
Asialo-rhuEPO as a Potential Neuroprotectant for Ischemic Stroke Treatment
Abstract
:1. Introduction
2. EPO and RhuEPOM
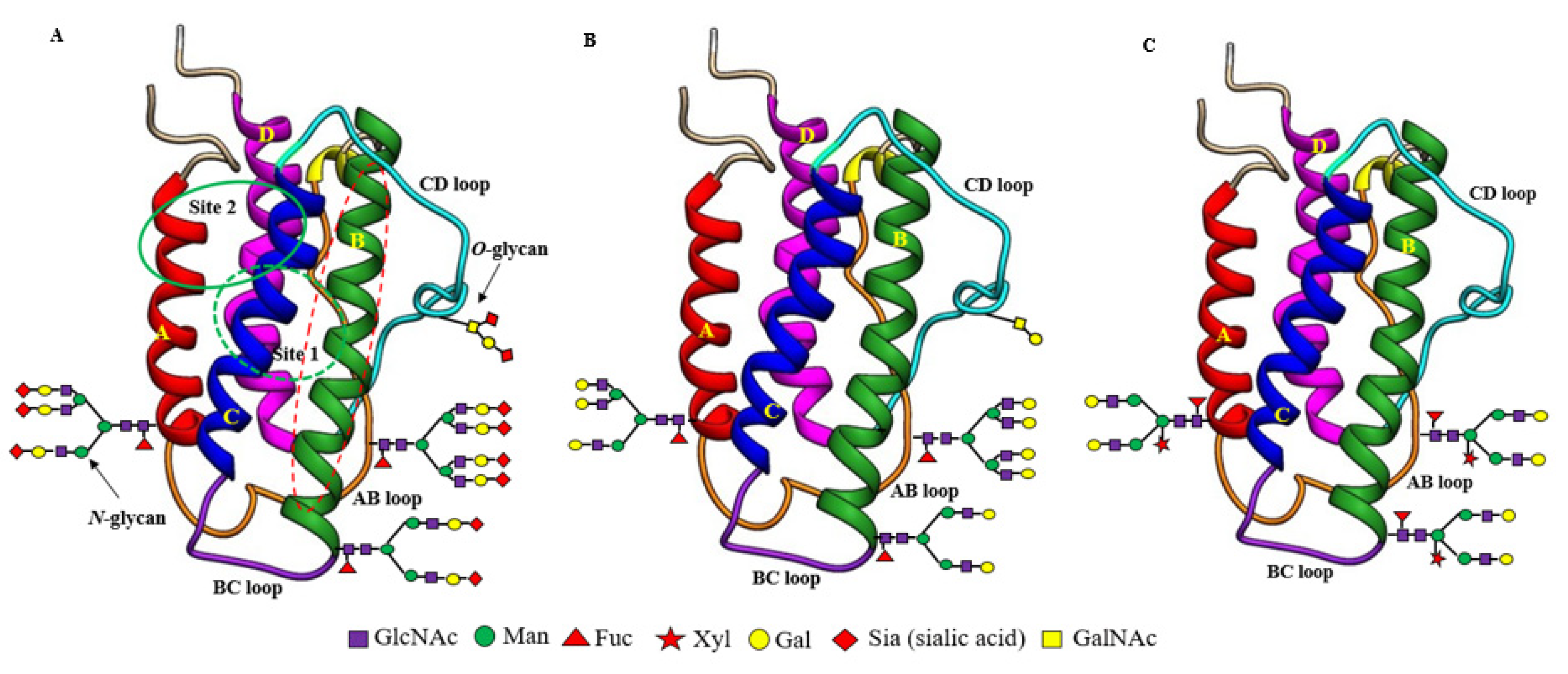
2.1. EPO Structures and Properties
2.2. Neuroprotective Function of RhuEPOM in Preclinical Studies
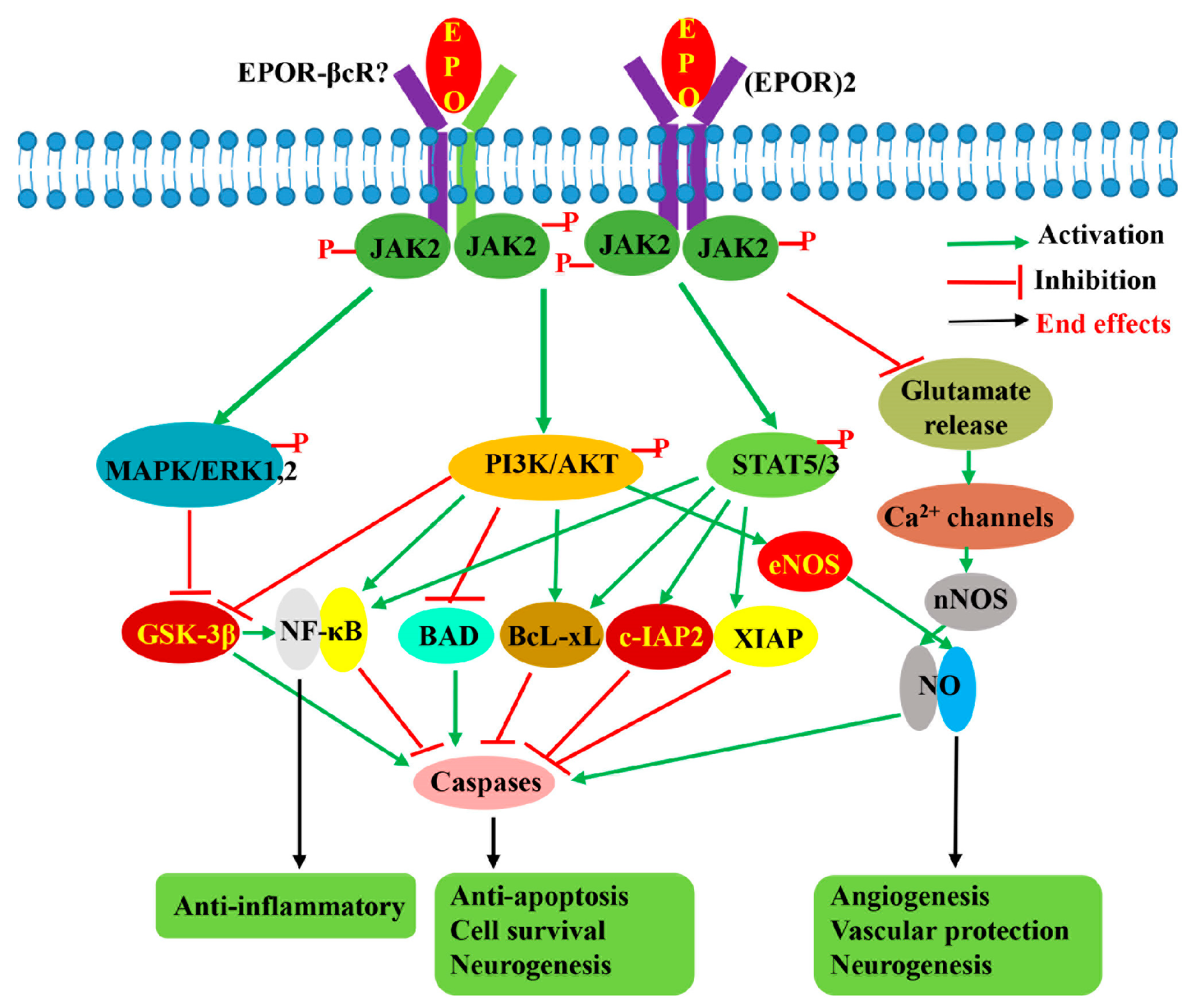
2.3. Neuroprotection Mechanisms of EPO
2.4. Clinical Trials of RhuEPOM in Acute IS Treatment
2.5. Possible Factors Responsible for Clinical Failure of RhuEPOM in Ischemic Stroke Treatment
3. Asialo-rhuEPO
3.1. Asialo-rhuEPO Structures and Properties
3.2. Methods of Asialo-rhuEPO Production
3.2.1. Enzymatic Method
3.2.2. Plant-Based Expression Method
3.3. Unique Properties of Asialo-rhuEPOP
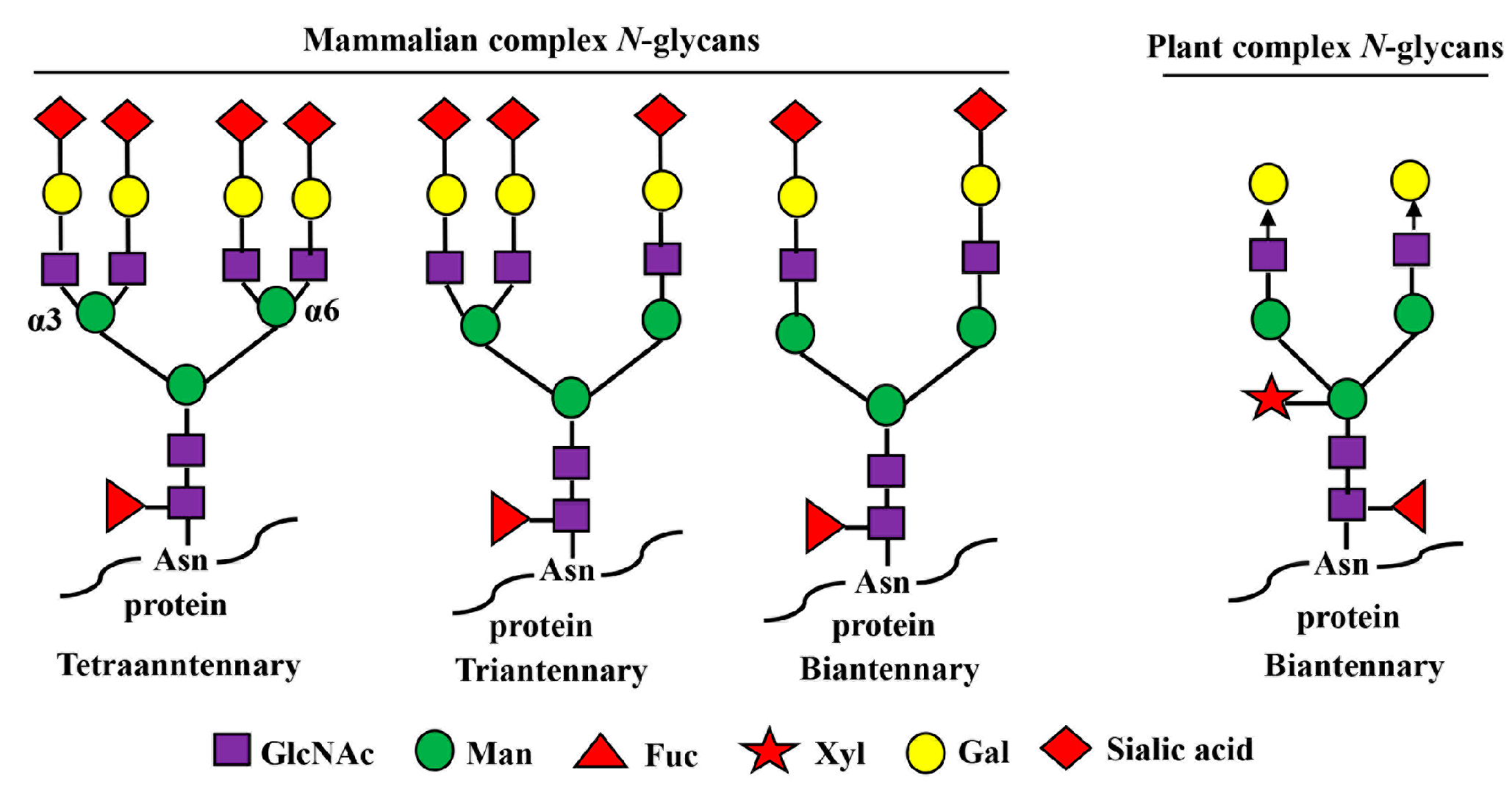
3.3.1. Asialo-rhuEPOP Carries Plant-Specific Biantennary N-Glycans
3.3.2. Asialo-rhuEPOP Is Non-Erythropoietic and Non-Immunogenic
3.4. Neuroprotective Effects of Asialo-rhuEPO
3.4.1. In Vitro and In Vivo Neuroprotective Effects of Asialo-rhuEPOP
3.4.2. Neuroprotective Mechanism of Asialo-rhuEPOP
4. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feigin, V.L.; Norrving, B.; Mensah, G.A. Global Burden of Stroke. Circ. Res. 2017, 120, 439–448. [Google Scholar] [CrossRef]
- Barthels, D.; Das, H. Current advances in ischemic stroke research and therapies. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1866, 165260. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.M.; Swanson, R.A. Opposing Effects of Glucose on Stroke and Reperfusion Injury. Stroke 2014, 45, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, X. Targeting O-GlcNAcylation in ischemic stroke. Neural Regen. Res. 2022, 17, 2427. [Google Scholar] [CrossRef]
- Ma, Y.; Zhou, Z.; Yang, G.-Y.; Ding, J.; Wang, X. The Effect of Erythropoietin and Its Derivatives on Ischemic Stroke Therapy: A Comprehensive Review. Front. Pharmacol. 2022, 13, 743926. [Google Scholar] [CrossRef]
- Xie, J.; Kittur, F.S.; Li, P.A.; Hung, C.-Y. Rethinking the necessity of low glucose intervention for cerebral ischemia/reperfusion injury. Neural Regen. Res. 2022, 17, 1397. [Google Scholar] [CrossRef]
- Pan, J.; Konstas, A.-A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2006, 49, 93–102. [Google Scholar] [CrossRef]
