
Synthesis, Fluorine-18 Radiolabeling, and In Vivo PET Imaging of a Hydrophilic Fluorosulfotetrazine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
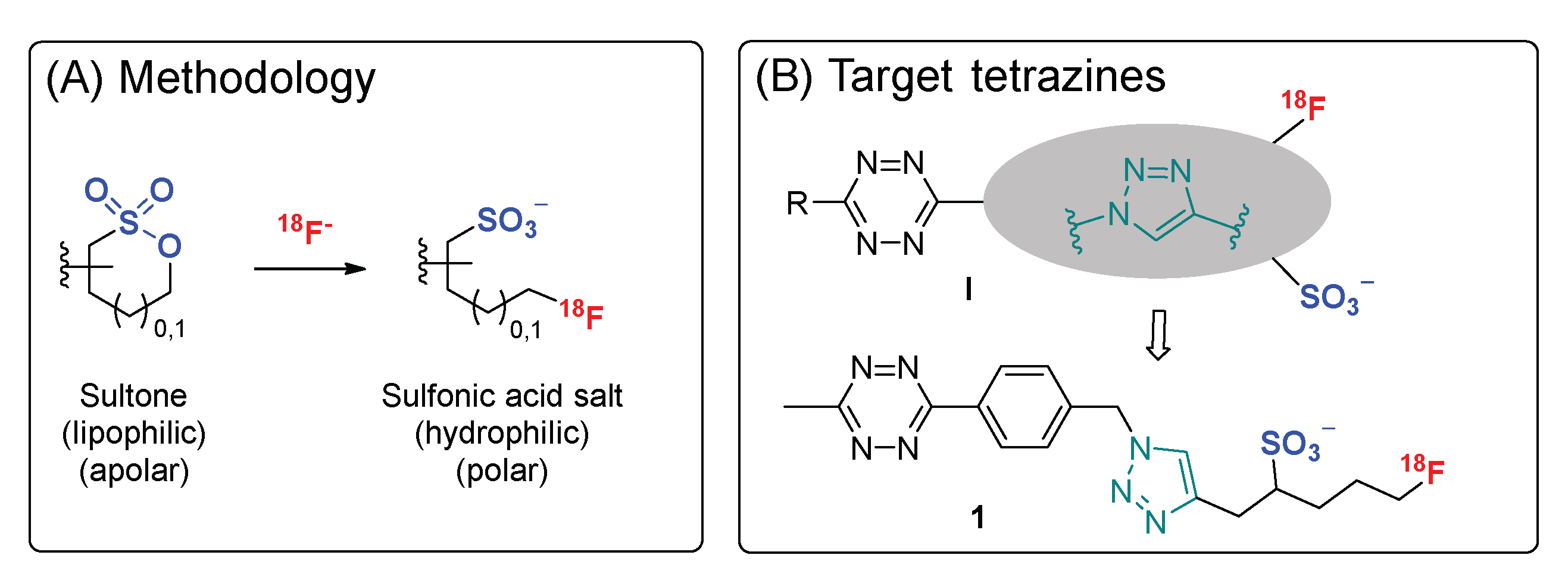
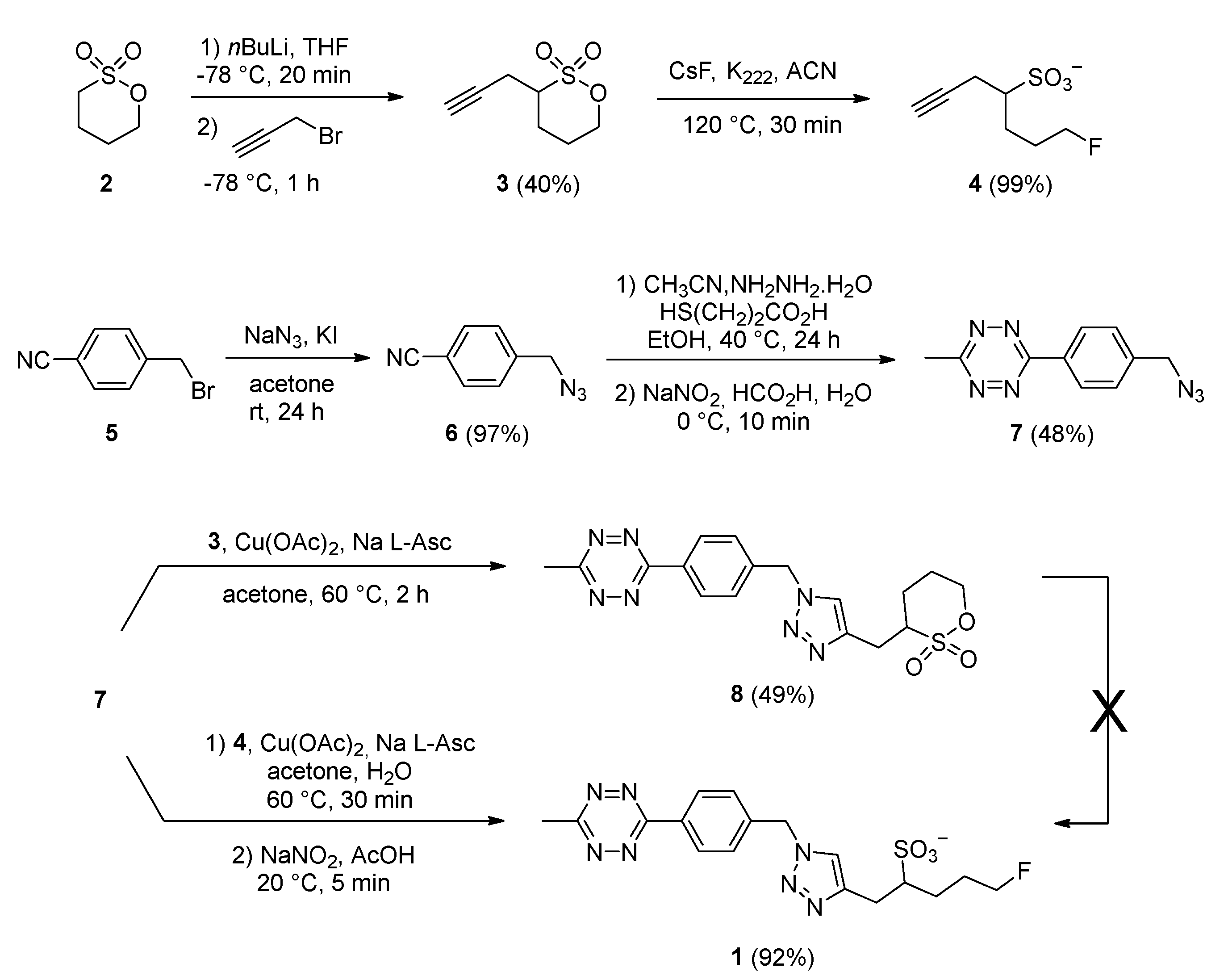
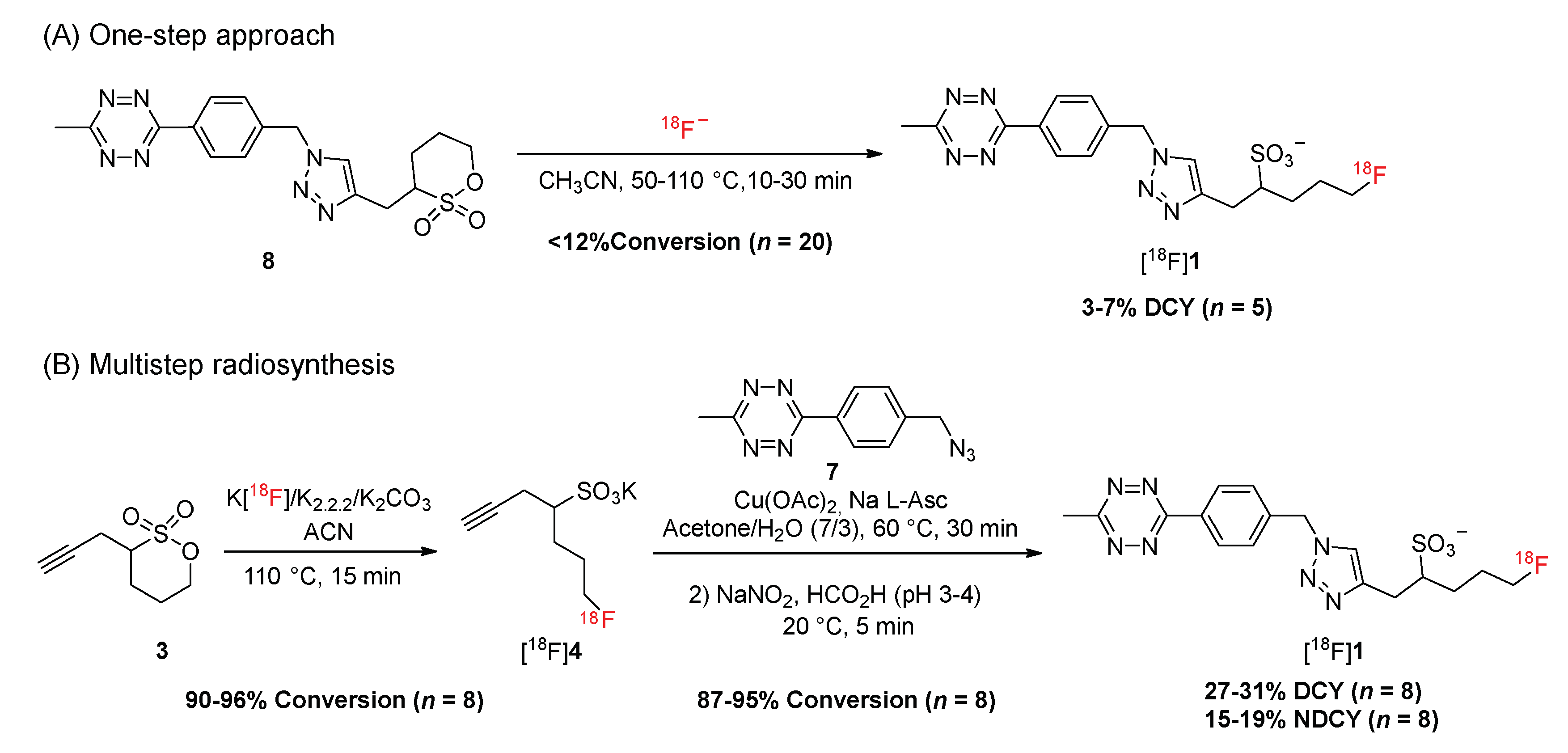
2.1. Synthesis of the Fluorosulfotetrazine 1
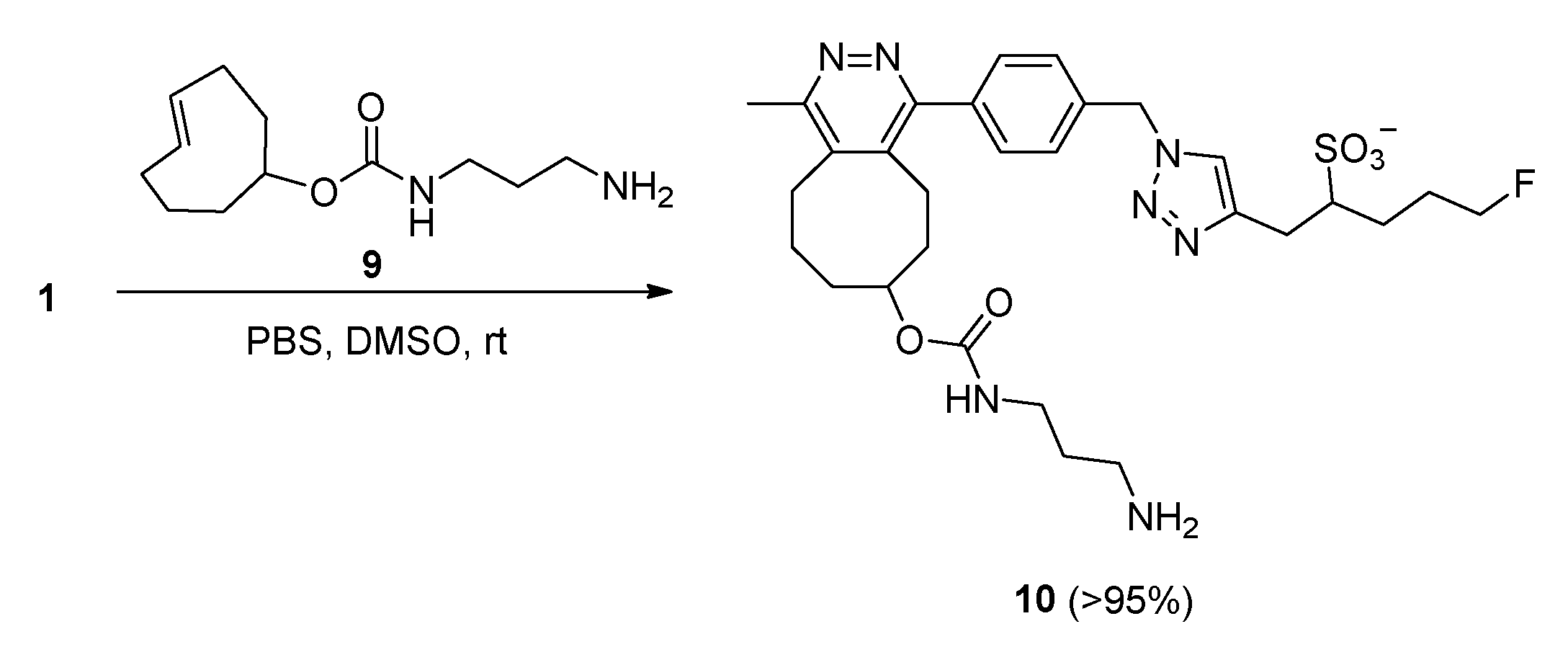
2.2. IEDDA Reaction of the Fluorosulfotetrazine 1 with TCO Reagent 9
2.3. Development of the Radiosynthesis of [18F]Fluorosulfotetrazine [18F]1
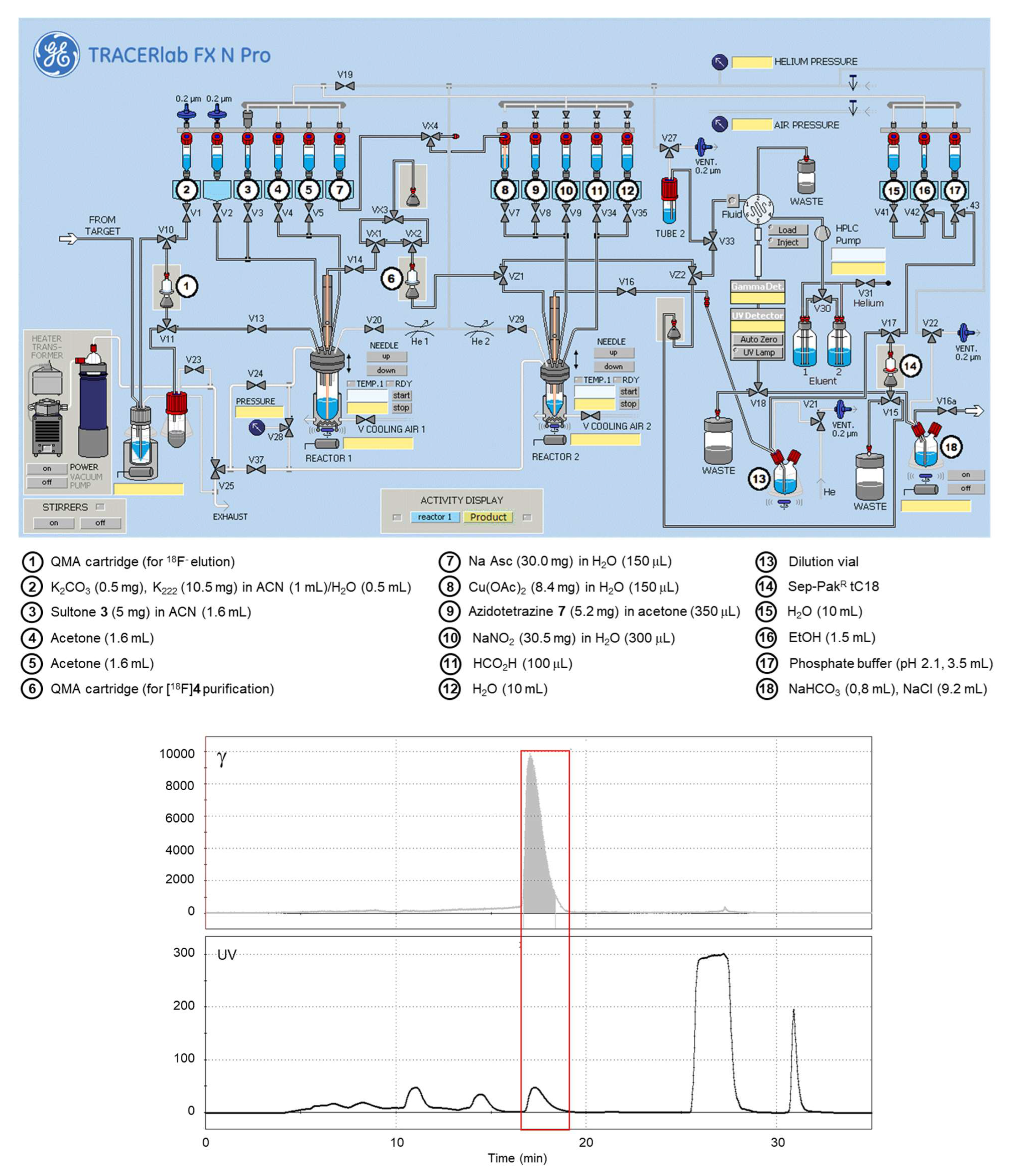
2.4. Automated Radiosynthesis of [18F]Fluorosulfotetrazine [18F]1
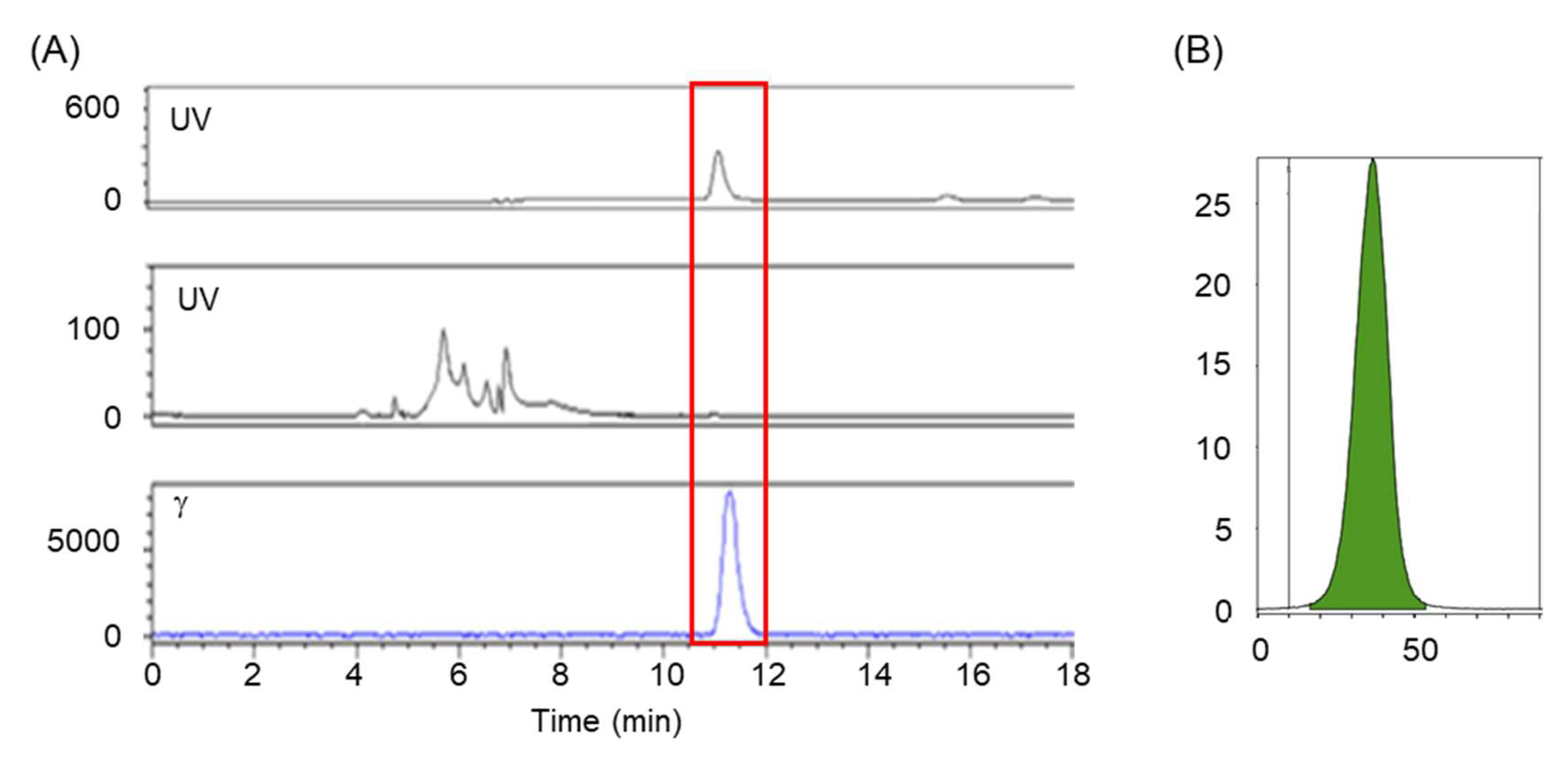
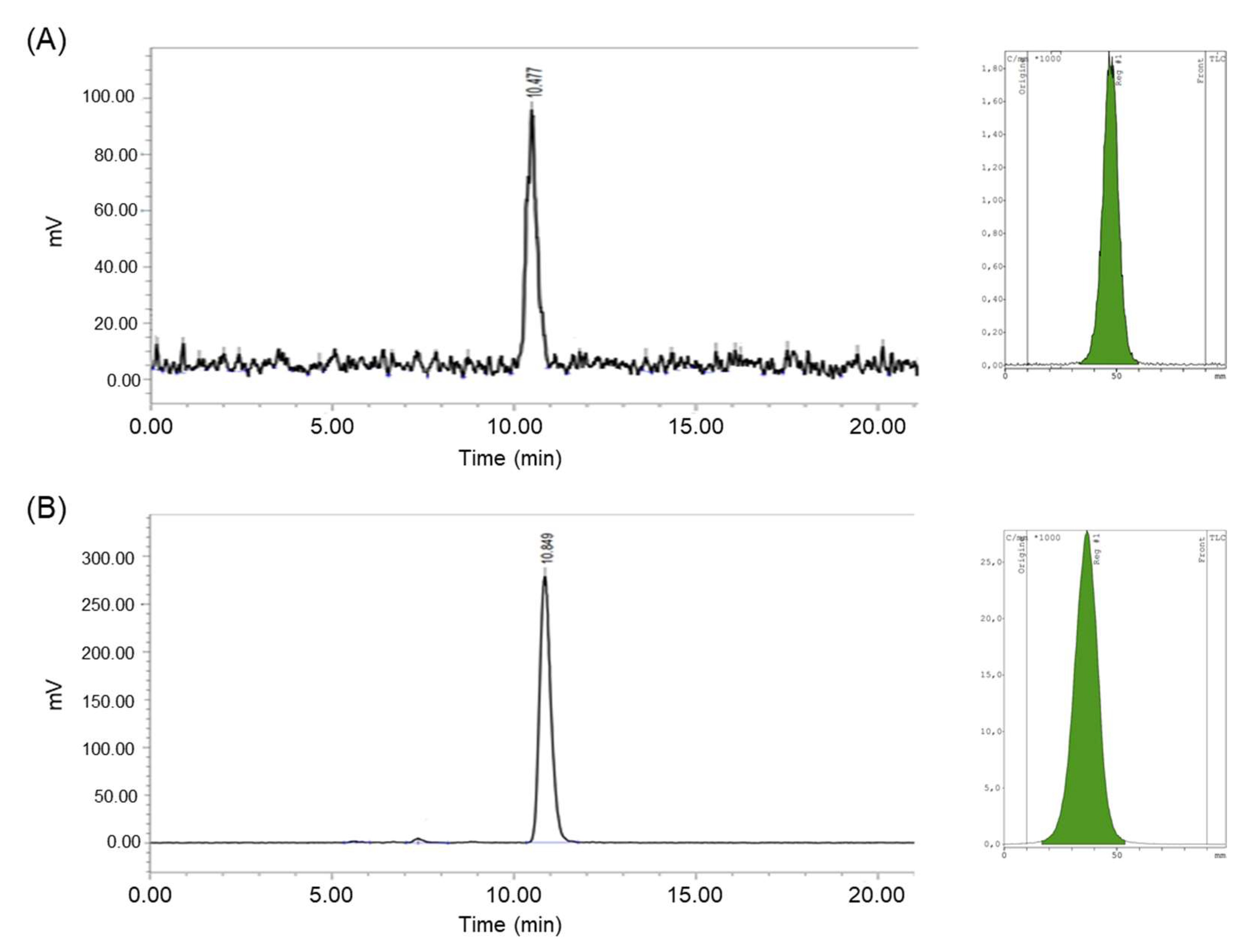
2.5. Quality Control
2.6. LogP and LogD7.4 Measurements
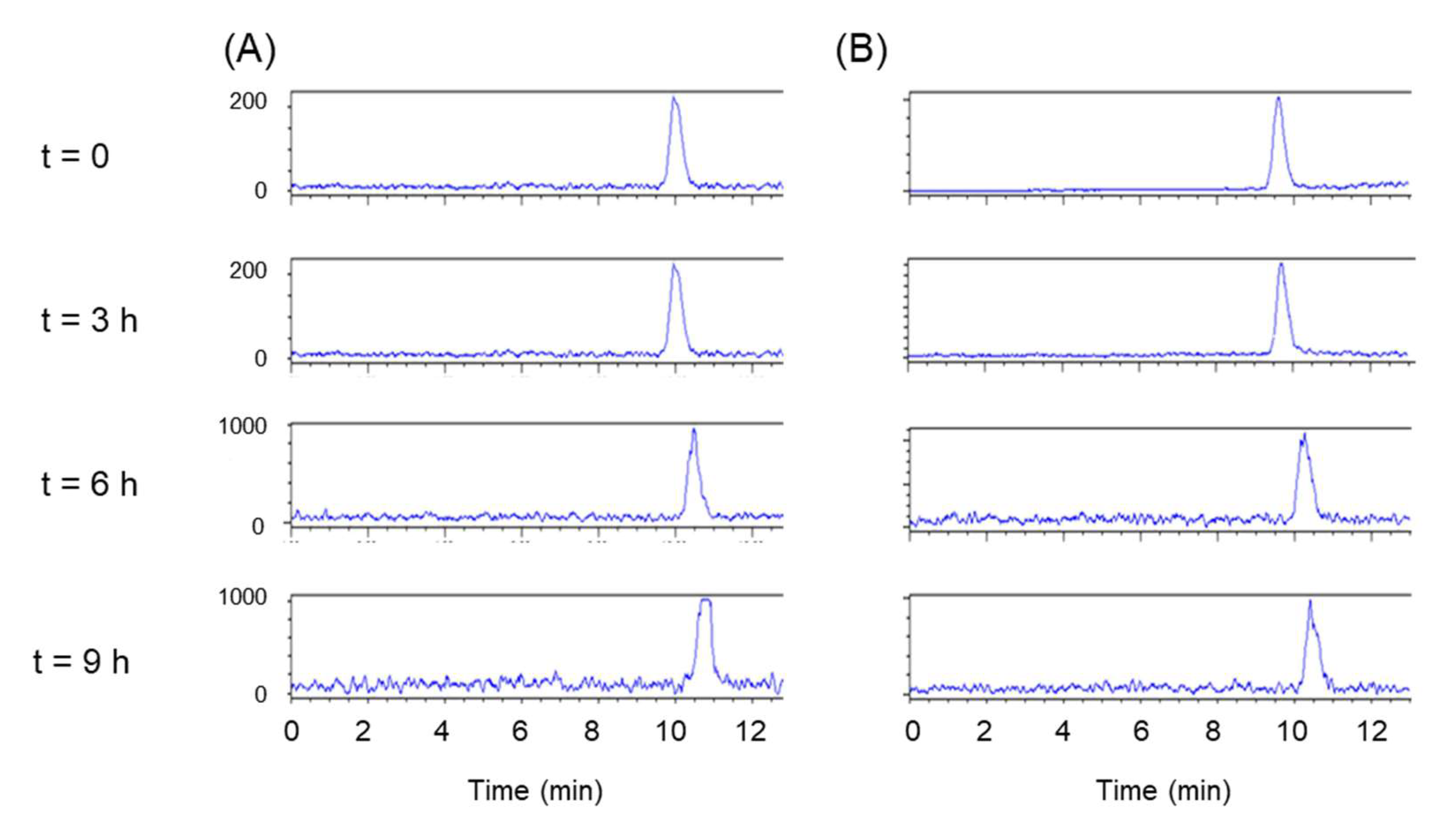
2.7. In Vitro Stability Studies
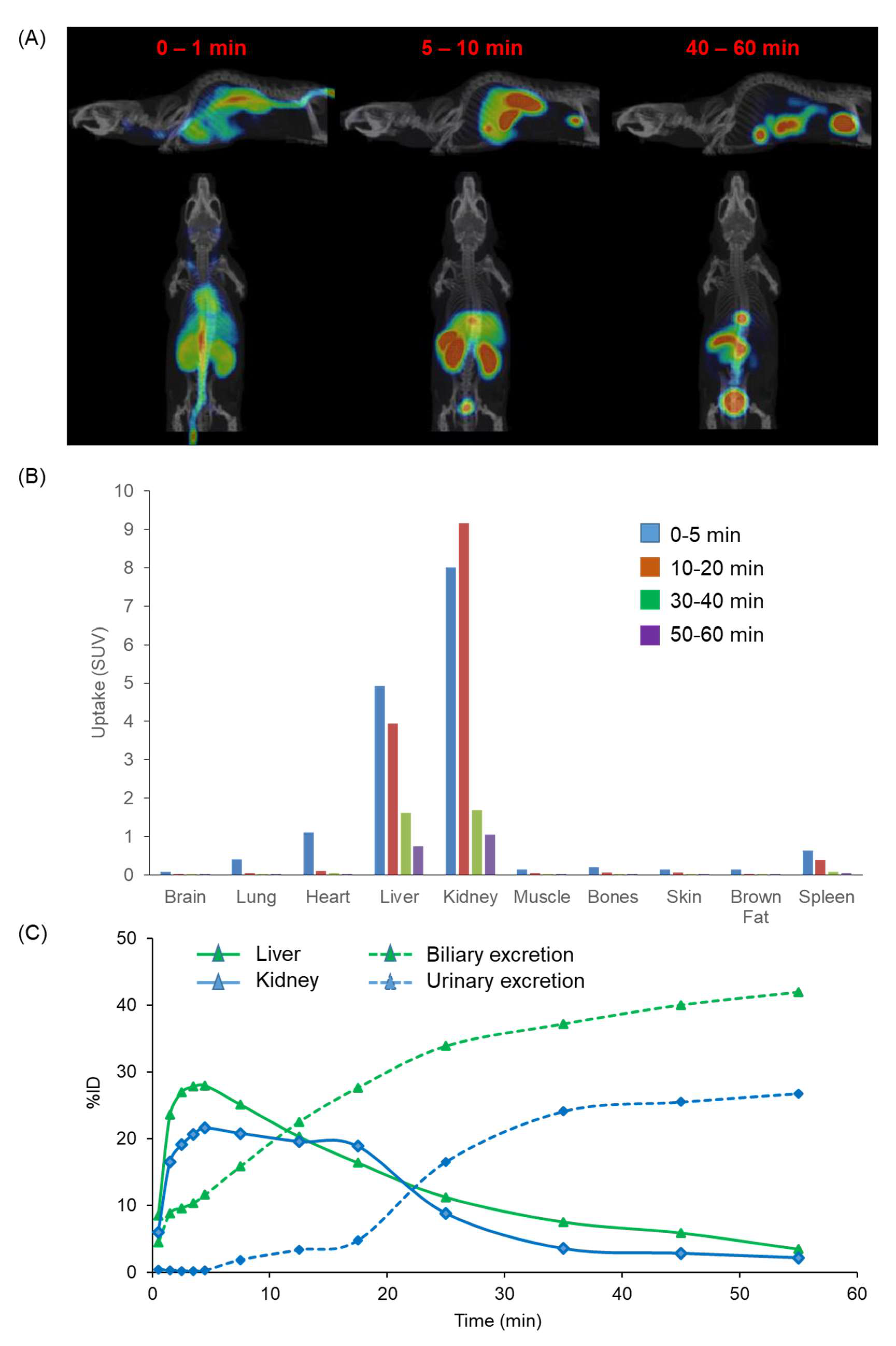
2.8. In Vivo Biodistribution Using PET/MR in Mice
2.9. Radiometabolite Analysis of Mouse Plasma Samples
3. Materials and Methods
3.1. Chemical Syntheses
3.1.1. General
3.1.2. 3-(Prop-2-yn-1-yl)-1,2-oxathiane 2,2-dioxide 3
3.1.3. 7-(Fluoro)hept-1-yne-4-sulfonic Acid 4
