
Engineering Rapalog-Inducible Genetic Switches Based on Split-T7 Polymerase to Regulate Oncolytic Virus-Driven Production of Tumour-Localized IL-12 for Anti-Cancer Immunotherapy
 , , ,
, , ,
Abstract
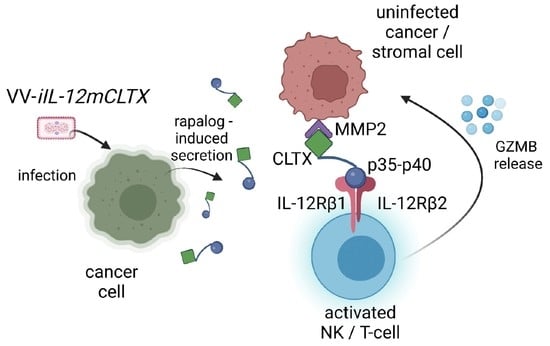
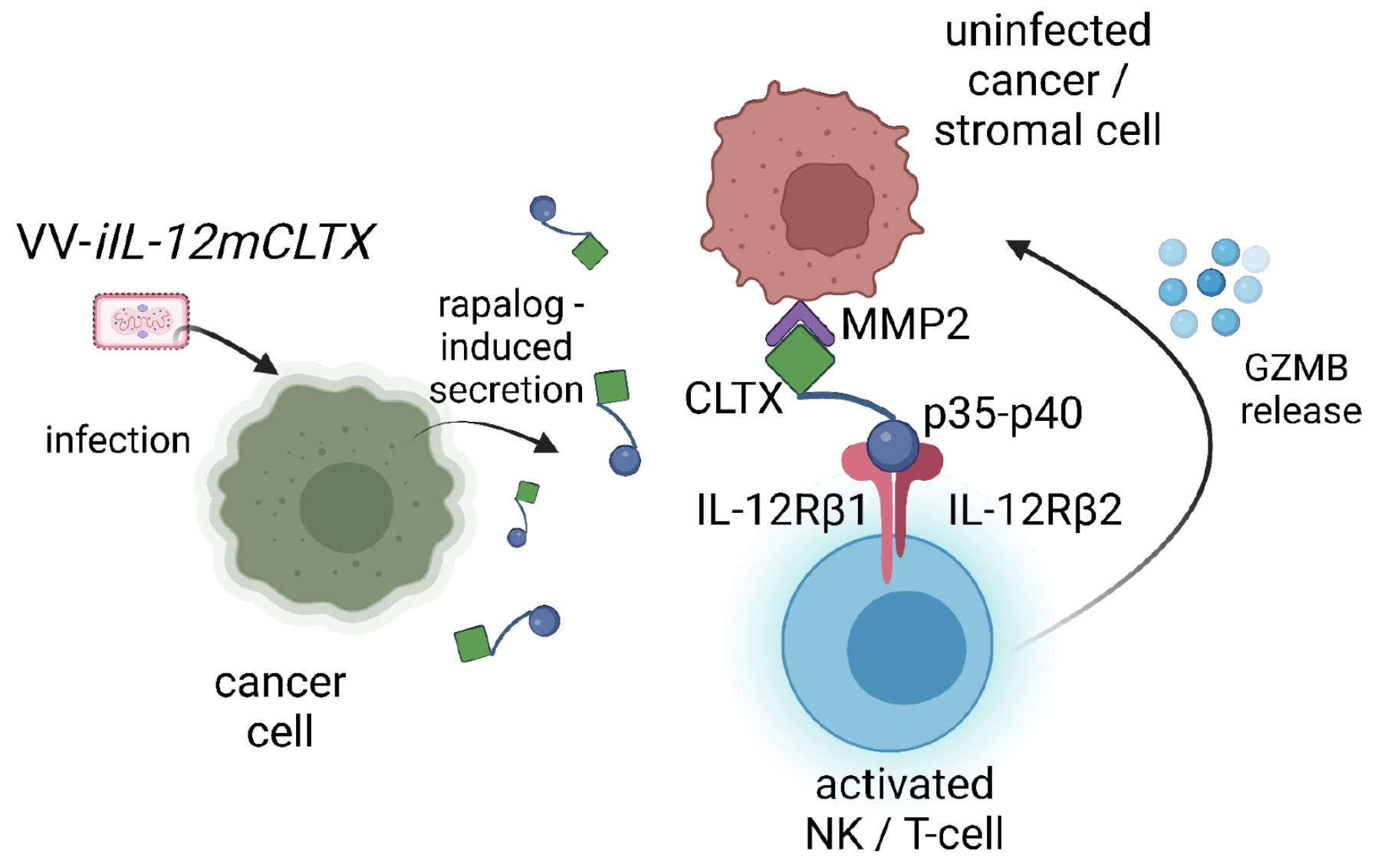
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
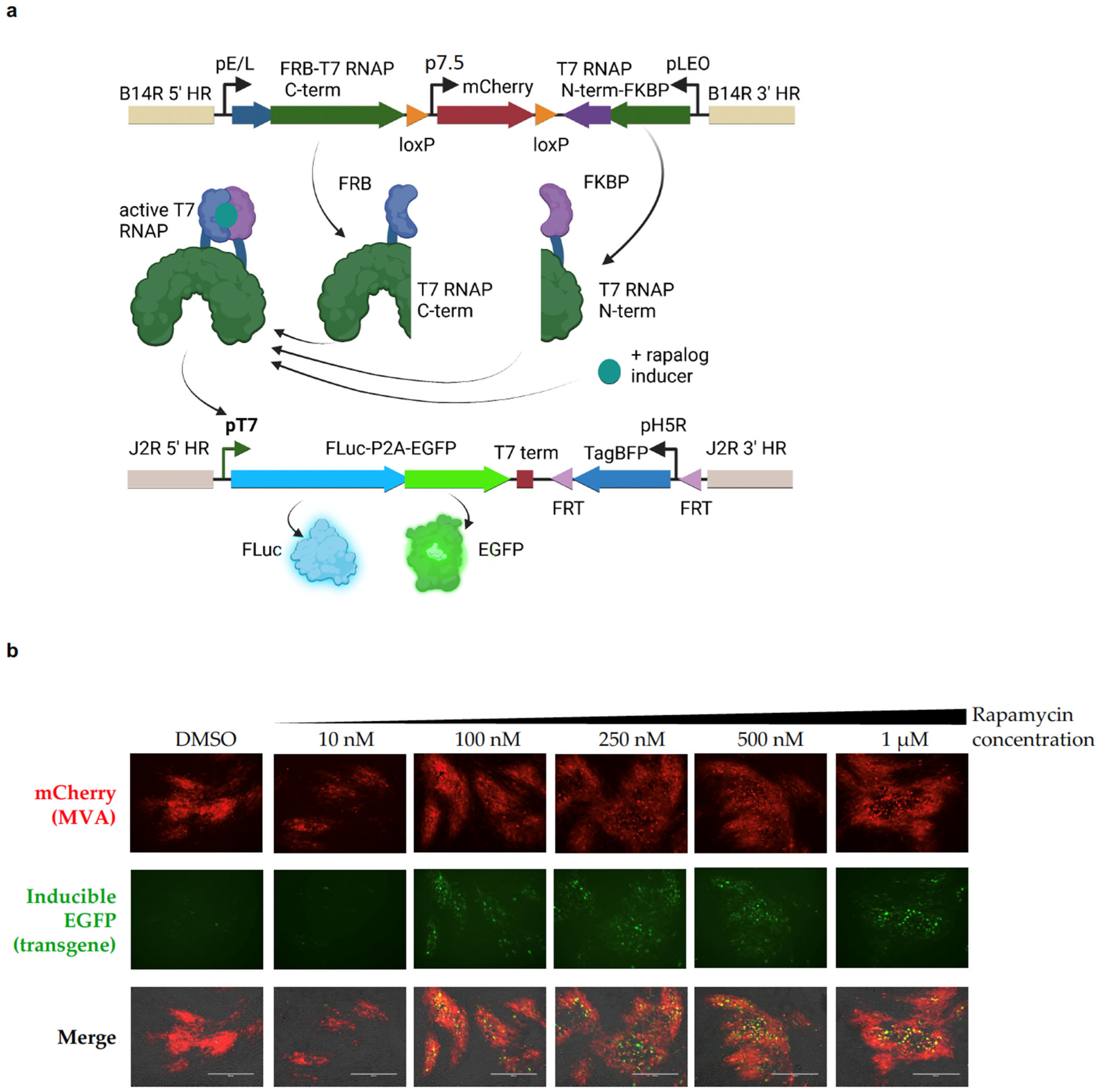
2.1. Split-T7 RNAP Can Be Encoded into the Genome of Poxviruses and Induces Transgene Expression When Supplemented with Rapamycin
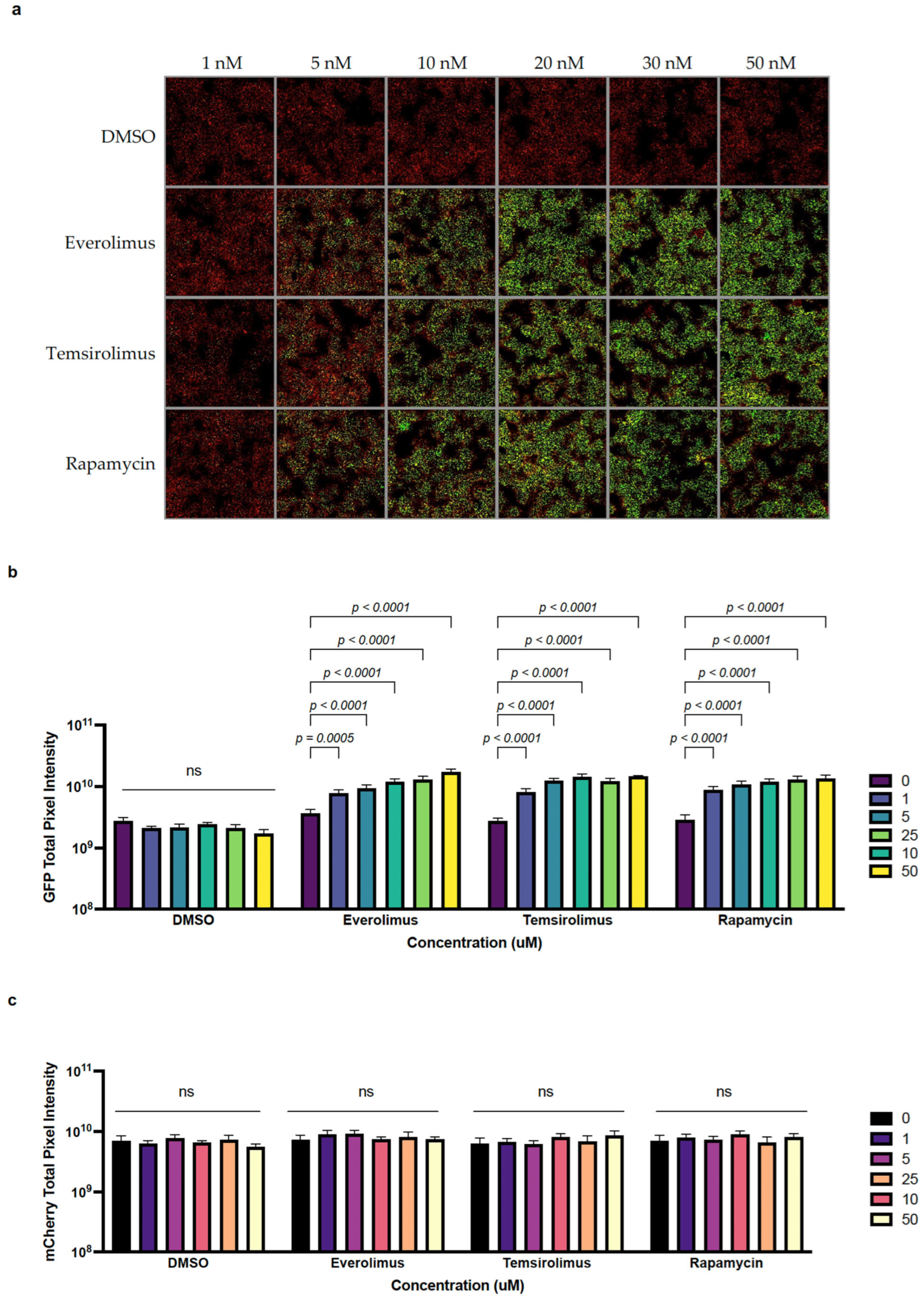
2.2. The Anti-Cancer Drugs Everolimus and Temsirolimus Can Be Used as Inducers of Transgene Expression
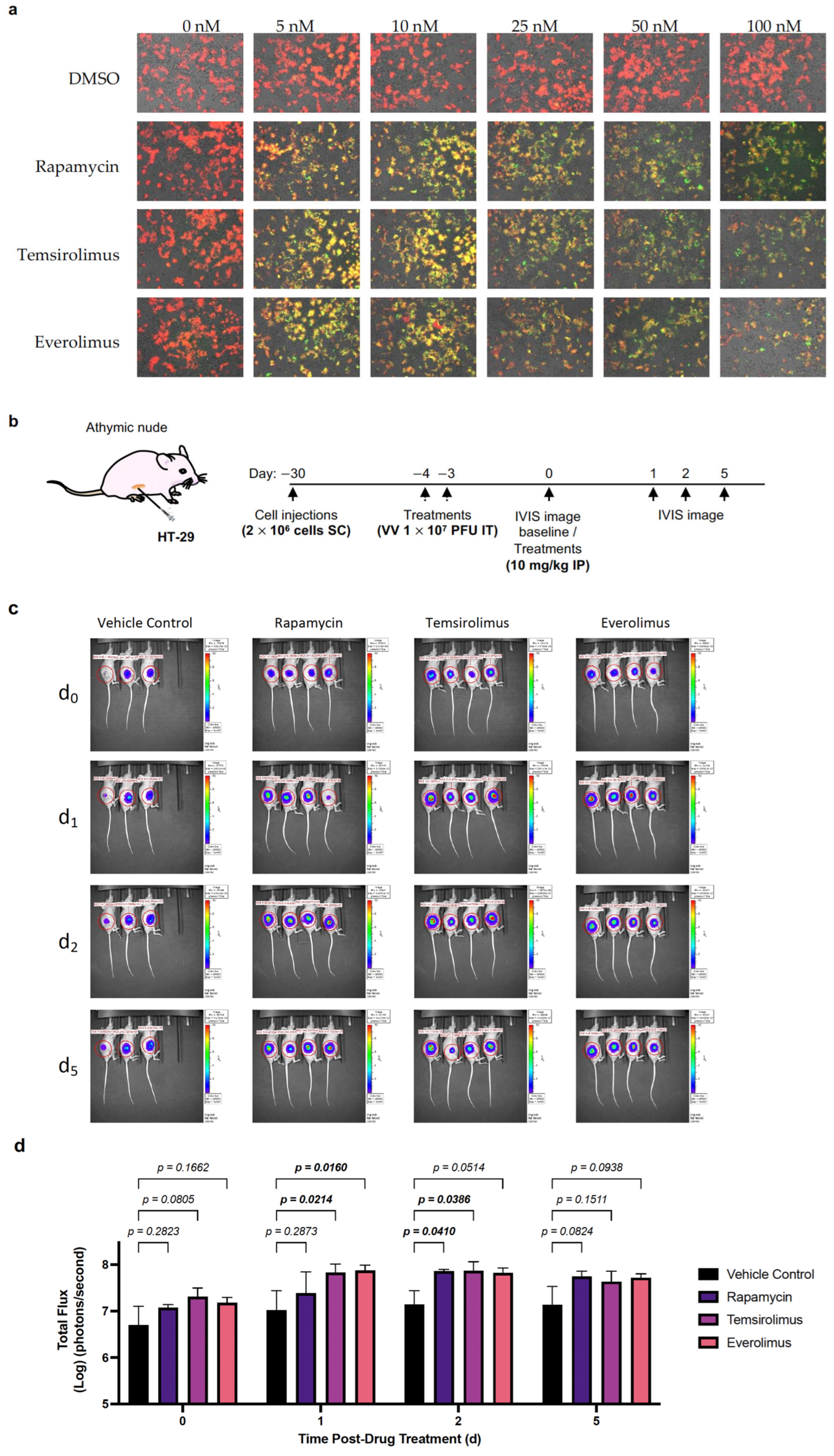
2.3. VV-iFlucEGFP Can Infect Cancer Cell Lines, and Transgene Expression Can Be Induced In Vitro and In Vivo
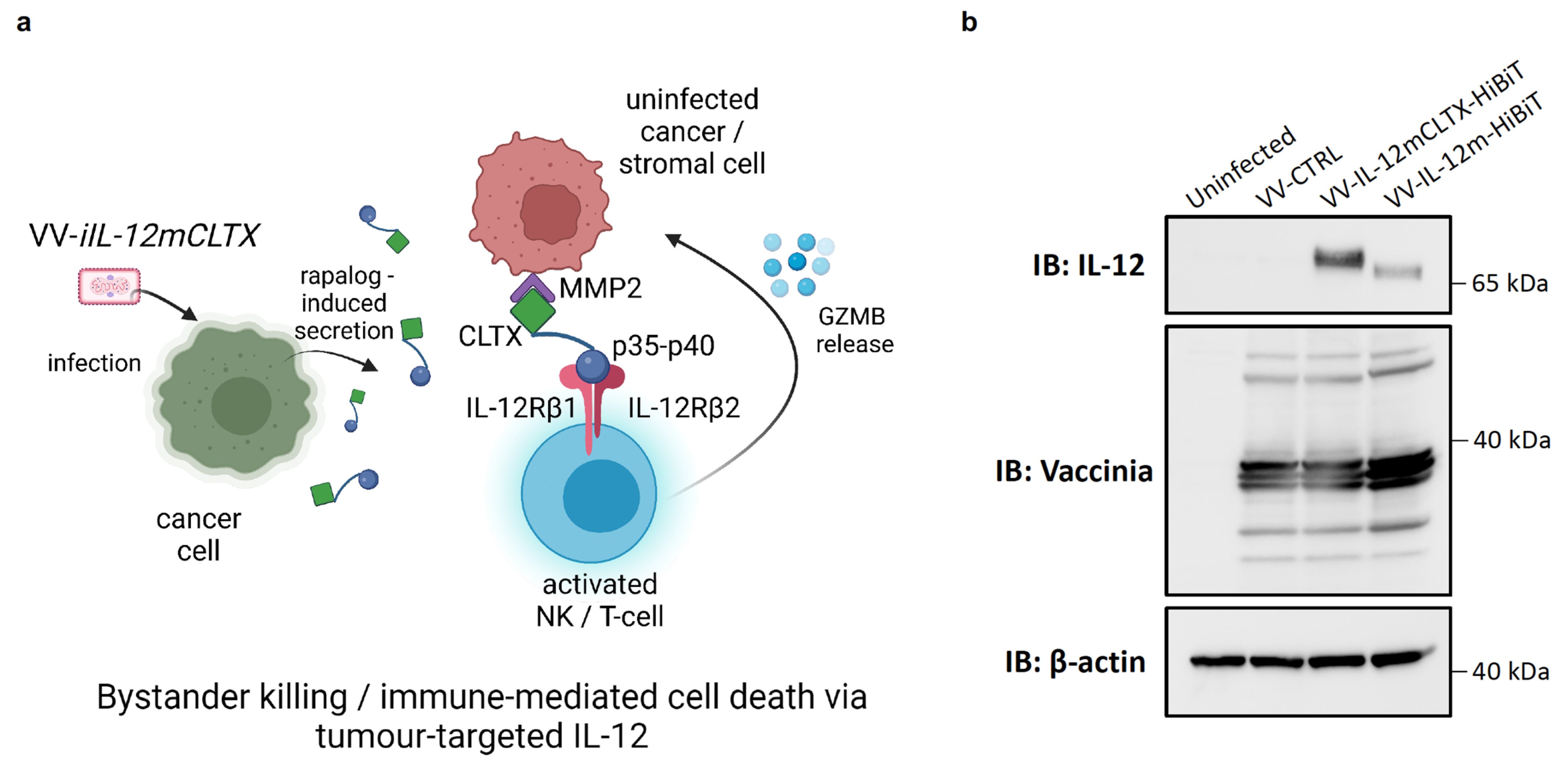
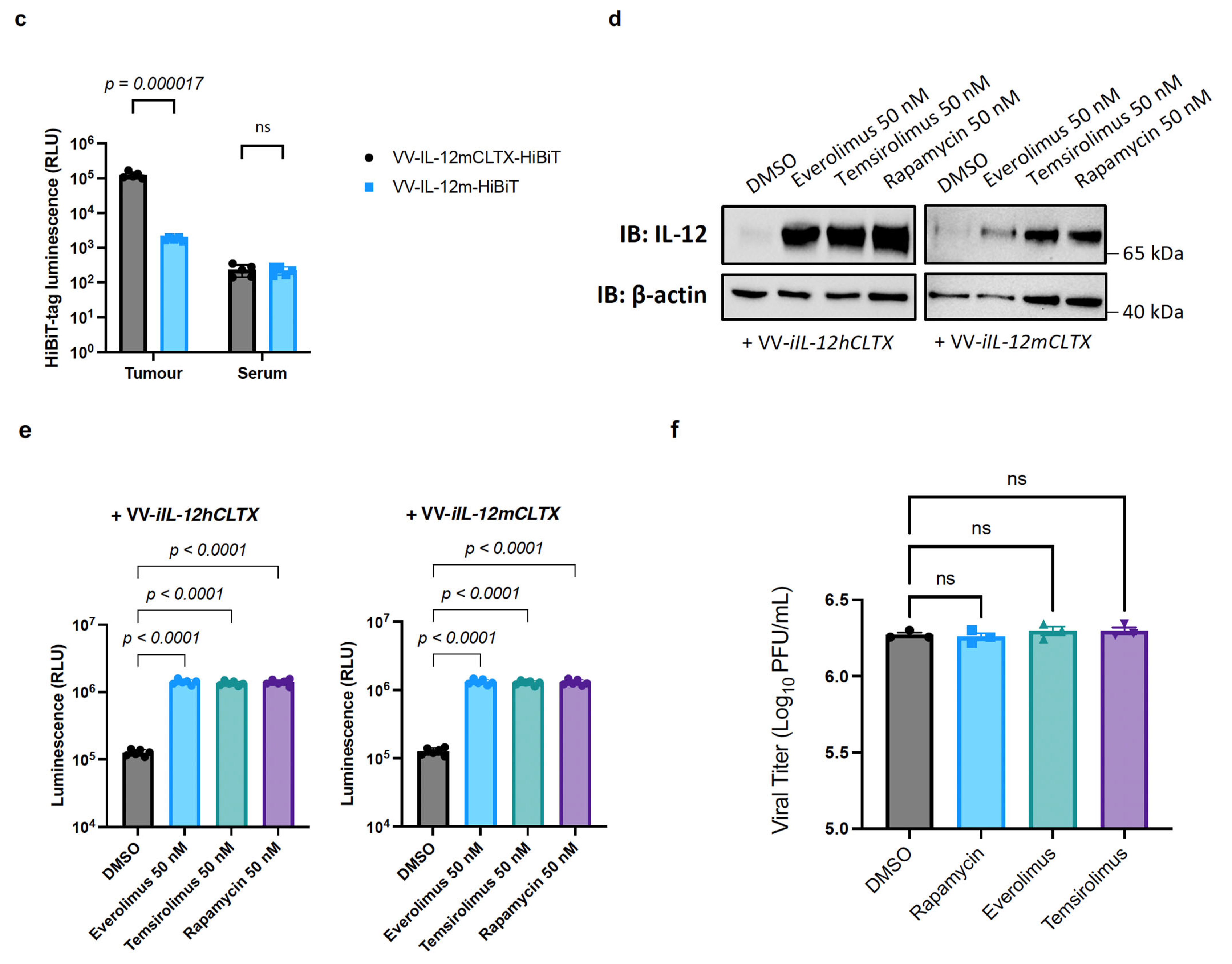
2.4. Design and Validation of Tumour-Targeted IL-12 for Expression of VV for Cancer Therapy
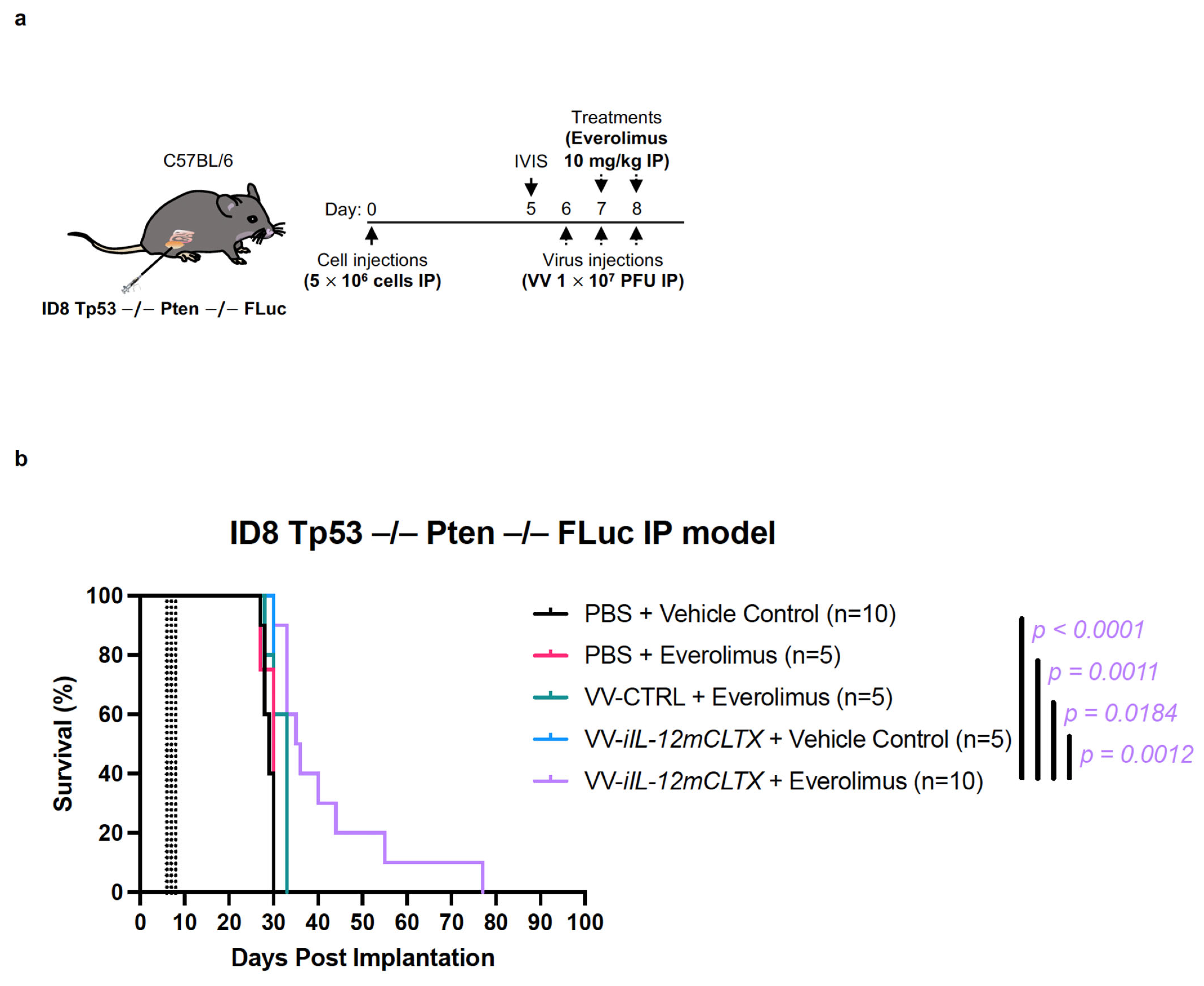
2.5. VV with Induced IL-12 Can Prolong Survival of Mice Bearing Ovarian Tumours
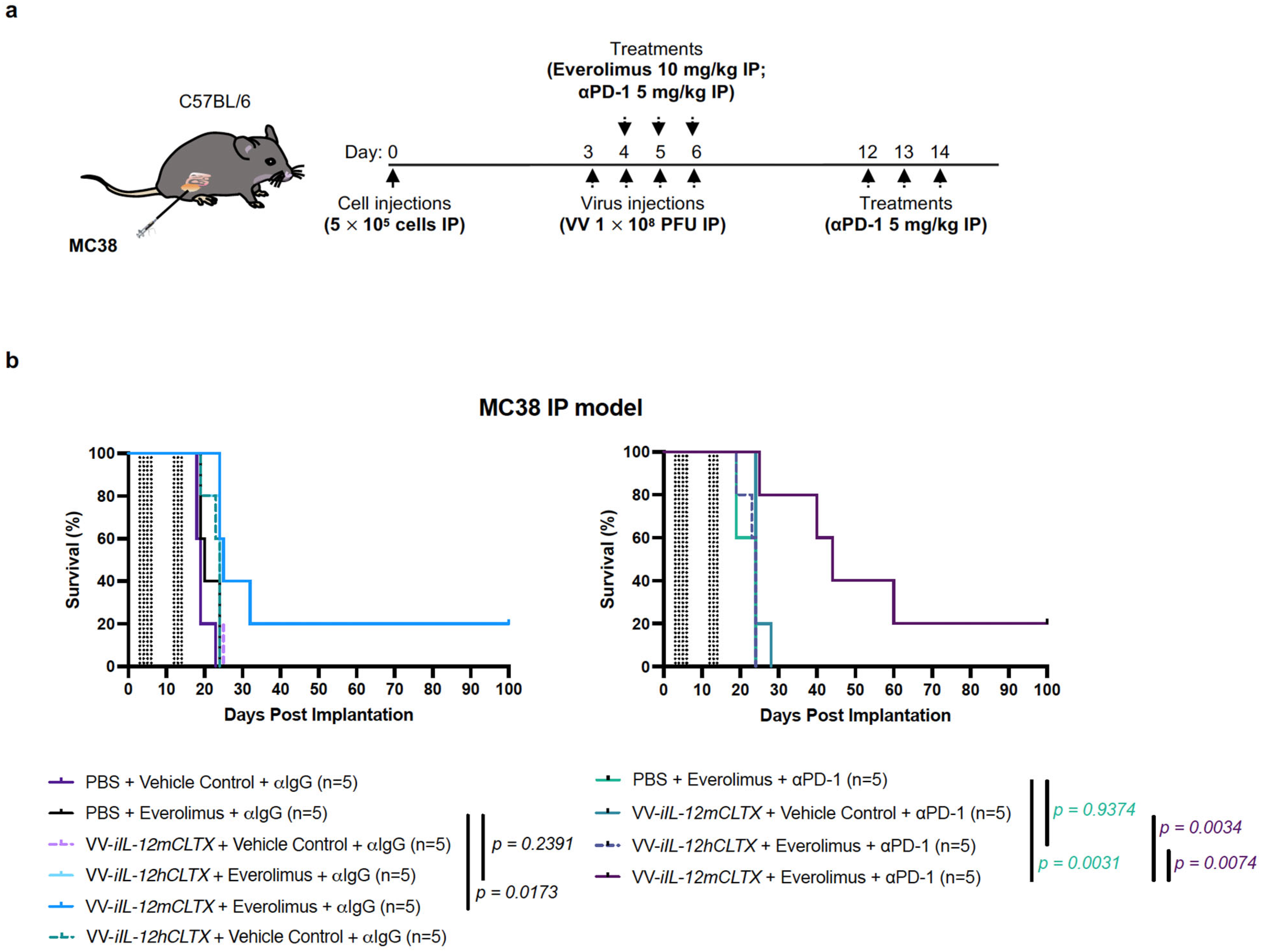
2.6. VV with Induced IL-12 Can Synergize with Immune Checkpoint Inhibition and Cure Mice with Peritoneal Carcinomatosis
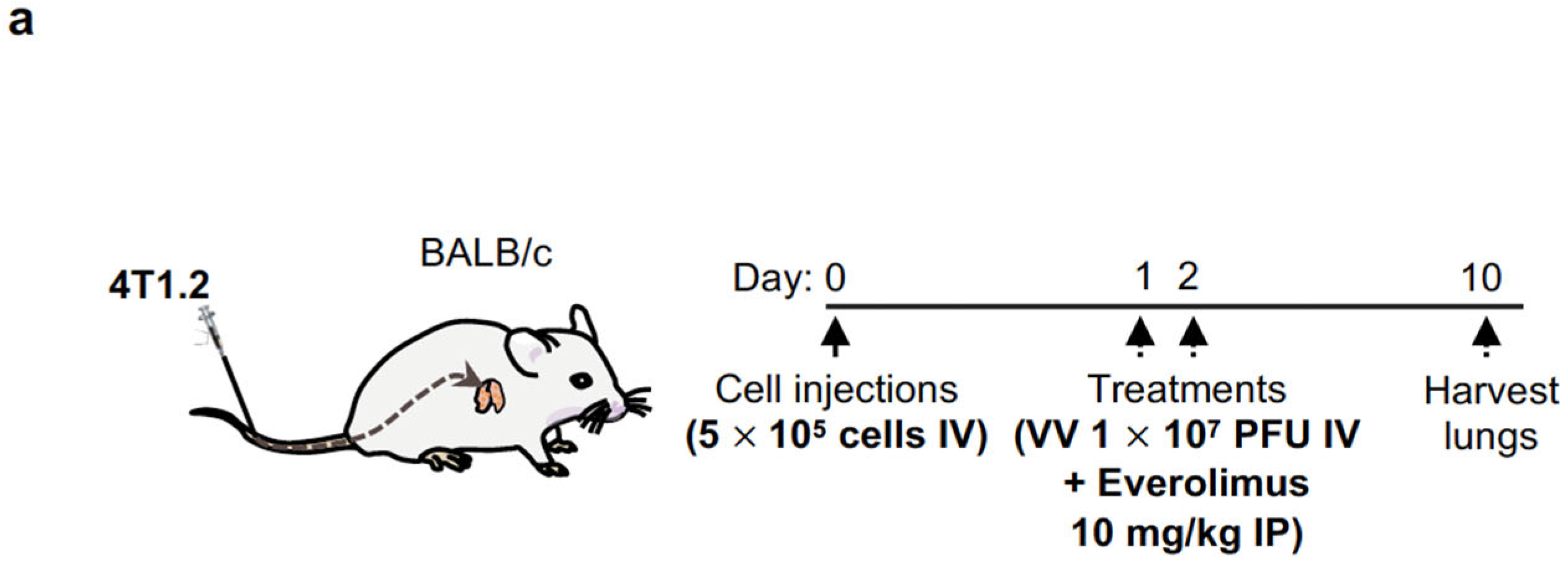
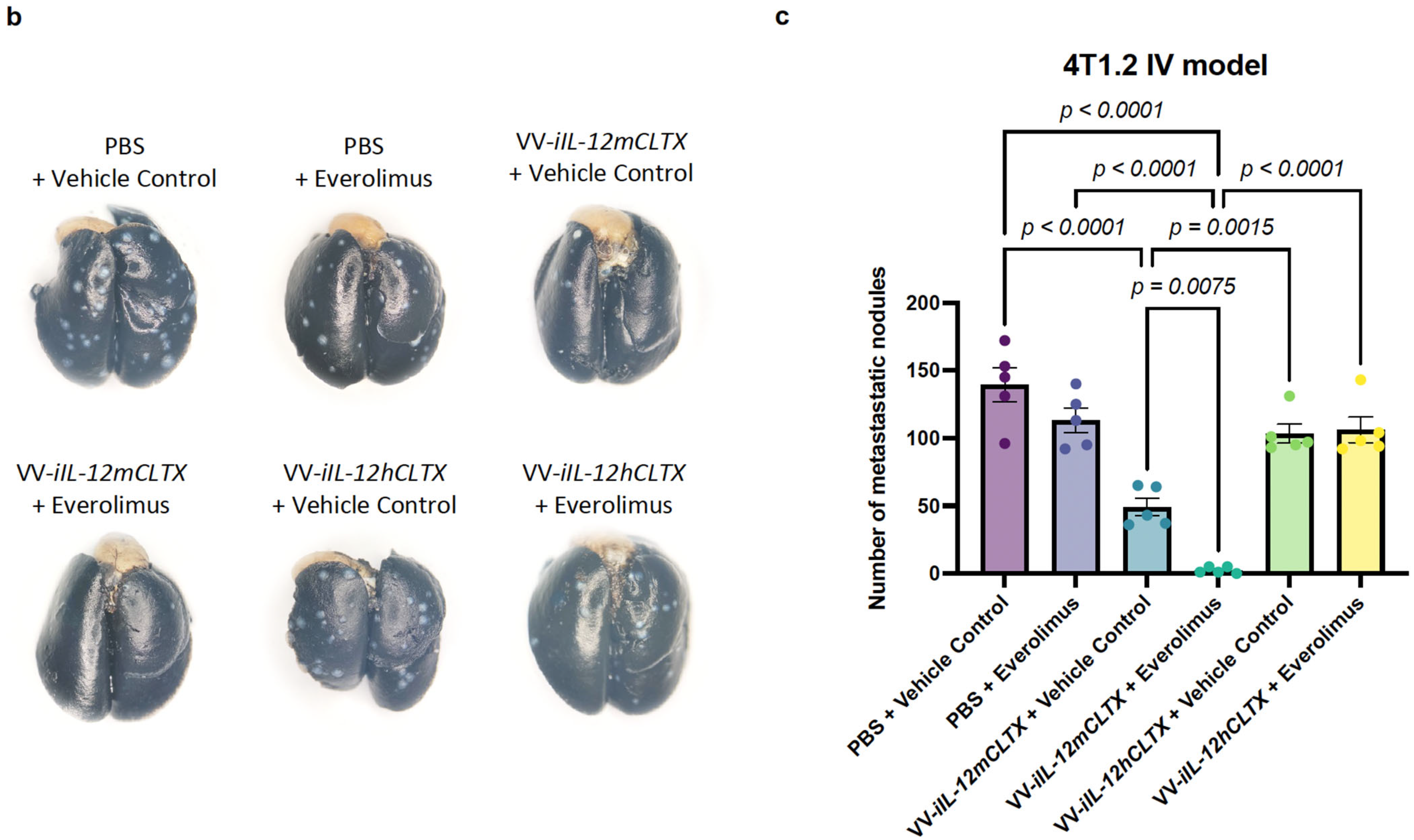
2.7. Intravenous Delivery of VV with Induced IL-12 Demonstrates Efficacy in Reducing Breast Cancer Metastases in Lungs
3. Discussion
4. Materials and Methods
4.1. Constructs
4.2. Cell Culture
4.3. Rapamycin and Rapalog Preparation
4.4. T7-Promoter Driven Reporter and Transgenes
4.5. Recombinant Oncolytic Vaccinia Virus
4.6. Oncolytic Virus Production and Titration
4.7. Immunoblotting
4.8. In Vitro Assays
4.9. Human Xenograft and Syngeneic Murine Model Studies
