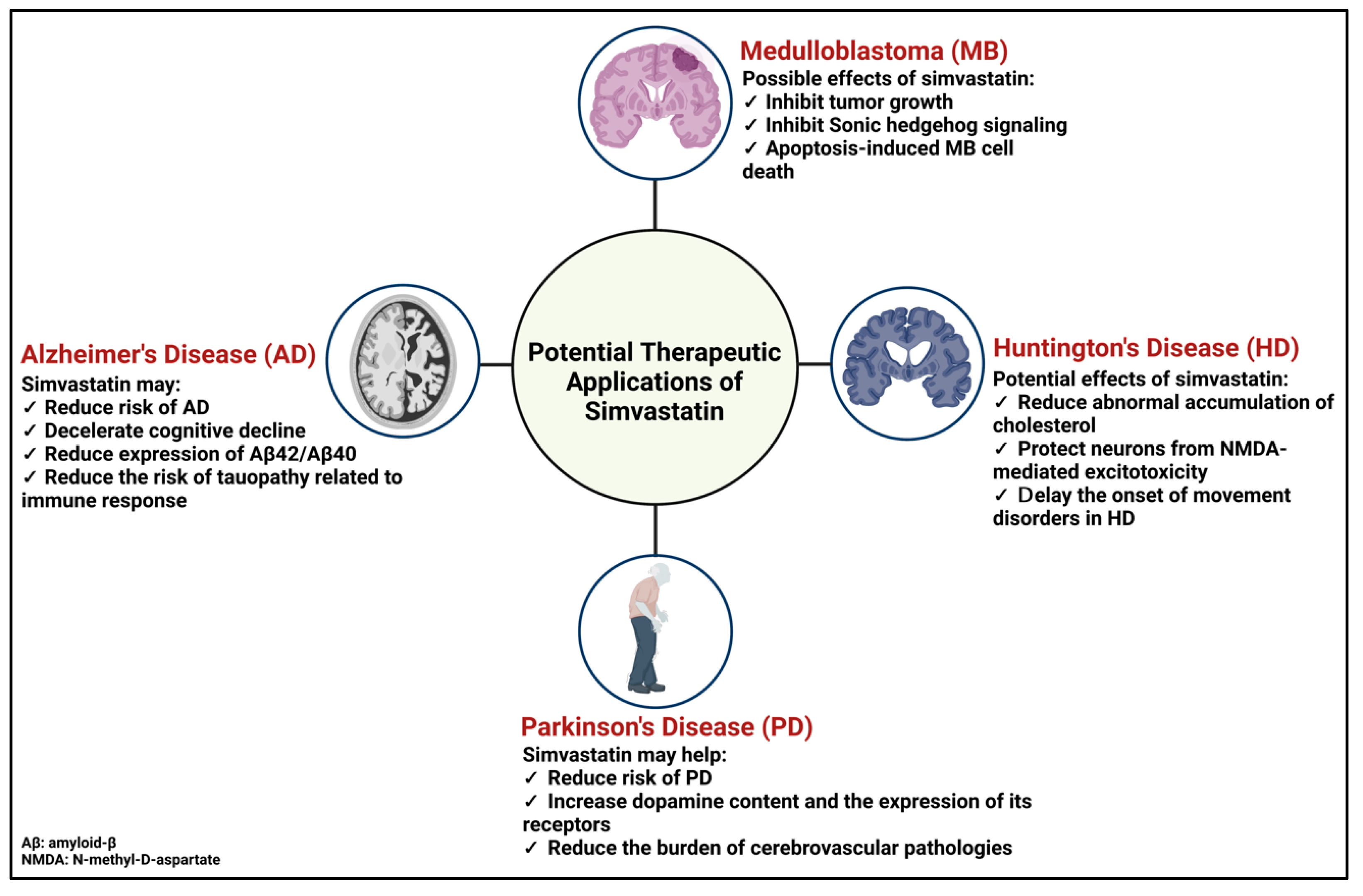
5. Potential Therapeutic Application of Simvastatin for Brain Complications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Model | Dosage | Result |
|---|---|---|---|
| Medulloblastoma [156] | Ptch1+/− mice | 40 mg/kg of body weight daily Duration: 2 weeks | Reduced size and growth rate of the tumor |
| Alzheimer’s disease [13] | Adult male guinea pigs | 0.5% of diet (diet ≈ 25 g/day) Duration: 3 weeks | Reduced levels of Aβ42 and Aβ40 in the cerebrospinal fluid and in the brain |
| Huntington’s disease [157] | Male Wistar rats (6 µmol of intrastriatal administration of malonic acid to induce Huntington-like symptoms) | 30 mg/kg of body weight daily Duration: 14 days | Significantly alleviated Huntington-like symptoms Restored mitochondrial activities Reduced neuro-inflammation (TNF-α and IL-6) |
| Condition | Model | Dosage | Result |
|---|---|---|---|
| Secondary progressive multiple sclerosis NCT00647348 [158,159] | 140 participants 18 to 65 years old Females and males Randomized, placebo-controlled | 80 mg daily Duration: 24 months | Phase 2 Well tolerated and safe Reduced annualized whole-brain atrophy rate. Improved frontal lobe function and physical quality of life |
| Alzheimer’s disease NCT00303277 [160] | 35 participants 18 to 90 years Females and males Randomized, controlled | 40 mg daily Duration: 12 weeks | Phase 4 Reduced cerebrospinal fluid level of phospho-tau-181 |
| Migraine NCT01225263 [161] | 89 participants 18 Years and older Females and males Randomized, double-blind, placebo-controlled | 20 mg, twice daily Dietary supplement: Vitamin D3, 1000 IU, twice daily Duration: 24 weeks | Phase 2 Effectively prevents episodic migraine |
| Stroke (acute) NCT01073007 [162] | 104 participants 18 years and older Females and males Multicentric, randomized, double-blind | 40 mg within 12 h from the onset Duration: 3 months | Phase 4, Decreased rate of bleeding complications Major neurological recovery |
| Subarachnoid hemorrhage-induced vasospasm NCT00235963 [163] | 104 participants 18 years and older Females and males Randomized, double-blind, placebo-controlled | 80 mg/day Duration: whether until discharge from the intensive care unit or a maximum of 21 days | Phase 1 Phase 2 Delayed cerebral ischemia |

5.1. Medulloblastoma
5.2. Alzheimer’s Disease
5.3. Parkinson’s Disease
5.4. Huntington’s Disease
6. Conclusions and Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Varolius, C.; Varolio, C. In De Nervis Opticis Nonnullisque Aliis Praeter Communem Opinionem in Humano Capite Observatis: Patavij 1573. Culture et Civilisation, 1969. Available online: https://books.google.ca/books/about/De_nervis_opticis_nonnullisque_aliis_pra.html?id=KIXqpwAACAAJ&redir_esc=y (accessed on 1 May 2023).