3.1.4. 4-(Azidomethyl)benzonitrile 6
3.1.5. 3-(4-(Azidomethyl)phenyl)-6-methyl-1,2,4,5-tetrazine 7
3.1.6. 3-((1-(4-(6-Methyl-1,2,4,5-tetrazin-3-yl)benzyl)-1H-1,2,3-triazol-4-yl)methyl)-1,2-oxathiane 2,2-dioxide 8
3.1.7. 5-(Fluoro)-1-(1-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)-1H-1,2,3-triazol-4-yl)pentane-2-sulfonic Acid 1
3.2. Automated Radiosynthesis of [18F]1
3.2.1. General
3.2.2. 5-([18F]Fluoro)-1-(1-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)-1H-1,2,3-triazol-4-yl)pentane-2-sulfonic Acid [18F]1
3.3. Radiochemical and Chemical Purities, and Molar Activity of [18F]1
3.4. LogP and LogD7.4 Calculations
3.5. In Vitro Stability Studies
3.6. Animal Studies
3.6.1. General Considerations
3.6.2. PET Imaging Experiments
3.6.3. In Vivo Stability Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lang, K.; Chin, J.W. Bioorthogonal Reactions for Labeling Proteins. ACS Chem. Biol. 2014, 9, 16–20. [Google Scholar] [CrossRef]
- Blackman, M.L.; Royzen, M.; Fox, J.M. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. [Google Scholar] [CrossRef]