5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dyck, L.; Mills, K.H.G. Immune Checkpoints and Their Inhibition in Cancer and Infectious Diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T Cells to Kill Cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Marchini, A.; Ilkow, C.S.; Melcher, A. Oncolytic Virus Immunotherapy. Cancers 2021, 13, 3672. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Harrington, K.; Vile, R. Oncolytic Virotherapy as Immunotherapy. Science 2021, 374, 1325–1326. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Chen, N.G.; Lu, J.; Martin, N.; Shen, Y.; Kim, S.-I.; Warner, S.G.; Woo, Y.; Fong, Y. A Chimeric Poxvirus with J2R (Thymidine Kinase) Deletion Shows Safety and Anti-Tumor Activity in Lung Cancer Models. Cancer Gene Ther. 2020, 27, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.-H.; Wechman, S.; McMasters, K.; Zhou, H. Oncolytic Replication of E1b-Deleted Adenoviruses. Viruses 2015, 7, 5767–5779. [Google Scholar] [CrossRef] [PubMed]
- Mejías-Pérez, E.; Carreño-Fuentes, L.; Esteban, M. Development of a Safe and Effective Vaccinia Virus Oncolytic Vector WR-Δ4 with a Set of Gene Deletions on Several Viral Pathways. Mol. Ther.-Oncolytics 2018, 8, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Bell, J.C. Oncolytic Virus Combination Therapy: Killing One Bird with Two Stones. Mol. Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef]
- de Graaf, J.F.; de Vor, L.; Fouchier, R.A.M.; van den Hoogen, B.G. Armed Oncolytic Viruses: A Kick-Start for Anti-Tumor Immunity. Cytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Fong, Y.; Warner, S.G. Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges. Cancers 2020, 12, 1699. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic Armed Oncolytic and Immunologic Therapy for Cancer with JX-594, a Targeted Poxvirus Expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Kowalsky, S.J.; Liu, Z.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Puig, J.M.; Lorenzo, M.M.; Blasco, R. A Vaccinia Virus Recombinant Transcribing an Alphavirus Replicon and Expressing Alphavirus Structural Proteins Leads to Packaging of Alphavirus Infectious Single Cycle Particles. PLoS ONE 2013, 8, e75574. [Google Scholar] [CrossRef]