- Tubbs, R.S.; Loukas, M.; Shoja, M.M.; Apaydin, N.; Ardalan, M.R.; Shokouhi, G.; Oakes, W.J. Costanzo Varolio (Constantius Varolius 1543–1575) and the Pons Varolli. Neurosurgery 2008, 62, 734–737. [Google Scholar] [CrossRef]
- Goritz, C.; Mauch, D.H.; Pfrieger, F.W. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell. Neurosci. 2005, 29, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Saher, G.; Brugger, B.; Lappe-Siefke, C.; Mobius, W.; Tozawa, R.; Wehr, M.C.; Wieland, F.; Ishibashi, S.; Nave, K.A. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005, 8, 468–475. [Google Scholar] [CrossRef]
- Ferris, H.A.; Perry, R.J.; Moreira, G.V.; Shulman, G.I.; Horton, J.D.; Kahn, C.R. Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc. Natl. Acad. Sci. USA 2017, 114, 1189–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, P.; Ramseier, A.; Gudat, F.; Mihatsch, M.J.; Polasek, W. Normal weight of the brain in adults in relation to age, sex, body height and weight. Pathologe 1994, 15, 165–170. [Google Scholar] [CrossRef]
- Djelti, F.; Braudeau, J.; Hudry, E.; Dhenain, M.; Varin, J.; Bieche, I.; Marquer, C.; Chali, F.; Ayciriex, S.; Auzeil, N.; et al. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 2015, 138 Pt 8, 2383–2398. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Sanz, P.; MFG Aerts, J.; Moratalla, R. The Role of Cholesterol in α-Synuclein and Lewy Body Pathology in GBA1 Parkinson’s Disease. Mov. Disord. 2021, 36, 1070–1085. [Google Scholar] [CrossRef]
- Kacher, R.; Mounier, C.; Caboche, J.; Betuing, S. Altered Cholesterol Homeostasis in Huntington’s Disease. Front. Aging Neurosci. 2022, 14, 797220. [Google Scholar] [CrossRef]
- Turri, M.; Marchi, C.; Adorni, M.P.; Calabresi, L.; Zimetti, F. Emerging role of HDL in brain cholesterol metabolism and neurodegenerative disorders. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159123. [Google Scholar] [CrossRef]
- Fracassi, A.; Marangoni, M.; Rosso, P.; Pallottini, V.; Fioramonti, M.; Siteni, S.; Segatto, M. Statins and the Brain: More than Lipid Lowering Agents? Curr. Neuropharmacol. 2019, 17, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [Green Version]
- Bjorkhem, I.; Lutjohann, D.; Diczfalusy, U.; Stahle, L.; Ahlborg, G.; Wahren, J. Cholesterol homeostasis in human brain: Turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 1998, 39, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Beffert, U.; Stolt, P.C.; Herz, J. Functions of lipoprotein receptors in neurons. J. Lipid Res. 2004, 45, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J. Biol. Chem. 2004, 279, 14009–14015. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.V.; Wang, C.; Tran, A.C.; Tabor, G.T.; Mahan, T.E.; Francis, C.M.; Finn, M.B.; Spellman, R.; Manis, M.; Tanzi, R.E.; et al. Lack of hepatic apoE does not influence early Abeta deposition: Observations from a new APOE knock-in model. Mol. Neurodegener. 2019, 14, 37. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiesberger, T.; Trommsdorff, M.; Howell, B.W.; Goffinet, A.; Mumby, M.C.; Cooper, J.A.; Herz, J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 1999, 24, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Frotscher, M. Cajal-Retzius cells, Reelin, and the formation of layers. Curr. Opin. Neurobiol. 1998, 8, 570–575. [Google Scholar] [CrossRef]
- Lutjohann, D.; von Bergmann, K. 24S-hydroxycholesterol: A marker of brain cholesterol metabolism. Pharmacopsychiatry 2003, 36 (Suppl. S2), S102–S106. [Google Scholar]
- Lund, E.G.; Xie, C.; Kotti, T.; Turley, S.D.; Dietschy, J.M.; Russell, D.W. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 2003, 278, 22980–22988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayciriex, S.; Djelti, F.; Alves, S.; Regazzetti, A.; Gaudin, M.; Varin, J.; Langui, D.; Bieche, I.; Hudry, E.; Dargere, D.; et al. Neuronal Cholesterol Accumulation Induced by Cyp46a1 Down-Regulation in Mouse Hippocampus Disrupts Brain Lipid Homeostasis. Front. Mol. Neurosci. 2017, 10, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abildayeva, K.; Jansen, P.J.; Hirsch-Reinshagen, V.; Bloks, V.W.; Bakker, A.H.; Ramaekers, F.C.; de Vente, J.; Groen, A.K.; Wellington, C.L.; Kuipers, F.; et al. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J. Biol. Chem. 2006, 281, 12799–12808. [Google Scholar] [CrossRef] [Green Version]
- Esposito, M.; Dubbioso, R.; Tozza, S.; Iodice, R.; Aiello, M.; Nicolai, E.; Cavaliere, C.; Salvatore, M.; Santoro, L.; Manganelli, F. In Vivo evidence of cortical amyloid deposition in the adult form of Niemann Pick type C. Heliyon 2019, 5, e02776. [Google Scholar] [CrossRef]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef]
- Lewis, P.M.; Dunn, M.P.; McMahon, J.A.; Logan, M.; Martin, J.F.; St-Jacques, B.; McMahon, A.P. Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell 2001, 105, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Marti, E.; Bumcrot, D.A.; Takada, R.; McMahon, A.P. Requirement of 19K form of Sonic hedgehog for induction of distinct ventral cell types in CNS explants. Nature 1995, 375, 322–325. [Google Scholar] [CrossRef]
- Ericson, J.; Morton, S.; Kawakami, A.; Roelink, H.; Jessell, T.M. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 1996, 87, 661–673. [Google Scholar] [CrossRef] [Green Version]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, E.E.; Chaudhry, A.; Farah, M.H.; Eberhart, C.G. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am. J. Pathol. 2007, 170, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Hooper, N.M. Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae. Mol. Membr. Biol. 1999, 16, 145–156. [Google Scholar] [CrossRef]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef] [Green Version]
- Boussicault, L.; Kacher, R.; Lamaziere, A.; Vanhoutte, P.; Caboche, J.; Betuing, S.; Potier, M.C. CYP46A1 protects against NMDA-mediated excitotoxicity in Huntington’s disease: Analysis of lipid raft content. Biochimie 2018, 153, 70–79. [Google Scholar] [CrossRef]
- Fuentealba, R.A.; Liu, Q.; Zhang, J.; Kanekiyo, T.; Hu, X.; Lee, J.M.; LaDu, M.J.; Bu, G. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Abeta42 uptake and lysosomal trafficking. PLoS ONE 2010, 5, e11884. [Google Scholar] [CrossRef] [Green Version]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabelo, N.; Martin, V.; Santpere, G.; Marin, R.; Torrent, L.; Ferrer, I.; Diaz, M. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef]