- Mallet, R.T.; Ryou, M.-G. Erythropoietin: Endogenous Protection of Ischemic Brain. Vitam. Horm. 2017, 105, 197–232. [Google Scholar] [CrossRef]
- Xiong, X.-Y.; Liu, L.; Yang, Q.-W. Refocusing Neuroprotection in Cerebral Reperfusion Era: New Challenges and Strategies. Front. Neurol. 2018, 9, 249. [Google Scholar] [CrossRef]
- Lin, L.; Wang, X.; Yu, Z. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem. Pharmacol. Open Access 2016, 5, 213. [Google Scholar] [CrossRef]
- Gou, X.; Xu, D.; Li, F.; Hou, K.; Fang, W.; Li, Y. Pyroptosis in stroke-new insights into disease mechanisms and therapeutic strategies. J. Physiol. Biochem. 2021, 77, 511–529. [Google Scholar] [CrossRef] [PubMed]
- Muresanu, D.F.; Strilciuc, S.; Stan, A. Current Drug Treatment of Acute Ischemic Stroke: Challenges and Opportunities. CNS Drugs 2019, 33, 841–847. [Google Scholar] [CrossRef]
- Fisher, M.; Dávalos, A.; Rogalewski, A.; Schneider, A.; Ringelstein, E.B.; Schabitz, W.-R. Toward a Multimodal Neuroprotective Treatment of Stroke. Stroke 2006, 37, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.G.; Chopp, M. The neurovascular unit and combination treatment strategies for stroke. Trends Pharmacol. Sci. 2012, 33, 415–422. [Google Scholar] [CrossRef]
- Jin, X.-C.; Liang, L.-J.; Yang, J.-M. Cocktail treatment, a promising strategy to treat acute cerebral ischemic stroke. Med. Gas Res. 2016, 6, 33–38. [Google Scholar] [CrossRef]
- Jelkmann, W. Erythropoietin: Structure, control of production, and function. Physiol. Rev. 1992, 72, 449–489. [Google Scholar] [CrossRef]
- Brines, M.L.; Ghezzi, P.; Keenan, S.; Agnello, D.; de Lanerolle, N.C.; Cerami, C.; Itri, L.M.; Cerami, A. Erythropoietin crosses the blood–brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 2000, 97, 10526–10531. [Google Scholar] [CrossRef]
- Calvillo, L.; Latini, R.; Kajstura, J.; Leri, A.; Anversa, P.; Ghezzi, P.; Salio, M.; Cerami, A.; Brines, M. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc. Natl. Acad. Sci. USA 2003, 100, 4802–4806. [Google Scholar] [CrossRef]
- Vesey, D.A. Erythropoietin protects against ischaemic acute renal injury. Nephrol. Dial. Transplant. 2004, 19, 348–355. [Google Scholar] [CrossRef]