- Stritzker, J.; Huppertz, S.; Zhang, Q.; Geissinger, U.; Härtl, B.; Gentschev, I.; Szalay, A.A. Inducible Gene Expression in Tumors Colonized by Modified Oncolytic Vaccinia Virus Strains. J. Virol. 2014, 88, 11556–11567. [Google Scholar] [CrossRef]
- Poutou, J.; Bunuales, M.; Gonzalez-Aparicio, M.; German, B.; Zugasti, I.; Hernandez-Alcoceba, R. Adaptation of Vectors and Drug-Inducible Systems for Controlled Expression of Transgenes in the Tumor Microenvironment. J. Control. Release 2017, 268, 247–258. [Google Scholar] [CrossRef]
- Grigg, P.; Titong, A.; Jones, L.A.; Yilma, T.D.; Verardi, P.H. Safety Mechanism Assisted by the Repressor of Tetracycline (SMART) Vaccinia Virus Vectors for Vaccines and Therapeutics. Proc. Natl. Acad. Sci. USA 2013, 110, 15407–15412. [Google Scholar] [CrossRef]
- Gossen, M.; Bujard, H. Tight Control of Gene Expression in Mammalian Cells by Tetracycline-Responsive Promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef]
- Das, T.A.; Tenenbaum, L.; Berkhout, B. Tet-On Systems For Doxycycline-Inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, J.; Verfaillie, C. Doxycycline Inducible Overexpression Systems: How to Induce Your Gene of Interest without Inducing Misinterpretations. Mol. Biol. Cell 2021, 32, 1517–1522. [Google Scholar] [CrossRef]
- Scialo, F.; Sriram, A.; Stefanatos, R.; Sanz, A. Practical Recommendations for the Use of the GeneSwitch Gal4 System to Knock-Down Genes in Drosophila Melanogaster. PLoS ONE 2016, 11, e0161817. [Google Scholar] [CrossRef]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef]