- Valencia, A.; Reeves, P.B.; Sapp, E.; Li, X.; Alexander, J.; Kegel, K.B.; Chase, K.; Aronin, N.; DiFiglia, M. Mutant huntingtin and glycogen synthase kinase 3-beta accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington’s disease. J. Neurosci. Res. 2010, 88, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Ference, B.A.; Severin, T.; Blom, D.; Nicholls, S.J.; Shiba, M.H.; Almahmeed, W.; Alonso, R.; Daccord, M.; Ezhov, M.; et al. World Heart Federation Cholesterol Roadmap 2022. Glob. Heart 2022, 17, 75. [Google Scholar] [CrossRef]
- Jones, P.J. Regulation of cholesterol biosynthesis by diet in humans. Am. J. Clin. Nutr. 1997, 66, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Luchoomun, J.; Zhou, Z.; Bakillah, A.; Jamil, H.; Hussain, M.M. Assembly and secretion of VLDL in nondifferentiated Caco-2 cells stably transfected with human recombinant ApoB48 cDNA. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2955–2963. [Google Scholar] [CrossRef]
- Cooper, A.D. Hepatic uptake of chylomicron remnants. J. Lipid Res. 1997, 38, 2173–2192. [Google Scholar] [CrossRef] [PubMed]
- Cortes, V.; Eckel, R.H. Insulin and Bile Acids in Cholesterol Homeostasis: New Players in Diabetes-Associated Atherosclerosis. Circulation 2022, 145, 983–986. [Google Scholar] [CrossRef]
- Turley, S.D.; Dietschy, J.M. The intestinal absorption of biliary and dietary cholesterol as a drug target for lowering the plasma cholesterol level. Prev. Cardiol. 2003, 6, 29–33, 64. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N.; Packard, C.J.; Chapman, M.J.; Boren, J.; Aguilar-Salinas, C.A.; Averna, M.; Ference, B.A.; Gaudet, D.; Hegele, R.A.; Kersten, S.; et al. Triglyceride-rich lipoproteins and their remnants: Metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur. Heart J. 2021, 42, 4791–4806. [Google Scholar] [CrossRef]
- Zanoni, P.; Velagapudi, S.; Yalcinkaya, M.; Rohrer, L.; von Eckardstein, A. Endocytosis of lipoproteins. Atherosclerosis 2018, 275, 273–295. [Google Scholar] [CrossRef] [PubMed]
- Dubland, J.A.; Francis, G.A. Lysosomal acid lipase: At the crossroads of normal and atherogenic cholesterol metabolism. Front. Cell Dev. Biol. 2015, 3, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Zhang, H. Lysosomal Acid Lipase in Lipid Metabolism and Beyond. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 850–856. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.B.; Horst, M.; Young, M.; Pascua, C.; Puffenberger, E.G.; Brigatti, K.W.; Gonzaga-Jauregui, C.; Shuldiner, A.R.; Gidding, S.; Strauss, K.A.; et al. Clinical characterization of familial hypercholesterolemia due to an amish founder mutation in Apolipoprotein, B. BMC Cardiovasc. Disord. 2022, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Mundal, L.; Sarancic, M.; Ose, L.; Iversen, P.O.; Borgan, J.K.; Veierod, M.B.; Leren, T.P.; Retterstol, K. Mortality among patients with familial hypercholesterolemia: A registry-based study in Norway, 1992–2010. J. Am. Heart Assoc. 2014, 3, e001236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Brown, M.S. Familial hypercholesterolemia: Identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc. Natl. Acad. Sci. USA 1973, 70, 2804–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambon, D.; Quintana, M.; Mata, P.; Alonso, R.; Benavent, J.; Cruz-Sanchez, F.; Gich, J.; Pocovi, M.; Civeira, F.; Capurro, S.; et al. Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am. J. Med. 2010, 123, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaste, M.; Koivisto, P. Risk of brain infarction in familial hypercholesterolemia. Stroke 1988, 19, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef]
- Vuu, Y.M.; Roberts, C.T.; Rastegar, M. MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism. Int. J. Mol. Sci. 2023, 24, 4218. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Tangirala, R.K.; Chun, S.; Usher, D.; Pure, E.; Rader, D.J. Hepatic expression of apolipoprotein E inhibits progression of atherosclerosis without reducing cholesterol levels in LDL receptor-deficient mice. Mol. Ther. 2000, 1, 189–194. [Google Scholar] [CrossRef]
- Johnson, L.A.; Olsen, R.H.; Merkens, L.S.; DeBarber, A.; Steiner, R.D.; Sullivan, P.M.; Maeda, N.; Raber, J. Apolipoprotein E-low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol. Dis. 2014, 64, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Cuchillo-Ibanez, I.; Lennol, M.P.; Escamilla, S.; Mata-Balaguer, T.; Valverde-Vozmediano, L.; Lopez-Font, I.; Ferrer, I.; Saez-Valero, J. The apolipoprotein receptor LRP3 compromises APP levels. Alzheimers Res. Ther. 2021, 13, 181. [Google Scholar] [CrossRef]
- Li, Y.; Lu, W.; Marzolo, M.P.; Bu, G. Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J. Biol. Chem. 2001, 276, 18000–18006. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Dana, S.E.; Faust, J.R.; Beaudet, A.L.; Brown, M.S. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J. Biol. Chem. 1975, 250, 8487–8495. [Google Scholar] [CrossRef]
- May, P.; Rohlmann, A.; Bock, H.H.; Zurhove, K.; Marth, J.D.; Schomburg, E.D.; Noebels, J.L.; Beffert, U.; Sweatt, J.D.; Weeber, E.J.; et al. Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol. Cell. Biol. 2004, 24, 8872–8883. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Cirrito, J.R.; Liu, C.C.; Shinohara, M.; Li, J.; Schuler, D.R.; Shinohara, M.; Holtzman, D.M.; Bu, G. Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J. Neurosci. 2013, 33, 19276–19283. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef]
- Benhayon, D.; Magdaleno, S.; Curran, T. Binding of purified Reelin to ApoER2 and VLDLR mediates tyrosine phosphorylation of Disabled-1. Mol. Brain Res. 2003, 112, 33–45. [Google Scholar] [CrossRef]
- Llorente-Cortes, V.; Otero-Vinas, M.; Sanchez, S.; Rodriguez, C.; Badimon, L. Low-density lipoprotein upregulates low-density lipoprotein receptor-related protein expression in vascular smooth muscle cells: Possible involvement of sterol regulatory element binding protein-2-dependent mechanism. Circulation 2002, 106, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Lutjohann, D.; Breuer, O.; Ahlborg, G.; Nennesmo, I.; Siden, A.; Diczfalusy, U.; Bjorkhem, I. Cholesterol homeostasis in human brain: Evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. USA 1996, 93, 9799–9804. [Google Scholar] [CrossRef] [Green Version]
