
Unveiling the Potential of BenzylethyleneAryl–Urea Scaffolds for the Design of New Onco Immunomodulating Agents


Abstract
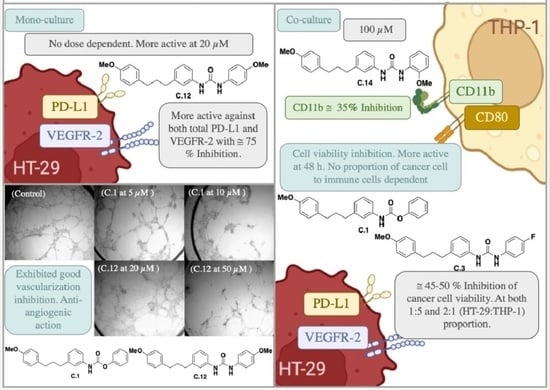
:
1. Introduction
2. Results and Discussion
2.1. Synthetic Strategy for the Obtention of Urea-Bearing Compounds
2.2. Biological Evaluation
2.2.1. Cell Proliferation Inhibition
2.2.2. Effect on Cellular PD-L1 and VEGFR-2 in Cancer Cell Lines
2.2.3. Study of the Action on Microvessel Formation on Matrigel
2.2.4. Effect on Cancer Cell Proliferation in Co-Cultures with Monocytes THP-1
2.2.5. Effect on Immune Cell Proliferation in Co-Cultures of HT-29/THP-1
3. Discussion
4. Materials and Methods
4.1. Symthetic Protocols
4.1.1. General Techniques
4.1.2. Experimental Procedure for the Synthesis of Ureas C.2–C.14
4.2. Biological Studies
4.2.1. Cell Culture
4.2.2. Cell Proliferation Assay
4.2.3. PD-L1 and VEGFR-2 Relative Quantification by Flow Cytometry
4.2.4. Microvessel Formation Inhibition Assay
4.2.5. Cancer and Immune Cell Proliferation Test in Co-Cultures
4.2.6. CD11b and CD80 Detection THP-1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–409. [Google Scholar] [CrossRef]
- Sitohy, B.; Nagy, J.A.; Dvorak, H.F. Anti-VEGF/VEGFR therapy for cancer: Reassessing the target. Cancer Res. 2012, 72, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Anastasiya, S.; Poltavets, L.; Polina, A.; Vishnyakova, A.; Gennady, T.; Sukhikh, L.; Fatkhudinov, T. Macrophage Modification Strategies for Efficient Cell Therapy. Cells 2020, 9, 1535. [Google Scholar] [CrossRef]
- Ansari, M.J.; Bokov, D.; Markov, A.; Jalil, A.T.; Shalaby, M.N.; Suksatan, W.; Chupradit, S.; Al-Ghamdi, H.S.; Shomali, N.; Zamani, A.; et al. Cancer combination therapies by angiogenesis inhibitors; a comprehensive review. Cell Commun. Signal. 2022, 20, 49. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Jonasch, E.; Atkins, M.B.; Chowdhury, S.; Mainwaring, P. Combination of Anti-Angiogenics and Checkpoint Inhibitors for Renal Cell Carcinoma: Is the Whole Greater Than the Sum of Its Parts? Cancers 2022, 14, 644. [Google Scholar] [CrossRef]
- Pla-López, A.; Castillo, R.; Cejudo-Marín, R.; García-Pedrero, O.; Bakir-Laso, M.; Falomir, E.; Carda, M. Synthesis and Bio-logical Evaluation of Small Molecules as Potential Anticancer Multitarget Agents. Int. J. Mol. Sci. 2022, 23, 7049. [Google Scholar] [CrossRef]
- Gil-Edo, R.; Espejo, S.; Falomir, E.; Carda, M. Synthesis and Biological Evaluation of Potential Oncoimmunomodulator Agents. Int. J. Mol. Sci. 2023, 24, 2614. [Google Scholar] [CrossRef]
- Martín-Beltrán, C.; Gil-Edo, R.; Hernández-Ribelles, G.; Agut, R.; Marí-Mezquita, P.; Carda, M.; Falomir, E. Aryl Urea Based Scaffolds for Multitarget Drug Discovery in Anticancer Immunotherapies. Pharmaceuticals 2021, 14, 337. [Google Scholar] [CrossRef]