- Elkrief, A.; Derosa, L.; Kroemer, G.; Zitvogel, L.; Routy, B. The Negative Impact of Antibiotics on Outcomes in Cancer Patients Treated with Immunotherapy: A New Independent Prognostic Factor? Ann. Oncol. 2019, 30, 1572–1579. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut Microbiome Influences Efficacy of PD-1–Based Immunotherapy against Epithelial Tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Gramenitskaya, D.; Altmann, D.M.; Boyton, R.J.; Mullish, B.H.; Marchesi, J.R.; Bower, M. Antibiotic Therapy and Outcome from Immune-Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 287. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, X.; Wei, J.; Li, B.; Shao, D.; Li, Y.; Liu, K.; Shi, Y.; Zhou, B.; Qiu, Y.; et al. Antiviral Activity of Doxycycline against Vesicular Stomatitis Virus in Vitro. FEMS Microbiol. Lett. 2015, 362, fnv195. [Google Scholar] [CrossRef]
- Alonso, M.M.; Jiang, H.; Yokoyama, T.; Xu, J.; Bekele, N.B.; Lang, F.F.; Kondo, S.; Gomez-Manzano, C.; Fueyo, J. Delta-24-RGD in Combination With RAD001 Induces Enhanced Anti-Glioma Effect via Autophagic Cell Death. Mol. Ther. 2008, 16, 487–493. [Google Scholar] [CrossRef]
- Pu, J.; Zinkus-Boltz, J.; Dickinson, B.C. Evolution of a Split RNA Polymerase as a Versatile Biosensor Platform. Nat. Chem. Biol. 2017, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Pulido, M.; Roubaud, G.; Cazeau, A.-L.; Mahammedi, H.; Vedrine, L.; Joly, F.; Mourey, L.; Pfister, C.; Goberna, A.; Lortal, B.; et al. Safety and Efficacy of Temsirolimus as Second Line Treatment for Patients with Recurrent Bladder Cancer. BMC Cancer 2018, 18, 194. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Faivre, S.; Serova, M.; Raymond, E. MTORC1 Inhibitors: Is Temsirolimus in Renal Cancer Telling Us How They Really Work? Br. J. Cancer 2008, 99, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, R.; Santoni, M.; Verzoni, E.; Grassi, P.; Testa, I.; de Braud, F.; Cascinu, S.; Procopio, G. Everolimus and Temsirolimus Are Not the Same Second-Line in Metastatic Renal Cell Carcinoma. A Systematic Review and Meta-Analysis of Literature Data. Clin. Genitourin. Cancer 2015, 13, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Chao, E.; Da, L.