- Konings, S.C.; Torres-Garcia, L.; Martinsson, I.; Gouras, G.K. Astrocytic and Neuronal Apolipoprotein E Isoforms Differentially Affect Neuronal Excitability. Front. Neurosci. 2021, 15, 734001. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.P.; Ioannou, Y.A. Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J. Biol. Chem. 2000, 275, 24367–24374. [Google Scholar] [PubMed] [Green Version]
- Kraft, M.L. Sphingolipid Organization in the Plasma Membrane and the Mechanisms That Influence It. Front. Cell Dev. Biol. 2016, 4, 154. [Google Scholar] [CrossRef] [Green Version]
- Geberhiwot, T.; Moro, A.; Dardis, A.; Ramaswami, U.; Sirrs, S.; Marfa, M.P.; Vanier, M.T.; Walterfang, M.; Bolton, S.; Dawson, C.; et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet. J. Rare Dis. 2018, 13, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cupidi, C.; Frangipane, F.; Gallo, M.; Clodomiro, A.; Colao, R.; Bernardi, L.; Anfossi, M.; Conidi, M.E.; Vasso, F.; Curcio, S.A.; et al. Role of Niemann-Pick Type C Disease Mutations in Dementia. J. Alzheimers Dis. 2017, 55, 1249–1259. [Google Scholar] [CrossRef]
- Kumar, S.; Balczarek, K.A.; Lai, Z.C. Evolution of the hedgehog gene family. Genetics 1996, 142, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.E.; Eisen, J.S. Hedgehog signaling is required for primary motoneuron induction in zebrafish. Development 2001, 128, 3485–3495. [Google Scholar] [CrossRef]
- Bumcrot, D.A.; Takada, R.; McMahon, A.P. Proteolytic processing yields two secreted forms of sonic hedgehog. Mol. Cell. Biol. 1995, 15, 2294–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, J.A.; Ekker, S.C.; Park, W.J.; von Kessler, D.P.; Young, K.E.; Chen, C.H.; Ma, Y.; Woods, A.S.; Cotter, R.J.; Koonin, E.V.; et al. Hedgehog patterning activity: Role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell 1996, 86, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, C.; Wolf, A.; Wagner, M.; Kuhlmann, J.; Waldmann, H. The cholesterol membrane anchor of the Hedgehog protein confers stable membrane association to lipid-modified proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 8531–8536. [Google Scholar] [CrossRef] [Green Version]
- Guy, R.K. Inhibition of sonic hedgehog autoprocessing in cultured mammalian cells by sterol deprivation. Proc. Natl. Acad. Sci. USA 2000, 97, 7307–7312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, J.A.; von Kessler, D.P.; Ekker, S.C.; Young, K.E.; Lee, J.J.; Moses, K.; Beachy, P.A. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature 1995, 374, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Boulet, A.M.; Moon, A.M.; Arenkiel, B.R.; Capecchi, M.R. The roles of Fgf4 and Fgf8 in limb bud initiation and outgrowth. Dev. Biol. 2004, 273, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Teran, M.; Piedra, M.E.; Kathiriya, I.S.; Srivastava, D.; Rodriguez-Rey, J.C.; Ros, M.A. Role of dHAND in the anterior-posterior polarization of the limb bud: Implications for the Sonic hedgehog pathway. Development 2000, 127, 2133–2142. [Google Scholar] [CrossRef]
- Bechtold, T.E.; Kurio, N.; Nah, H.D.; Saunders, C.; Billings, P.C.; Koyama, E. The Roles of Indian Hedgehog Signaling in TMJ Formation. Int. J. Mol. Sci. 2019, 20, 6300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Dilower, I.; Niloy, A.J.; Kumar, V.; Kothari, A.; Lee, E.B.; Rumi, M.A.K. Hedgehog Signaling in Gonadal Development and Function. Cells 2023, 12, 358. [Google Scholar] [CrossRef] [PubMed]
- Coltin, H.; Sundaresan, L.; Smith, K.S.; Skowron, P.; Massimi, L.; Eberhart, C.G.; Schreck, K.C.; Gupta, N.; Weiss, W.A.; Tirapelli, D.; et al. Subgroup and subtype-specific outcomes in adult medulloblastoma. Acta Neuropathol. 2021, 142, 859–871. [Google Scholar] [CrossRef]
- Alaamery, M.; Albesher, N.; Aljawini, N.; Alsuwailm, M.; Massadeh, S.; Wheeler, M.A.; Chao, C.C.; Quintana, F.J. Role of sphingolipid metabolism in neurodegeneration. J. Neurochem. 2021, 158, 25–35. [Google Scholar] [CrossRef]
- Hefter, D.; Ludewig, S.; Draguhn, A.; Korte, M. Amyloid, APP, and Electrical Activity of the Brain. Neuroscientist 2020, 26, 231–251. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Rudajev, V.; Novotny, J. Cholesterol as a key player in amyloid beta-mediated toxicity in Alzheimer’s disease. Front. Mol. Neurosci. 2022, 15, 937056. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.; Feightner, J.W.; Garcia, A.; Hsiung, G.Y.; MacKnight, C.; Sadovnick, A.D. Diagnosis and treatment of dementia: 1. Risk assessment and primary prevention of Alzheimer disease. CMAJ 2008, 178, 548–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y.; Kwon, O.H.; Park, M.K.; Kim, T.W.; Chung, S. Elevated cellular cholesterol in Familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of beta-amyloid precursor protein. PLoS ONE 2019, 14, e0210535. [Google Scholar]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [Green Version]
- Wahrle, S.; Das, P.; Nyborg, A.C.; McLendon, C.; Shoji, M.; Kawarabayashi, T.; Younkin, L.H.; Younkin, S.G.; Golde, T.E. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 2002, 9, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Urano, Y.; Hayashi, I.; Isoo, N.; Reid, P.C.; Shibasaki, Y.; Noguchi, N.; Tomita, T.; Iwatsubo, T.; Hamakubo, T.; Kodama, T. Association of active gamma-secretase complex with lipid rafts. J. Lipid Res. 2005, 46, 904–912. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- van Maarschalkerweerd, A.; Vetri, V.; Vestergaard, B. Cholesterol facilitates interactions between alpha-synuclein oligomers and charge-neutral membranes. FEBS Lett. 2015, 589 Pt B, 2661–2667. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. α-synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Benito, M.; Granado, N.; Garcia-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease with the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Man, W.K.; De Simone, A.; Barritt, J.D.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. A Role of Cholesterol in Modulating the Binding of alpha-Synuclein to Synaptic-Like Vesicles. Front. Neurosci. 2020, 14, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.Y.; Baumann, K.; Zhou, C.; Zhou, W.; Cuellar, A.E.; Xue, H. Trends in Use and Expenditures for Brand-name Statins after Introduction of Generic Statins in the US, 2002–2018. JAMA Netw. Open 2021, 4, e2135371. [Google Scholar] [CrossRef]
- Buist, M.; Fuss, D.; Rastegar, M. Transcriptional Regulation of MECP2E1-E2 Isoforms and BDNF by Metformin and Simvastatin through Analyzing Nascent RNA Synthesis in a Human Brain Cell Line. Biomolecules 2021, 11, 1253. [Google Scholar] [CrossRef]