- Conesa-Milián, L.; Falomir, E.; Murga, J.; Carda, M.; Marco, J.A. Novel multitarget inhibitors with antiangiogenic and immunomodulator properties. Eur. J. Med. Chem. 2019, 148, 87–98. [Google Scholar] [CrossRef]
- Conesa-Milián, L.; Falomir, E.; Murga, J.; Carda, M.; Marco, J.A. Synthesis and biological evaluation as antiangiogenic agents of ureas derived from 3′-aminocombretastatin A-4. Eur. J. Med. Chem. 2019, 162, 781–792. [Google Scholar] [CrossRef]
- Iyer, R.; Fetterly, G.; Lugade, A.; Thanavala, Y. Sorafenib: A clinical and pharmacologic review. Expert Opin. Pharmacother. 2010, 11, 1943–1955. [Google Scholar] [CrossRef]
- Kumari, N.; Choi, S.H. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]
- Radha Abbas Hasoon, M.; Jawad Kadhim, N. Improvement of the Selectivity Index (SI) and Cytotoxicity Activity of Doxorubicin Drug by Panax ginseng Plant Extract. Arch. Razi Inst. 2021, 76, 659–666. [Google Scholar] [CrossRef]
- Jeong, S.; Jeon, Y.; Park, S.J. Inhibitory effects of dieckol on hypoxia-induced epithelial-mesenchymal transition of HT-29 human colorectal cancer cells. Mol. Med. Rep. 2016, 14, 5148–5154. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, S.; Hushmandi, K.; Entezari, M.; Bahonar, A.; Raei, M.; Akbari, M.E. Mesenchymal Stem Cells Trigger Epithelial to Mesenchymal Transition in the HT-29 Colorectal Cancer Cell Line. J. Adv. Med. Biomed. Res. 2022, 143, 477–485. [Google Scholar] [CrossRef]
- Marcuello, M.; Mayol, X.; Felipe-Fumero, E.; Costa, J.; López-Hierro, L.; Salvans, S.; Alonso, S.; Pascual, M.; Grande, L.; Pera, M. Modulation of the colon cancer cell phenotype by pro-inflammatory macrophages: A preclinical model of surgery-associated inflammation and tumor recurrence. PLoS ONE 2018, 13, e0192958. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.-C.; Hao, C.-Y.; Fan, Y.-Z.; Liu, D.; Qiao, Y.-F.; Yao, J.-B.; Li, C.-Z.; Yu, Y. Altered Membrane Expression and Function of CD11b Play a Role in the Immunosuppressive Effects of Morphine on Macrophages at the Nanomolar Level. Pharmaceuticals 2023, 16, 282. [Google Scholar] [CrossRef]
- Mesri, M.; Plescia, J.; Altieri, D.C. Dual Regulation of Ligand Binding by CD11b I Domain: Inhibition of Intercellular Adhesion and Monocyte Procoagulant Activity by a Factor X-Derived Peptide. J. Biol. Chem. 1998, 273, 744–748. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | HT-29 | A-549 | HEK-293 | JURKAT | HMEC-1 |
|---|---|---|---|---|---|
| Sorafenib | 17 ± 4 | 27 ± 2 | 5.0 ± 0.7 | --- | 34 ± 3 |
| BMS-8 | 19 ± 2 | 6 ± 1 | 60 ± 10 | >100 | --- |
| C.1. | >100 | >100 | >100 | >100 | >100 |
| C.2. | 25 ± 5 | >100 | 12 ± 1 | >100 | >100 |
| C.3. | >100 | >100 | >100 | >100 | >100 |
| C.4. | >100 | >100 | 22 ± 5 | >100 | >100 |
| C.5. | 58 ± 16 | 29 ± 5 | 24 ± 4 | >100 | >100 |
| C.6. | 4 ± 1 | 20 ± 10 | 4 ± 1 | >100 | >100 |
| C.7. | 14 ± 3 | 19 ± 1 | 14 ± 3 | 17 ± 1 | 18 ± 8 |
| C.8. | 47 ± 2 | >100 | 37 ± 8 | >100 | >100 |
| C.9. | 1.9 ± 0.6 | 7 ± 1 | 2.1 ± 0.2 | >100 | >100 |