-T.; Yu, J. A Viral T7 RNA Polymerase Ratcheting Along DNA With Fidelity Control. Comput. Struct. Biotechnol. J. 2019, 17, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Fuerst, T.R.; Niles, E.G.; Studier, F.W.; Moss, B. Eukaryotic Transient-Expression System Based on Recombinant Vaccinia Virus That Synthesizes Bacteriophage T7 RNA Polymerase. Proc. Natl. Acad. Sci. USA 1986, 83, 8122–8126. [Google Scholar] [CrossRef]
- Fuerst, T.R.; Earl, P.L.; Moss, B. Use of a Hybrid Vaccinia Virus-T7 RNA Polymerase System for Expression of Target Genes. Mol. Cell Biol. 1987, 7, 299. [Google Scholar]
- Borkotoky, S.; Murali, A. The Highly Efficient T7 RNA Polymerase: A Wonder Macromolecule in Biological Realm. Int. J. Biol. Macromol. 2018, 118, 49–56. [Google Scholar] [CrossRef]
- Tahirov, T.H.; Temiakov, D.; Anikin, M.; Patlan, V.; McAllister, W.T.; Vassylyev, D.G.; Yokoyama, S. Structure of a T7 RNA Polymerase Elongation Complex at 2.9 Å Resolution. Mol. Cell Biol. 2002, 420, 1012. [Google Scholar] [CrossRef]
- Mirlekar, B.; Pylayeva-Gupta, Y. IL-12 Family Cytokines in Cancer and Immunotherapy. Cancers 2021, 13, 167. [Google Scholar] [CrossRef]
- Agliardi, G.; Liuzzi, A.R.; Hotblack, A.; De Feo, D.; Núñez, N.; Stowe, C.L.; Friebel, E.; Nannini, F.; Rindlisbacher, L.; Roberts, T.A.; et al. Intratumoral IL-12 Delivery Empowers CAR-T Cell Immunotherapy in a Pre-Clinical Model of Glioblastoma. Nat. Commun. 2021, 12, 444. [Google Scholar] [CrossRef]
- Gately, M.K.; Renzetti, L.M.; Magram, J.; Stern, A.S.; Adorini, L.; Gubler, U.; Presky, D.H. THE INTERLEUKIN-12/INTERLEUKIN-12-RECEPTOR SYSTEM: Role in Normal and Pathologic Immune Responses. Annu. Rev. Immunol. 1998, 16, 495–521. [Google Scholar] [CrossRef]