- Sheikholeslami, K.; Ali Sher, A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin Induces Apoptosis in Medulloblastoma Brain Tumor Cells via Mevalonate Cascade Prenylation Substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wei, X.; Wang, Q.; Li, W.; Yang, T. Inverse screening of Simvastatin kinase targets from glioblastoma druggable kinome. Comput. Biol. Chem. 2020, 86, 107243. [Google Scholar]
- Yang, S.; Xie, C.; Guo, T.; Li, H.; Li, N.; Zhou, S.; Wang, X.; Xie, C. Simvastatin Inhibits Tumor Growth and Migration by Mediating Caspase-1-Dependent Pyroptosis in Glioblastoma Multiforme. World Neurosurg. 2022, 165, e12–e21. [Google Scholar] [CrossRef]
- Stevens, K.N.; Creanor, S.; Jeffery, A.; Whone, A.; Zajicek, J.; Foggo, A.; Jones, B.; Chapman, R.; Cocking, L.; Wilks, J.; et al. Evaluation of Simvastatin as a Disease-Modifying Treatment for Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2022, 79, 1232–1241. [Google Scholar] [CrossRef]
- Mueller, A.M.; Liakoni, E.; Schneider, C.; Burkard, T.; Jick, S.S.; Krahenbuhl, S.; Meier, C.R.; Spoendlin, J. The Risk of Muscular Events Among New Users of Hydrophilic and Lipophilic Statins: An Observational Cohort Study. J. Gen. Intern. Med. 2021, 36, 2639–2647. [Google Scholar] [CrossRef]
- Climent, E.; Benaiges, D.; Pedro-Botet, J. Hydrophilic or Lipophilic Statins? Front. Cardiovasc. Med. 2021, 8, 687585. [Google Scholar] [CrossRef]
- Pedersen, T.R.; Tobert, J.A. Simvastatin: A review. Expert Opin. Pharmacother. 2004, 5, 2583–2596. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.; Lurie, Y.; Groskop, I.; Weintrob, M. Cholesterol-lowering effects of a 10 mg daily dose of lovastatin in patients with initial total cholesterol levels 200 to 240 mg/dl (5.18 to 6.21 mmol/liter). Am. J. Cardiol. 1991, 68, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, G.; Cassinotti, M.; Vecchio, G.; Gianfranceschi, G.; Pazzucconi, F.; Murakami, T.; Sirtori, M.; D’Acquarica, A.L.; Sirtori, C.R. Pravastatin effectively lowers LDL cholesterol in familial combined hyperlipidemia without changing LDL subclass pattern. Arterioscler. Thromb. 1994, 14, 1569–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, J.W.; Weiss, S.R.; Davidson, M.H.; Sprecher, D.L.; Schwartz, S.L.; Lupien, P.J.; Jones, P.H.; Haber, H.E.; Black, D.M. Reduction of LDL cholesterol by 25% to 60% in patients with primary hypercholesterolemia by atorvastatin, a new HMG-CoA reductase inhibitor. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 678–682. [Google Scholar] [CrossRef]
- Tran, D.T.; Palfrey, D.; Welsh, R. The Healthcare Cost Burden in Adults with High Risk for Cardiovascular Disease. Pharmacoecon. Open. 2021, 5, 425–435. [Google Scholar] [CrossRef]
- De Giorgi, R.; De Crescenzo, F.; Rizzo Pesci, N.; Martens, M.; Howard, W.; Cowen, P.J.; Harmer, C.J. Statins for major depressive disorder: A systematic review and meta-analysis of randomized controlled trials. PLoS ONE 2021, 16, e0249409. [Google Scholar] [CrossRef]
- Sheng, Z.; Otani, H.; Brown, M.S.; Goldstein, J.L. Independent regulation of sterol regulatory element-binding proteins 1 and 2 in hamster liver. Proc. Natl. Acad. Sci. USA 1995, 92, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Matusewicz, L.; Meissner, J.; Toporkiewicz, M.; Sikorski, A.F. The effect of statins on cancer cells—Review. Tumour. Biol. 2015, 36, 4889–4904. [Google Scholar] [CrossRef]
- Espano, E.; Nam, J.H.; Song, E.J.; Song, D.; Lee, C.K.; Kim, J.K. Lipophilic statins inhibit Zika virus production in Vero cells. Sci. Rep. 2019, 9, 11461. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, L.; Davis, C.R.; Roach, C.; Nguyen, D.K.; Aldrich, T.; McAda, P.C.; Reeves, C.D. Lovastatin biosynthesis in Aspergillus terreus: Characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. Chem. Biol. 1999, 6, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. J. Antibiot. 1976, 29, 1346–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberts, A.W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, W.F.; Alberts, A.W.; Anderson, P.S.; Chen, J.S.; Smith, R.L.; Willard, A.K. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. 4. Side chain ester derivatives of mevinolin. J. Med. Chem. 1986, 29, 849–852. [Google Scholar] [CrossRef]
- Armitage, J.; Bowman, L.; Wallendszus, K.; Bulbulia, R.; Rahimi, K.; Haynes, R.; Parish, S.; Peto, R.; Collins, R. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: A double-blind randomised trial. Lancet 2010, 376, 1658–1669. [Google Scholar] [PubMed] [Green Version]
- Heart Protection Study Collaborative, Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Pedersen, R.T.; Kjekshus, J.; Berg, K.; Haghfelt, T.; Faergeman, O.; Thorgeirsson, G.; Pyorala, K.; Miettinen, T.; Wilhelmsen, L.; Olsson, G.A.; et al. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Morofuji, Y.; Nakagawa, S.; Ujifuku, K.; Fujimoto, T.; Otsuka, K.; Niwa, M.; Tsutsumi, K. Beyond Lipid-Lowering: Effects of Statins on Cardiovascular and Cerebrovascular Diseases and Cancer. Pharmaceuticals 2022, 15, 151. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [Green Version]
- Svec, A.; Adameova, A. Facts and ideas on statins with respect to their lipophilicity: A focus on skeletal muscle cells and bone besides known cardioprotection. Mol. Cell. Biochem. 2023, 478, 1661–1667. [Google Scholar] [CrossRef]
- McGowan, M.P.; Hosseini Dehkordi, S.H.; Moriarty, P.M.; Duell, P.B. Diagnosis and Treatment of Heterozygous Familial Hypercholesterolemia. J. Am. Heart Assoc. 2019, 8, e013225. [Google Scholar] [CrossRef]
- Roglans, N.; Verd, J.C.; Peris, C.; Alegret, M.; Vazquez, M.; Adzet, T.; Diaz, C.; Hernandez, G.; Laguna, J.C.; Sanchez, R.M. High doses of atorvastatin and simvastatin induce key enzymes involved in VLDL production. Lipids 2002, 37, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Frascaroli, C.; Senkeev, R.; Cavalot, F.; Russo, I. Simvastatin Effects on Inflammation and Platelet Activation Markers in Hypercholesterolemia. Biomed. Res. Int. 2018, 2018, 6508709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehghankelishadi, P.; Maritz, M.F.; Dmochowska, N.; Badiee, P.; Cheah, E.; Kempson, I.; Berbeco, R.I.; Thierry, B. Formulation of simvastatin within high density lipoprotein enables potent tumour radiosensitisation. J. Control. Release 2022, 346, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Zendehdel Baher, S.; Yaqoubi, S.; Asare-Addo, K.; Hamishehkar, H.; Nokhodchi, A. Dry Powder Formulation of Simvastatin Nanoparticles for Potential Application in Pulmonary Arterial Hypertension. Pharmaceutics 2022, 14, 895. [Google Scholar] [CrossRef]