- Sharples, E.; Yaqoob, M. Erythropoietin in Experimental Acute Renal Failure. Nephron Exp. Nephrol. 2006, 104, e83–e88. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.; Bellomo, R. Erythropoietin (EPO) in acute kidney injury. Ann. Intensiv. Care 2011, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Marti, H.H.; Gassmann, M.; Wenger, R.H.; Kvietikova, I.; Morganti-Kossmann, M.C.; Kossmann, T.; Trentz, O.; Bauer, C. Detection of erythropoietin in human liquor: Intrinsic erythropoietin production in the brain. Kidney Int. 1997, 51, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Li, F.; Chong, Z.Z. Erythropoietin in the brain: Can the promise to protect be fulfilled? Trends Pharmacol. Sci. 2004, 25, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, M.; Wen, T.-C.; Matsuda, S.; Morishita, E.; Nagao, M.; Sasaki, R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 1998, 95, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.S.; Nandra, K.K.; Thiemermann, C. Bench-to-bedside review: Erythropoietin and its derivatives as therapies in critical care. Crit. Care 2012, 16, 229. [Google Scholar] [CrossRef]
- Souvenir, R.; Doycheva, D.; Zhang, J.H.; Tang, J. Erythropoietin in stroke therapy: Friend or foe. Curr. Med. Chem. 2015, 22, 1205–1213. [Google Scholar] [CrossRef]
- Brines, M.; Cerami, A. Emerging biological roles for erythropoietin in the nervous system. Nat. Rev. Neurosci. 2005, 6, 484–494. [Google Scholar] [CrossRef]
- Brines, M.; Cerami, A. Erythropoietin-mediated tissue protection: Reducing collateral damage from the primary injury response. J. Intern. Med. 2008, 264, 405–432. [Google Scholar] [CrossRef]
- Sølling, C. Organ-Protective and Immunomodulatory Effects of Erythropoietin—An Update on Recent Clinical Trials. Basic Clin. Pharmacol. Toxicol. 2011, 110, 113–121. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Hasselblatt, M.; Dembowski, C.; Cepek, L.; Lewczuk, P.; Stiefel, M.; Rustenbeck, H.-H.; Breiter, N.; Jacob, S.; Knerlich, F.; et al. Erythropoietin Therapy for Acute Stroke Is Both Safe and Beneficial. Mol. Med. 2002, 8, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Weissenborn, K.; Prange, H.; Schneider, D.; Weimar, C.; Wartenberg, K.; Schellinger, P.D.; Bohn, M.; Becker, H.; Wegrzyn, M.; et al. Recombinant Human Erythropoietin in the Treatment of Acute Ischemic Stroke. Stroke 2009, 40, e647–e656. [Google Scholar] [CrossRef] [PubMed]