- Nguyen, K.G.; Vrabel, M.R.; Mantooth, S.M.; Hopkins, J.J.; Wagner, E.S.; Gabaldon, T.A.; Zaharoff, D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020, 11, 575597. [Google Scholar] [CrossRef]
- Mamelak, A.N.; Jacoby, D.B. Targeted Delivery of Antitumoral Therapy to Glioma and Other Malignancies with Synthetic Chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Starr, R.; Chang, W.-C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-Directed CAR T Cells for Specific and Effective Targeting of Glioblastoma. Sci. Transl. Med. 2020, 12, eaaw2672. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.A.; O’Neal, J.; Sontheimer, H. Chlorotoxin, a Scorpion-Derived Peptide, Specifically Binds to Gliomas and Tumors of Neuroectodermal Origin. Glia 2002, 39, 162–173. [Google Scholar] [CrossRef]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin Inhibits Glioma Cell Invasion via Matrix Metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar] [CrossRef]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2017; Volume 97, pp. 187–243. ISBN 978-0-12-811801-6. [Google Scholar]
- Sutter, G.; Moss, B. Nonreplicating Vaccinia Vector Efficiently Expresses Recombinant Genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Sisler, J.R.; Moss, B. Compact, Synthetic, Vaccinia Virus Early/Late Promoter for Protein Expression. BioTechniques 1997, 23, 1094–1097. [Google Scholar] [CrossRef]
- Alharbi, N.K. Poxviral Promoters for Improving the Immunogenicity of MVA Delivered Vaccines. Hum. Vaccines Immunother. 2019, 15, 203–209. [Google Scholar] [CrossRef]
- Puhlmann, M.; Gnant, M.; Brown, C.K.; Alexander, H.R.; Bartlett, D.L. Thymidine Kinase-Deleted Vaccinia Virus Expressing Purine Nucleoside Phosphorylase as a Vector for Tumor-Directed Gene Therapy. Hum. Gene Ther. 1999, 10, 649–657. [Google Scholar] [CrossRef]