- Tulbah, A.S.; Ong, H.X.; Morgan, L.; Colombo, P.; Young, P.M.; Traini, D. Dry powder formulation of simvastatin. Expert Opin. Drug Deliv. 2015, 12, 857–868. [Google Scholar] [CrossRef]
- Vree, T.B.; Dammers, E.; Ulc, I.; Horkovics-Kovats, S.; Ryska, M.; Merkx, I. Differences between lovastatin and simvastatin hydrolysis in healthy male and female volunteers:gut hydrolysis of lovastatin is twice that of simvastatin. Sci. World J. 2003, 3, 1332–1343. [Google Scholar] [CrossRef] [Green Version]
- Corsini, A.; Bellosta, S.; Baetta, R.; Fumagalli, R.; Paoletti, R.; Bernini, F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol. Ther. 1999, 84, 413–428. [Google Scholar] [CrossRef]
- Kee, P.S.; Chin, P.K.L.; Kennedy, M.A.; Maggo, S.D.S. Pharmacogenetics of Statin-Induced Myotoxicity. Front. Genet. 2020, 11, 575678. [Google Scholar] [CrossRef]
- Kirby, B.J.; Collier, A.C.; Kharasch, E.D.; Whittington, D.; Thummel, K.E.; Unadkat, J.D. Complex drug interactions of HIV protease inhibitors 1: Inactivation, induction, and inhibition of cytochrome P450 3A by ritonavir or nelfinavir. Drug Metab. Dispos. 2011, 39, 1070–1078. [Google Scholar] [CrossRef] [Green Version]
- Loue, C.; Tod, M. Reliability and extension of quantitative prediction of CYP3A4-mediated drug interactions based on clinical data. AAPS J. 2014, 16, 1309–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, V.F.; MacDonald, J.L. Simvastatin: A review of its pharmacology and clinical use. DICP 1991, 25, 257–264. [Google Scholar] [CrossRef]
- Boccuzzi, S.J.; Bocanegra, T.S.; Walker, J.F.; Shapiro, D.R.; Keegan, M.E. Long-term safety and efficacy profile of simvastatin. Am. J. Cardiol. 1991, 68, 1127–1131. [Google Scholar] [CrossRef]
- Safitri, N.; Alaina, M.F.; Pitaloka, D.A.E.; Abdulah, R. A Narrative Review of Statin-Induced Rhabdomyolysis: Molecular Mechanism, Risk Factors, and Management. Drug Healthc. Patient Saf. 2021, 13, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Bruckert, E.; Hayem, G.; Dejager, S.; Yau, C.; Begaud, B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients--the PRIMO study. Cardiovasc. Drugs Ther. 2005, 19, 403–414. [Google Scholar] [CrossRef]
- Turner, R.M.; Pirmohamed, M. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J. Clin. Med. 2019, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skottheim, I.B.; Gedde-Dahl, A.; Hejazifar, S.; Hoel, K.; Asberg, A. Statin induced myotoxicity: The lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur. J. Pharm. Sci. 2008, 33, 317–325. [Google Scholar] [CrossRef]
- Ovbiagele, B.; Kidwell, C.S.; Saver, J.L. Expanding indications for statins in cerebral ischemia: A quantitative study. Arch. Neurol. 2005, 62, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Scott, M.P. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8408–8413. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Mahmud, I.; Pokhrel, R.; Murad, R.; Yuan, M.; Stapleton, S.; Bettegowda, C.; Jallo, G.; Eberhart, C.G.; Garrett, T.; et al. Medulloblastoma cerebrospinal fluid reveals metabolites and lipids indicative of hypoxia and cancer-specific RNAs. Acta Neuropathol. Commun. 2022, 10, 25. [Google Scholar] [CrossRef]
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779. [Google Scholar] [CrossRef]
- Huang, X.; Sterling, N.W.; Du, G.; Sun, D.; Stetter, C.; Kong, L.; Zhu, Y.; Neighbors, J.; Lewis, M.M.; Chen, H.; et al. Brain cholesterol metabolism and Parkinson’s disease. Mov. Disord. 2019, 34, 386–395. [Google Scholar] [CrossRef]
- Carroll, C.B.; Wyse, R.K.H. Simvastatin as a Potential Disease-Modifying Therapy for Patients with Parkinson’s Disease: Rationale for Clinical Trial, and Current Progress. J. Park. Dis. 2017, 7, 545–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreilaus, F.; Spiro, A.S.; McLean, C.A.; Garner, B.; Jenner, A.M. Evidence for altered cholesterol metabolism in Huntington’s disease post mortem brain tissue. Neuropathol. Appl. Neurobiol. 2016, 42, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.E.; Zhang, L.; Peri, S.; Kuo, Y.M.; Du, F.; Egleston, B.L.; Ng, J.M.Y.; Andrews, A.J.; Astsaturov, I.; Curran, T.; et al. Statins Synergize with Hedgehog Pathway Inhibitors for Treatment of Medulloblastoma. Clin. Cancer Res. 2018, 24, 1375–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Sharma, N.; Mishra, J.; Kalonia, H. Synergistical neuroprotection of rofecoxib and statins against malonic acid induced Huntington’s disease like symptoms and related cognitive dysfunction in rats. Eur. J. Pharmacol. 2013, 709, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Binks, S.; Nicholas, J.M.; Frost, C.; Cardoso, M.J.; Ourselin, S.; Wilkie, D.; Nicholas, R.; Chataway, J. Effect of high-dose simvastatin on cognitive, neuropsychiatric, and health-related quality-of-life measures in secondary progressive multiple sclerosis: Secondary analyses from the MS-STAT randomised, placebo-controlled trial. Lancet Neurol. 2017, 16, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Chataway, J.; Schuerer, N.; Alsanousi, A.; Chan, D.; MacManus, D.; Hunter, K.; Anderson, V.; Bangham, C.R.; Clegg, S.; Nielsen, C.; et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): A randomised, placebo-controlled, phase 2 trial. Lancet 2014, 383, 2213–2221. [Google Scholar] [CrossRef] [Green Version]
- Riekse, R.G.; Li, G.; Petrie, E.C.; Leverenz, J.B.; Vavrek, D.; Vuletic, S.; Albers, J.J.; Montine, T.J.; Lee, V.M.; Lee, M.; et al. Effect of statins on Alzheimer’s disease biomarkers in cerebrospinal fluid. J. Alzheimers Dis. 2006, 10, 399–406. [Google Scholar] [CrossRef]
- Buettner, C.; Nir, R.R.; Bertisch, S.M.; Bernstein, C.; Schain, A.; Mittleman, M.A.; Burstein, R. Simvastatin and vitamin D for migraine prevention: A randomized, controlled trial. Ann. Neurol. 2015, 78, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Montaner, J.; Bustamante, A.; Garcia-Matas, S.; Martinez-Zabaleta, M.; Jimenez, C.; de la Torre, J.; Rubio, F.R.; Segura, T.; Masjuan, J.; Canovas, D.; et al. Combination of Thrombolysis and Statins in Acute Stroke Is Safe: Results of the STARS Randomized Trial (Stroke Treatment with Acute Reperfusion and Simvastatin). Stroke 2016, 47, 2870–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.H.; Smith, E.E.; Badjatia, N.; Nogueira, R.G.; Sims, J.R., II; Ogilvy, C.S.; Rordorf, G.A.; Ayata, C. A randomized, double-blind, placebo-controlled pilot study of simvastatin in aneurysmal subarachnoid hemorrhage. Stroke 2008, 39, 2891–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Gong, T.; Zheng, C.; Ng, J.M.Y.; Chen, J.; Myers, C.; Hensley, H.; Curran, T.; Yang, Z.J. Statins repress hedgehog signaling in medulloblastoma with no bone toxicities. Oncogene 2021, 40, 2258–2272. [Google Scholar] [CrossRef] [PubMed]