- Devaraj, N.K.; Weissleder, R.; Hilderbrand, S.A. Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjug. Chem. 2008, 19, 2297–2299. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [PubMed]
- Karver, M.R.; Weissleder, R.; Hilderbrand, S.A. Synthesis and Evaluation of a Series of 1,2,4,5-Tetrazines for Bioorthogonal Conjugation. Bioconjug. Chem. 2011, 22, 2263–2270. [Google Scholar] [CrossRef]
- García-Vázquez, R.; Battisti, U.M.; Herth, M.M. Recent Advances in the Development of Tetrazine Ligation Tools for Pretargeted Nuclear Imaging. Pharmaceuticals 2022, 15, 685. [Google Scholar] [CrossRef] [PubMed]
- Handula, M.; Chen, K.-T.; Seimbille, Y. IEDDA: An Attractive Bioorthogonal Reaction for Biomedical Applications. Molecules 2021, 26, 4640. [Google Scholar] [CrossRef]
- Denk, C.; Svatunek, D.; Filip, T.; Wanek, T.; Lumpi, D.; Fröhlich, J.; Kuntner, C.; Mikula, H. Development of a 18F-Labeled Tetrazine with Favorable Pharmacokinetics for Bioorthogonal PET Imaging. Angew. Chem. Int. Ed. 2014, 53, 9655–9659. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Keliher, E.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.; Edens, J.G.; Tas, J.M.G.; Victora, G.; et al. The Use of 18F-2-fluorodeoxyglucose (FDG) to Label Antibody Fragments for Immuno-PET of Pancreatic Cancer. ACS Cent Sci. 2015, 1, 142–147. [Google Scholar] [CrossRef]