- Tang, X.; Liu, X.; Tao, G.; Qin, M.; Yin, G.; Suo, J.; Suo, X. “Self-Cleaving” 2A Peptide from Porcine Teschovirus-1 Mediates Cleavage of Dual Fluorescent Proteins in Transgenic Eimeria Tenella. Vet. Res. 2016, 47, 68. [Google Scholar] [CrossRef]
- Rosel, J.L. Conserved TAAATG Sequence at the Transcriptional and Translational Initiation Sites of Vaccinia Virus Late Genes Deduced by Structural and Functional Analysis of the Hindlll H Genome Fragment. J. Virol. 1986, 60, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.; Poutou, J.; Martin, N.T.; Azad, T.; Singaravelu, R.; Crupi, M.J.F.; Jamieson, T.; He, X.; Marius, R.; Petryk, J.; et al. Single-Dose Replicating Poxvirus Vector-Based RBD Vaccine Drives Robust Humoral and T Cell Immune Response against SARS-CoV-2 Infection. Mol. Ther. 2022, 30, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Pelin, A.; Boulton, S.; Tamming, L.A.; Bell, J.C.; Singaravelu, R. Engineering Vaccinia Virus as an Immunotherapeutic Battleship to Overcome Tumor Heterogeneity. Expert Opin. Biol. Ther. 2020, 20, 1083–1097. [Google Scholar] [CrossRef]
- Crupi, M.J.F.; Taha, Z.; Janssen, T.J.A.; Petryk, J.; Boulton, S.; Alluqmani, N.; Jirovec, A.; Kassas, O.; Khan, S.T.; Vallati, S.; et al. Oncolytic Virus Driven T-Cell-Based Combination Immunotherapy Platform for Colorectal Cancer. Front. Immunol. 2022, 13, 1029269. [Google Scholar] [CrossRef]
- Cao, X.; Liang, Y.; Hu, Z.; Li, H.; Yang, J.; Hsu, E.J.; Zhu, J.; Zhou, J.; Fu, Y.-X. Next Generation of Tumor-Activating Type I IFN Enhances Anti-Tumor Immune Responses to Overcome Therapy Resistance. Nat. Commun. 2021, 12, 5866. [Google Scholar] [CrossRef]