- Torrandell-Haro, G.; Branigan, G.L.; Vitali, F.; Geifman, N.; Zissimopoulos, J.M.; Brinton, R.D. Statin therapy and risk of Alzheimer’s and age-related neurodegenerative diseases. Alzheimers Dement. (NY) 2020, 6, e12108. [Google Scholar] [CrossRef]
- Geifman, N.; Brinton, R.D.; Kennedy, R.E.; Schneider, L.S.; Butte, A.J. Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Ting, W.L.; Yang, H.; Wong, P.T. High doses of simvastatin upregulate dopamine D1 and D2 receptor expression in the rat prefrontal cortex: Possible involvement of endothelial nitric oxide synthase. Br. J. Pharmacol. 2005, 144, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Tang, X.N.; Wang, L.; Yenari, M.A.; Ying, W.; Goh, B.C.; Lee, H.S.; Wilder-Smith, E.P.; Wong, P.T. Effects of high dose of simvastatin on levels of dopamine and its reuptake in prefrontal cortex and striatum among SD rats. Neurosci. Lett. 2006, 408, 189–193. [Google Scholar] [CrossRef] [PubMed]
- del Toro, D.; Xifro, X.; Pol, A.; Humbert, S.; Saudou, F.; Canals, J.M.; Alberch, J. Altered cholesterol homeostasis contributes to enhanced excitotoxicity in Huntington’s disease. J. Neurochem. 2010, 115, 153–167. [Google Scholar] [CrossRef]
- Schultz, J.L.; Nopoulos, P.C.; Killoran, A.; Kamholz, J.A. Statin use and delayed onset of Huntington’s disease. Mov. Disord. 2019, 34, 281–285. [Google Scholar] [CrossRef]
- Mduma, E.; Awuor, A.; Lugina, E.L. Adult medulloblastoma: A case report. J. Med. Case Rep. 2022, 16, 330. [Google Scholar] [CrossRef]
- Vriend, J.; Rastegar, M. Ubiquitin ligases and medulloblastoma: Genetic markers of the four consensus subgroups identified through transcriptome datasets. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165839. [Google Scholar] [CrossRef]
- Marzban, H.; Del Bigio, M.R.; Alizadeh, J.; Ghavami, S.; Zachariah, R.M.; Rastegar, M. Cellular commitment in the developing cerebellum. Front. Cell. Neurosci. 2015, 8, 450. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Cushing, H.W. Medulloblastoma cerebelli: A common type of midcerebellar glioma of childhood. J. Nerv. Ment. Dis. 1925, 14, 192–224. [Google Scholar] [CrossRef]
- Wang, X.; Dubuc, A.M.; Ramaswamy, V.; Mack, S.; Gendoo, D.M.; Remke, M.; Wu, X.; Garzia, L.; Luu, B.; Cavalli, F.; et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015, 129, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millard, N.E.; De Braganca, K.C. Medulloblastoma. J. Child Neurol. 2016, 31, 1341–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funakoshi, Y.; Sugihara, Y.; Uneda, A.; Nakashima, T.; Suzuki, H. Recent advances in the molecular understanding of medulloblastoma. Cancer Sci. 2023, 114, 741–749. [Google Scholar] [CrossRef]
- Fuccillo, M.; Joyner, A.L.; Fishell, G. Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 772–783. [Google Scholar] [CrossRef]
- Brownell, I.; Guevara, E.; Bai, C.B.; Loomis, C.A.; Joyner, A.L. Nerve-derived sonic hedgehog defines a niche for hair follicle stem cells capable of becoming epidermal stem cells. Cell Stem Cell 2011, 8, 552–565. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Patel, R.; Trofka, A.; Harfe, B.D.; Mackem, S. Sonic hedgehog is not a limb morphogen but acts as a trigger to specify all digits in mice. Dev. Cell 2022, 57, 2048–2062.e4. [Google Scholar] [CrossRef]
- Peng, Y.C.; Levine, C.M.; Zahid, S.; Wilson, E.L.; Joyner, A.L. Sonic hedgehog signals to multiple prostate stromal stem cells that replenish distinct stromal subtypes during regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 20611–20616. [Google Scholar] [CrossRef] [Green Version]
- Shin, K.; Lee, J.; Guo, N.; Kim, J.; Lim, A.; Qu, L.; Mysorekar, I.U.; Beachy, P.A. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature 2011, 472, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Hallahan, A.R.; Pritchard, J.I.; Hansen, S.; Benson, M.; Stoeck, J.; Hatton, B.A.; Russell, T.L.; Ellenbogen, R.G.; Bernstein, I.D.; Beachy, P.A.; et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004, 64, 7794–7800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachtergaele, S.; Mydock, L.K.; Krishnan, K.; Rammohan, J.; Schlesinger, P.H.; Covey, D.F.; Rohatgi, R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 2012, 8, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Delcuve, G.P.; Rastegar, M.; Davie, J.R. Epigenetic control. J. Cell. Physiol. 2009, 219, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Shevkoplyas, D.; Vuu, Y.M.; Davie, J.R.; Rastegar, M. The Chromatin Structure at the MECP2 Gene and In Silico Prediction of Potential Coding and Non-Coding MECP2 Splice Variants. Int. J. Mol. Sci. 2022, 23, 15643. [Google Scholar] [CrossRef]
- Pejhan, S.; Rastegar, M. Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules 2021, 11, 75. [Google Scholar] [CrossRef]
- Bin Akhtar, G.; Buist, M.; Rastegar, M. MeCP2 and transcriptional control of eukaryotic gene expression. Eur. J. Cell Biol. 2022, 101, 151237. [Google Scholar] [CrossRef]
- Pejhan, S.; Siu, V.M.; Ang, L.C.; Del Bigio, M.R.; Rastegar, M. Differential brain region-specific expression of MeCP2 and BDNF in Rett Syndrome patients: A distinct grey-white matter variation. Neuropathol. Appl. Neurobiol. 2020, 46, 735–750. [Google Scholar] [CrossRef]
- Buist, M.; El Tobgy, N.; Shevkoplyas, D.; Genung, M.; Sher, A.A.; Pejhan, S.; Rastegar, M. Differential Sensitivity of the Protein Translation Initiation Machinery and mTOR Signaling to MECP2 Gain- and Loss-of-Function Involves MeCP2 Isoform-Specific Homeostasis in the Brain. Cells 2022, 11, 1442. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Ali Sher, A.; Del Bigio, M.R.; Sztainberg, Y.; Siu, V.M.; et al. MECP2 Mutation Interrupts Nucleolin-mTOR-P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pejhan, S.; Del Bigio, M.R.; Rastegar, M. The MeCP2E1/E2-BDNF-miR132 Homeostasis Regulatory Network Is Region-Dependent in the Human Brain and Is Impaired in Rett Syndrome Patients. Front. Cell Dev Biol. 2020, 8, 763. [Google Scholar] [CrossRef]