- Keinänen, O.; Li, X.-G.; Chenna, N.K.; Lumen, D.; Ott, J.; Molthoff, C.F.M.; Sarparanta, M.; Helariutta, K.; Vuorinen, T.; Windhorst, A.D.; et al. A New Highly Reactive and Low Lipophilicity Fluorine-18 Labeled Tetrazine Derivative for Pretargeted PET Imaging. ACS Med. Chem. Lett. 2016, 7, 62–66. [Google Scholar] [CrossRef]
- Stéen, E.J.L.; Jørgensen, J.T.; Denk, C.; Battisti, U.M.; Nørregaard, K.; Edem, P.E.; Bratteby, K.; Shalgunov, V.; Wilkovitsch, M.; Svatunek, D.; et al. Lipophilicity and Click Reactivity Determine the Performance of Bioorthogonal Tetrazine Tools in Pretargeted In Vivo Chemistry. ACS Pharmacol. Transl. Sci. 2021, 4, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Bratteby, K.; Shalgunov, V.; Battisti, U.M.; Petersen, I.N.; Lopes van den Broek, S.; Ohlsson, T.; Gillings, N.; Erlandsson, M.; Herth, M.M. Insights into Elution of Anion Exchange Cartridges: Opening the Path toward Aliphatic 18F-Radiolabeling of Base-Sensitive Tracers. ACS Pharmacol. Transl. Sci. 2021, 4, 1556–1566. [Google Scholar] [CrossRef]