- Crupi, M.J.F.; Bell, J.C.; Singaravelu, R. Concise Review: Targeting Cancer Stem Cells and Their Supporting Niche Using Oncolytic Viruses. Stem Cells 2019, 37, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Woerly, G.; Brooks, N.; Ryffel, B. Effect of Rapamycin on the Expression of the IL-2 Receptor (CD25). Clin. Exp. Immunol. 2003, 103, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Disare, M.; Dickinson, B.C. Evolution of C-Terminal Modification Tolerance in Full-Length and Split T7 RNA Polymerase Biosensors. ChemBioChem 2019, 20, 1547–1553. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Thaker, P.H.; Bradley, W.H.; Leath, C.A.; Gunderson Jackson, C.; Borys, N.; Anwer, K.; Musso, L.; Matsuzaki, J.; Bshara, W.; Odunsi, K.; et al. GEN-1 in Combination with Neoadjuvant Chemotherapy for Patients with Advanced Epithelial Ovarian Cancer: A Phase I Dose-Escalation Study. Clin. Cancer Res. 2021, 27, 5536–5545. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, N.T.; Crupi, M.J.F.; Taha, Z.; Poutou, J.; Whelan, J.T.; Vallati, S.; Petryk, J.; Marius, R.; Austin, B.; Azad, T.; et al. Engineering Rapalog-Inducible Genetic Switches Based on Split-T7 Polymerase to Regulate Oncolytic Virus-Driven Production of Tumour-Localized IL-12 for Anti-Cancer Immunotherapy. Pharmaceuticals 2023, 16, 709. https://doi.org/10.3390/ph16050709
Martin NT, Crupi MJF, Taha Z, Poutou J, Whelan JT, Vallati S, Petryk J, Marius R, Austin B, Azad T, et al. Engineering Rapalog-Inducible Genetic Switches Based on Split-T7 Polymerase to Regulate Oncolytic Virus-Driven Production of Tumour-Localized IL-12 for Anti-Cancer Immunotherapy. Pharmaceuticals. 2023; 16(5):709. https://doi.org/10.3390/ph16050709
Chicago/Turabian StyleMartin, Nikolas T., Mathieu J. F. Crupi, Zaid Taha, Joanna Poutou, Jack T. Whelan, Sydney Vallati, Julia Petryk, Ricardo Marius, Bradley Austin, Taha Azad, and et al. 2023. "Engineering Rapalog-Inducible Genetic Switches Based on Split-T7 Polymerase to Regulate Oncolytic Virus-Driven Production of Tumour-Localized IL-12 for Anti-Cancer Immunotherapy" Pharmaceuticals 16, no. 5: 709. https://doi.org/10.3390/ph16050709