- Nejati-Koshki, K.R.C.; Babaei, G.; Rastegar, M. The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology. Cancers 2023, 15, 2683. [Google Scholar] [CrossRef]
- Ashford, M.T.; Veitch, D.P.; Neuhaus, J.; Nosheny, R.L.; Tosun, D.; Weiner, M.W. The search for a convenient procedure to detect one of the earliest signs of Alzheimer’s disease: A systematic review of the prediction of brain amyloid status. Alzheimers Dement. 2021, 17, 866–887. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Yu, W.; Senda, T.; Yanagisawa, K.; Michikawa, M. Cholesterol-dependent modulation of tau phosphorylation in cultured neurons. J. Neurochem. 2001, 76, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Lutjohann, D.; Papassotiropoulos, A.; Bjorkhem, I.; Locatelli, S.; Bagli, M.; Oehring, R.D.; Schlegel, U.; Jessen, F.; Rao, M.L.; von Bergmann, K.; et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J. Lipid Res. 2000, 41, 195–198. [Google Scholar] [CrossRef]
- Schonknecht, P.; Lutjohann, D.; Pantel, J.; Bardenheuer, H.; Hartmann, T.; von Bergmann, K.; Beyreuther, K.; Schroder, J. Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer’s disease compared to healthy controls. Neurosci. Lett. 2002, 324, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Katzov, H.; Zetterberg, H.; Feuk, L.; Johansson, B.; Bogdanovic, N.; Andreasen, N.; Lenhard, B.; Brookes, A.J.; Pedersen, N.L.; et al. Variants of CYP46A1 may interact with age and APOE to influence CSF Abeta42 levels in Alzheimer’s disease. Hum. Genet. 2004, 114, 581–587. [Google Scholar]
- Lerner, A.J.; Arnold, S.E.; Maxfield, E.; Koenig, A.; Toth, M.E.; Fortin, B.; Mast, N.; Trombetta, B.A.; Denker, J.; Pieper, A.A.; et al. CYP46A1 activation by low-dose efavirenz enhances brain cholesterol metabolism in subjects with early Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 198. [Google Scholar] [CrossRef]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Choudhury, P.; Ramanan, V.K.; Boeve, B.F. APOE Allele Testing and Alzheimer Disease-Reply. JAMA 2021, 325, 2211. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Firulyova, M.; Manis, M.; Herz, J.; Smirnov, I.; Aladyeva, E.; Wang, C.; Bao, X.; Finn, M.B.; Hu, H.; et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023, 615, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. The effects of statins on microglial cells to protect against neurodegenerative disorders: A mechanistic review. Biofactors 2020, 46, 309–325. [Google Scholar] [CrossRef]
- Li, B.; Mahmood, A.; Lu, D.; Wu, H.; Xiong, Y.; Qu, C.; Chopp, M. Simvastatin attenuates microglial cells and astrocyte activation and decreases interleukin-1beta level after traumatic brain injury. Neurosurgery 2009, 65, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, M.F.; Bologna, M. The pathophysiology of Parkinson’s disease tremor. J. Neurol. Sci. 2022, 435, 120196. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Yagi, N.; Aoyama, K.; Choong, C.J.; Hayakawa, H.; Fujimura, H.; Nagai, Y.; Goto, Y.; Mochizuki, H. Parkinson’s disease is a type of amyloidosis featuring accumulation of amyloid fibrils of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2019, 116, 17963–17969. [Google Scholar] [CrossRef] [Green Version]
- Brekk, O.R.; Honey, J.R.; Lee, S.; Hallett, P.J.; Isacson, O. Cell type-specific lipid storage changes in Parkinson’s disease patient brains are recapitulated by experimental glycolipid disturbance. Proc. Natl. Acad. Sci. USA 2020, 117, 27646–27654. [Google Scholar] [CrossRef]
- Willis, A.W.; Roberts, E.; Beck, J.C.; Fiske, B.; Ross, W.; Savica, R.; Van Den Eeden, S.K.; Tanner, C.M.; Marras, C.; Parkinson’s Foundation P4 Group. Incidence of Parkinson disease in North America. NPJ Park. Dis. 2022, 8, 170. [Google Scholar] [CrossRef]
- Hurh, K.; Park, M.; Jang, S.I.; Park, E.C.; Jang, S.Y. Association between serum lipid levels over time and risk of Parkinson’s disease. Sci. Rep. 2022, 12, 21020. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Wang, M.; Sterling, N.W.; Du, G.; Lewis, M.M.; Yao, T.; Mailman, R.B.; Li, R.; Huang, X. Circulating Cholesterol Levels May Link to the Factors Influencing Parkinson’s Risk. Front. Neurol. 2017, 8, 501. [Google Scholar] [CrossRef] [Green Version]
- Oveisgharan, S.; Yu, L.; Barnes, L.L.; Agrawal, S.; Schneider, J.A.; Bennett, D.A.; Buchman, A.S. Association of Statins with Cerebral Atherosclerosis and Incident Parkinsonism in Older Adults. Neurology 2022, 98, e1976–e1984. [Google Scholar] [CrossRef] [PubMed]
- Huntington, G. On chorea. J. Neuropsychiatry Clin. Neurosci. 2003, 15, 109–112. [Google Scholar] [CrossRef]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Aronin, N.; Chase, K.; Young, C.; Sapp, E.; Schwarz, C.; Matta, N.; Kornreich, R.; Landwehrmeyer, B.; Bird, E.; Beal, M.F.; et al. CAG expansion affects the expression of mutant Huntingtin in the Huntington’s disease brain. Neuron 1995, 15, 1193–1201. [Google Scholar] [CrossRef] [Green Version]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov. Disord. 2022, 37, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.R.; Broadhurst, D.; Dunn, W.B.; Ellis, D.I.; Michell, A.W.; Vacher, C.; Mosedale, D.E.; Kell, D.B.; Barker, R.A.; Grainger, D.J.; et al. Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain 2006, 129 Pt 4, 877–986. [Google Scholar] [CrossRef] [Green Version]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Fielding, C.J.; Bist, A.; Fielding, P.E. Caveolin mRNA levels are up-regulated by free cholesterol and down-regulated by oxysterols in fibroblast monolayers. Proc. Natl. Acad. Sci. USA 1997, 94, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Li, V.; Wang, Y.T. Molecular mechanisms of NMDA receptor-mediated excitotoxicity: Implications for neuroprotective therapeutics for stroke. Neural Regen. Res. 2016, 11, 1752–1753. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuu, Y.M.; Kadar Shahib, A.; Rastegar, M. The Potential Therapeutic Application of Simvastatin for Brain Complications and Mechanisms of Action. Pharmaceuticals 2023, 16, 914. https://doi.org/10.3390/ph16070914
Vuu YM, Kadar Shahib A, Rastegar M. The Potential Therapeutic Application of Simvastatin for Brain Complications and Mechanisms of Action. Pharmaceuticals. 2023; 16(7):914. https://doi.org/10.3390/ph16070914
Chicago/Turabian StyleVuu, Yen My, Ashraf Kadar Shahib, and Mojgan Rastegar. 2023. "The Potential Therapeutic Application of Simvastatin for Brain Complications and Mechanisms of Action" Pharmaceuticals 16, no. 7: 914. https://doi.org/10.3390/ph16070914