- Battisti, U.M.; Bratteby, K.; Jørgensen, J.T.; Hvass, L.; Shalgunov, V.; Mikula, H.; Kjær, A.; Herth, M.M. Development of the First Aliphatic 18F-Labeled Tetrazine Suitable for Pretargeted PET Imaging—Expanding the Bioorthogonal Tool Box. J. Med. Chem. 2021, 64, 15297–15312. [Google Scholar] [CrossRef]
- García-Vázquez, R.; Jørgensen, J.T.; Bratteby, K.E.; Shalgunov, V.; Hvass, L.; Herth, M.M.; Kjær, A.; Battisti, U.M. Development of 18F-Labeled Bispyridyl Tetrazines for In Vivo Pretargeted PET Imaging. Pharmaceuticals 2022, 15, 245. [Google Scholar] [CrossRef]
- Rashidian, M.; Wang, L.; Edens, J.G.; Jacobsen, J.T.; Hossain, I.; Wang, Q.; Victora, G.D.; Vasdev, N.; Ploegh, H.; Liang, S.H. Enzyme-Mediated Modification of Single-Domain Antibodies for Imaging Modalities with Different Characteristics. Angew. Chem. Int. Ed. 2016, 128, 528–533. [Google Scholar] [CrossRef]
- García-Vázquez, R.; Battisti, U.M.; Jørgensen, J.T.; Shalgunov, V.; Hvass, L.; Stares, D.L.; Petersen, I.N.; Crestey, F.; Löffler, A.; Svatunek, D.; et al. Direct Cu-Mediated Aromatic 18F-Labeling of Highly Reactive Tetrazines for Pretargeted Bioorthogonal PET Imaging. Chem. Sci. 2021, 12, 11668–11675. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zalutsky, M.R.; Vaidyanathan, G. Labeling a TCO-functionalized Single domain antibody fragment with 18F via inverse electron demand Diels Alder cycloaddition using a fluoronicotinyl moiety-bearing tetrazine derivative. Bioorg. Med. Chem. 2020, 28, 115634. [Google Scholar] [CrossRef]
- Zhu, J.; Li, S.; Wängler, C.; Wängler, B.; Lennox, R.B.; Schirrmacher, R. Synthesis of 3-chloro-6-((4-(di-tert-butyl[18F]fluorosilyl)-benzyl)oxy)-1,2,4,5-tetrazine ([18F]SiFA-OTz) for rapid tetrazine-based 18F-radiolabeling. Chem. Commun. 2015, 51, 12415–12418. [Google Scholar] [CrossRef]
- Otaru, S.; Imlimthan, S.; Sarparanta, M.; Helariutta, K.; Wähälä, K.; Airaksinen, A.J. Evaluation of Organo [18F]Fluorosilicon Tetrazine as a Prosthetic Group for the Synthesis of PET Radiotracers. Molecules 2020, 25, 1208. [Google Scholar] [CrossRef]
- Da Pieve, C.; Allott, L.; Martins, C.D.; Vardon, A.; Ciobota, D.M.; Kramer-Marek, G.; Smith, G. Efficient [18F]AlF Radiolabeling of ZHER3:8698 Affibody Molecule for Imaging of HER3 Positive Tumors. Bioconjug. Chem. 2016, 27, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Xu, H.; Wang, H.; Du, W.