- Asadi, B.; Askari, G.R.; Khorvash, F.; Bagherpur, A.; Mehrabi, F.; Karimi, M.; Ghasemi, M.; Najjaran, A. Neuroprotective Effects of Erythropoietin in Acute Ischemic Stroke. Int. J. Prev. Med. 2013, 4, S306–S312. [Google Scholar] [PubMed]
- Tsai, T.-H.; Lu, C.-H.; Wallace, C.G.; Chang, W.-N.; Chen, S.-F.; Huang, C.-R.; Tsai, N.-W.; Lan, M.-Y.; Sung, P.-H.; Liu, C.-F.; et al. Erythropoietin improves long-term neurological outcome in acute ischemic stroke patients: A randomized, prospective, placebo-controlled clinical trial. Crit. Care 2015, 19, 1–9. [Google Scholar] [CrossRef]
- Erbayraktar, S.; Grasso, G.; Sfacteria, A.; Xie, Q.-W.; Coleman, T.; Kreilgaard, M.; Torup, L.; Sager, T.; Erbayraktar, Z.; Gokmen, N.; et al. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 6741–6746. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, C.; Wang, X.; Gerwien, J.G.; Schrattenholz, A.; Sandberg, M.; Leist, M.; Blomgren, K. The nonerythropoietic asialoerythropoietin protects against neonatal hypoxia-ischemia as potently as erythropoietin. J. Neurochem. 2004, 91, 900–910. [Google Scholar] [CrossRef]
- Grasso, G.; Sfacteria, A.; Erbayraktar, S.; Passalacqua, M.; Meli, F.; Gokmen, N.; Yilmaz, O.; La Torre, D.; Buemi, M.; Iacopino, D.G.; et al. Amelioration of spinal cord compressive injury by pharmacological preconditioning with erythropoietin and a nonerythropoietic erythropoietin derivative. J. Neurosurg. Spine 2006, 4, 310–318. [Google Scholar] [CrossRef]
- Price, C.D.; Yang, Z.; Karlnoski, R.; Kumar, D.; Chaparro, R.; Camporesi, E.M. Effect of continuous infusion of asialoerythropoietin on short-term changes in infarct volume, penumbra apoptosis and behaviour following middle cerebral artery occlusion in rats. Clin. Exp. Pharmacol. Physiol. 2010, 37, 185–192. [Google Scholar] [CrossRef]
- Yamashita, T.; Nonoguchi, N.; Ikemoto, T.; Miyatake, S.-I.; Kuroiwa, T. Asialoerythropoietin attenuates neuronal cell death in the hippocampal CA1 region after transient forebrain ischemia in a gerbil model. Neurol. Res. 2010, 32, 957–962. [Google Scholar] [CrossRef]