| C.10. | 11 ± 2 | 8 ± 2 | 9 ± 4 | 18 ± 7 | 17 ± 7 |
| C.11. | 15 ± 4 | 20 ± 5 | 27 ± 4 | 26 ± 18 | 25 ± 1 |
| C.12. | >100 | 1.2 ± 0.2 | >100 | >100 | >100 |
| C.13. | 8 ± 5 | 18 ± 5 | 15 ± 3 | 24 ± 4 | 20 ± 5 |
| C.14. | >100 | >100 | >100 | >100 | >100 |
| Comp. | SI (HT-29) (IC50HEK293/IC50HT-29) | SI (A-549) (IC50HEK293/A-549) |
|---|---|---|
| Sorafenib | 0.3 | 0.2 |
| BMS-8 | 3 | 10 |
| C.1 | No effect | No effect |
| C.2 | 0.5 | <2 |
| C.3 | No effect | No effect |
| C.4 | <0.2 | <0.2 |
| C.5 | 2.0 | 0.8 |
| C.6 | 1.0 | 0.2 |
| C.7 | 1.0 | 1.2 |
| C.8 | 0.8 | <0.4 |
| C.9 | 1.1 | 1.2 |
| C.10 | 0.8 | 1.1 |
| C.11 | 1.8 | 1.3 |
| C.12 | No effect | 83 |
| C.13 | 1.9 | 0.8 |
| C.14 | No effect | No effect |
| 20 μM | 100 μM | |||
|---|---|---|---|---|
| Comp. | PD-L1 (%) | VEGFR-2 (%) | PD-L1 (%) | VEGFR-2 (%) |
| Control | 100 | 100 | 100 | 100 |
| Sorafenib | 96 ± 49 | 132 ± 15 | 102 ± 10 | 95 ± 8 |
| BMS-8 | 82 ± 21 | 86 ± 21 | 67 ± 20 | - |
| C.1. | 70 ± 16 | 51 ± 10 | 91 ± 7 | 191 ± 87 |
| C.3. | 70 ± 4 | 38 ± 7 | 222 ± 69 | 40 ± 7 |
| C.12. | 25 ± 5 | 30 ± 10 | 96 ± 1 | 65 ± 45 |
| C.14. | 45 ± 11 | 228 ± 66 | 145 ± 8 | 32 ± 9 |
| Comp. | Minimum Active Conc. (µM) |
|---|---|
| Sunitinib | 3 |
| Sorafenib | 10 |
| C.1 | 5 |
| C.3 | 50 |
| C.12 | 20 |
| C.14 | >100 |
| 1:5 HT-29/THP-1 | 2:1 HT-29/THP-1 | |||
|---|---|---|---|---|
| Comp. | 24 h | 48 h | 24 h | 48 h |
| BMS-8 | 11 ± 6 | 21 ± 3 | 8 ± 1 | 16 ± 2 |
| C.1 | 48 ± 8 | 52 ± 3 | 70 ± 8 | 66 ± 8 |
| C.3 | 78 ± 8 | 64 ± 3 | 67 ± 9 | 57 ± 5 |
| C.12 | 84 ± 2 | 67 ± 11 | 93 ± 8 | 89 ± 10 |
| C.14 | 64 ± 14 | 68 ± 8 | 82 ± 7 | 58 ± 5 |
| 1:5 HT-29/THP-1 | 2:1 HT-29/THP-1 | |||
|---|---|---|---|---|
| Comp. | 24 h | 48 h | 24 h | 48 h |
| BMS-8 | 158 ± 6 | 124 ± 0 | 385 ± 12 | 186 ± 10 |
| C.1 | 104 ± 8 | 97 ± 8 | 124 ± 6 | 113 ± 7 |
| C.3 | 87 ± 8 | 78 ± 5 | 113 ± 26 | 91 ± 18 |
| C.12 | 95 ± 18 | 90 ± 6 | 94 ± 5 | 90 ± 2 |
| C.14 | 99 ± 10 | 91 ± 6 | 110 ± 7 | 86 ± 3 |
| 1:5 HT-29/THP-1; 24 h | ||
|---|---|---|
| Comp. | % CD80 | % CD11b |
| BMS-8 | 93 ± 3 | 79 ± 5 |
| C.1 | 96 ± 1 | 79 ± 9 |
| C.3 | 93 ± 2 | 67 ± 22 |
| C.12 | 89 ± 2 | 73 ± 4 |
| C.14 | 92 ± 1 | 66 ± 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil-Edo, R.; Royo, S.; Carda, M.; Falomir, E. Unveiling the Potential of BenzylethyleneAryl–Urea Scaffolds for the Design of New Onco Immunomodulating Agents. Pharmaceuticals 2023, 16, 808. https://doi.org/10.3390/ph16060808
Gil-Edo R, Royo S, Carda M, Falomir E. Unveiling the Potential of BenzylethyleneAryl–Urea Scaffolds for the Design of New Onco Immunomodulating Agents. Pharmaceuticals. 2023; 16(6):808. https://doi.org/10.3390/ph16060808
Chicago/Turabian StyleGil-Edo, Raquel, Santiago Royo, Miguel Carda, and Eva Falomir. 2023. "Unveiling the Potential of BenzylethyleneAryl–Urea Scaffolds for the Design of New Onco Immunomodulating Agents" Pharmaceuticals 16, no. 6: 808. https://doi.org/10.3390/ph16060808