-G.H.; Wang, N.; Xiong, H.; Gu, Y.; Noodleman, L.; Sharpless, K.B.; Yang, G.; et al. Sulfur [18F]Fluoride Exchange Click Chemistry Enabled Ultrafast Late-Stage Radiosynthesis. J. Am. Chem. Soc. 2021, 143, 3753–3763. [Google Scholar] [CrossRef]
- Otaru, S.; Paulus, A.; Imlimthan, S.; Kuurne, I.; Virtanen, H.; Liljenbäck, H.; Tolvanen, T.; Auchynnikava, T.; Roivainen, A.; Helariutta, K.; et al. Development of [18F]AmBF3 Tetrazine for Radiolabeling of Peptides: Preclinical Evaluation and PET Imaging of [18F]AmBF3-PEG7-Tyr3-Octreotide in an AR42J Pancreatic Carcinoma Model. Bioconjug. Chem. 2022, 33, 1393–1404. [Google Scholar] [CrossRef]
- Hempel, N.; Barnett, A.; Gamage, N.; McManus, M.E.; Negishi, M. Human SULT1A Sulfotransferases. In Human Cytosolic Sulfotransferases; Pacifici, G.M., Coughtrie, M.W.H., Eds.; CRC Press: London, UK, 2005; pp. 181–232. [Google Scholar]
- Schmitt, S.; Bouteiller, C.; Barré, L.; Perrio, C. Sultone Opening with [18F]Fluoride: An Efficient 18F-Labelling Strategy for PET Imaging. Chem. Commun. 2011, 47, 11465. [Google Scholar] [CrossRef] [PubMed]