- Ishii, T.; Asai, T.; Oyama, D.; Fukuta, T.; Yasuda, N.; Shimizu, K.; Minamino, T.; Oku, N. Amelioration of cerebral ischemia–reperfusion injury based on liposomal drug delivery system with asialo-erythropoietin. J. Control. Release 2012, 160, 81–87. [Google Scholar] [CrossRef]
- He, M.; Kittur, F.S.; Hung, C.-Y.; Zhang, J.; Jing, L.; Sane, D.C.; Li, P.A.; Xie, J. A Novel Plant-Produced Asialo-rhuEPO Protects Brain from Ischemic Damage Without Erythropoietic Action. Transl. Stroke Res. 2021, 13, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Sawada, T.; Kubota, K. Asialoerythropoietin Has Strong Renoprotective Effects Against Ischemia-Reperfusion Injury in a Murine Model. Transplantation 2007, 84, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Ogino, A.; Takemura, G.; Kawasaki, M.; Tsujimoto, A.; Kanamori, H.; Li, L.; Goto, K.; Maruyama, R.; Kawamura, I.; Takeyama, T.; et al. Erythropoietin Receptor Signaling Mitigates Renal Dysfunction-Associated Heart Failure by Mechanisms Unrelated to Relief of Anemia. J. Am. Coll. Cardiol. 2010, 56, 1949–1958. [Google Scholar] [CrossRef]
- Takeyama, T.; Takemura, G.; Kanamori, H.; Kawaguchi, T.; Ogino, A.; Watanabe, T.; Morishita, K.; Tsujimoto, A.; Goto, K.; Maruyama, R.; et al. Asialoerythropoietin, a Nonerythropoietic Derivative of Erythropoietin, Displays Broad Anti-Heart Failure Activity. Circ. Heart Fail. 2012, 5, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L.O.; Goldwasser, E.; Fried, W.; Plzak, L. Role of the Kidney in Erythropoiesis. Nature 1957, 179, 633–634. [Google Scholar] [CrossRef]
- Erslev, A.J.; Caro, J.; Kansu, E.; Silver, R. Renal and Extrarenal Erythropoietin Production in Anaemic Rats. Br. J. Haematol. 1980, 45, 65–72. [Google Scholar] [CrossRef]
- Marti, H.H.; Wenger, R.H.; Rivas, L.A.; Straumann, U.; Oigicaylioglu, M.; Henn, V.; Yonekawa, Y.; Bauer, C.; Gassmann, M. Erythropoietin Gene Expression in Human, Monkey and Murine Brain. Eur. J. Neurosci. 1996, 8, 666–676. [Google Scholar] [CrossRef]
- Recny, M.A.; Scoble, H.A.; Kim, Y. Structural characterization of natural human urinary and recombinant DNA-derived erythropoietin. Identification of des-arginine 166 erythropoietin. J. Biol. Chem. 1987, 262, 17156–17163. [Google Scholar] [CrossRef]
- Browne, J.; Cohen, A.; Egrie, J.; Lai, P.; Lin, F.-K.; Strickland, T.; Watson, E.; Stebbing, N. Erythropoietin: Gene Cloning, Protein Structure, and Biological Properties. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 693–702. [Google Scholar] [CrossRef]
- Jelkmann, W. Physiology and Pharmacology of Erythropoietin. Transfus. Med. Hemother. 2013, 40, 302–309. [Google Scholar] [CrossRef]
- Cheetham, J.C.; Smith, D.M.; Aoki, K.H.; Stevenson, J.L.; Hoeffel, T.J.; Syed, R.S.; Egrie, J.; Harvey, T.S. NMR structure of human erythropoietin and a comparison with its receptor bound conformation. Nat. Struct. Biol. 1998, 5, 861–866. [Google Scholar] [CrossRef]
- Syed, R.S.; Reid, S.W.; Li, C.; Cheetham, J.C.; Aoki, K.H.; Liu, B.; Zhan, H.; Osslund, T.D.; Chirino, A.J.; Zhang, J.; et al. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature 1998, 395, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Wasley, L.; Timony, G.; Murtha, P.; Stoudemire, J.; Dorner, A.; Caro, J.; Krieger, M.; Kaufman, R. The importance of N- and O-linked oligosaccharides for the biosynthesis and in vitro and in vivo biologic activities of erythropoietin. Blood 1991, 77, 2624–2632. [Google Scholar] [CrossRef]
- Thompson, N.K.; Wakarchuk, W. O-glycosylation and its role in therapeutic proteins. Biosci. Rep. 2022, 42, BSR20220094. [Google Scholar] [CrossRef]
- Dubé, S.; Fisher, J.W.; Powell, J.S. Glycosylation at specific sites of erythropoietin is essential for biosynthesis, secretion, and biological function. J. Biol. Chem. 1988, 263, 17516–17521. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.; Arakawa, T.; Aoki, K.; Elmore, R.; Rohde, M.; Boone, T.; Strickland, T. The effect of carbohydrate on the structure and stability of erythropoietin. J. Biol. Chem. 1991, 266, 23022–23026. [Google Scholar] [CrossRef] [PubMed]
- Lasne, F.; De Ceaurriz, J. Recombinant erythropoietin in urine. Nature 2000, 405, 635. [Google Scholar] [CrossRef] [PubMed]
- Skibeli, V.; Nissen-Lie, G.; Torjesen, P. Sugar profiling proves that human serum erythropoietin differs from recombinant human erythropoietin. Blood 2001, 98, 3626–3634. [Google Scholar] [CrossRef]