- Potts, K.T. The Chemistry of 1,2,4-Triazoles. Chem. Rev. 1961, 61, 87–127. [Google Scholar] [CrossRef]
- Mao, W.; Shi, W.; Li, J.; Su, D.; Wang, X.; Zhang, L.; Pan, L.; Wu, X.; Wu, H. Organocatalytic and Scalable Syntheses of Unsymmetrical 1,2,4,5-Tetrazines by Thiol-Containing Promotors. Angew. Chem. Int. Ed. 2019, 58, 1106–1109. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaufrez, J.; Guillouet, S.; Seimbille, Y.; Perrio, C. Synthesis, Fluorine-18 Radiolabeling, and In Vivo PET Imaging of a Hydrophilic Fluorosulfotetrazine. Pharmaceuticals 2023, 16, 636. https://doi.org/10.3390/ph16050636
Beaufrez J, Guillouet S, Seimbille Y, Perrio C. Synthesis, Fluorine-18 Radiolabeling, and In Vivo PET Imaging of a Hydrophilic Fluorosulfotetrazine. Pharmaceuticals. 2023; 16(5):636. https://doi.org/10.3390/ph16050636
Chicago/Turabian StyleBeaufrez, Jason, Stéphane Guillouet, Yann Seimbille, and Cécile Perrio. 2023. "Synthesis, Fluorine-18 Radiolabeling, and In Vivo PET Imaging of a Hydrophilic Fluorosulfotetrazine" Pharmaceuticals 16, no. 5: 636. https://doi.org/10.3390/ph16050636