- Takeuchi, M.; Inoue, N.; Strickland, T.W.; Kubota, M.; Wada, M.; Shimizu, R.; Hoshi, S.; Kozutsumi, H.; Takasaki, S.; Kobata, A. Relationship between sugar chain structure and biological activity of recombinant human erythropoietin produced in Chinese hamster ovary cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7819–7822. [Google Scholar] [CrossRef]
- Lukowsky, W.A.; Painter, R.H. Studies on the Role of Sialic Acid in the Physical and Biological Properties of Erythropoietin. Can. J. Biochem. 1972, 50, 909–917. [Google Scholar] [CrossRef]
- Goldwasser, E.; Kung, C.K.-H.; Eliason, J. On the Mechanism of Erythropoietin-induced Differentiation. J. Biol. Chem. 1974, 249, 4202–4206. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Sasaki, H.; Lopez, L. Survival of recombinant erythropoietin in the circulation: The role of carbohydrates. Blood 1989, 73, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.; Hogans, B. The in vivo metabolism of recombinant human erythropoietin in the rat. Blood 1989, 73, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. The enigma of the metabolic fate of circulating erythropoietin (Epo) in view of the pharmacokinetics of the recombinant drugs rhEpo and NESP. Eur. J. Haematol. 2002, 69, 265–274. [Google Scholar] [CrossRef]
- Leist, M.; Ghezzi, P.; Grasso, G.; Bianchi, R.; Villa, P.; Fratelli, M.; Savino, C.; Bianchi, M.; Nielsen, J.; Gerwien, J.; et al. Derivatives of Erythropoietin That Are Tissue Protective But Not Erythropoietic. Science 2004, 305, 239–242. [Google Scholar] [CrossRef]
- Gan, Y.; Xing, J.; Jing, Z.; Stetler, R.A.; Zhang, F.; Luo, Y.; Ji, X.; Gao, Y.; Cao, G. Mutant Erythropoietin Without Erythropoietic Activity Is Neuroprotective Against Ischemic Brain Injury. Stroke 2012, 43, 3071–3077. [Google Scholar] [CrossRef]
- Brines, M.; Patel, N.S.A.; Villa, P.; Brines, C.; Mennini, T.; De Paola, M.; Erbayraktar, Z.; Erbayraktar, S.; Sepodes, B.; Thiemermann, C.; et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc. Natl. Acad. Sci. USA 2008, 105, 10925–10930. [Google Scholar] [CrossRef]
- Marsh, J.T.; Brown, W.S.; Wolcott, D.; Carr, C.R.; Harper, R.; Schweitzer, S.V.; Nissenson, A.R. rHuEPO treatment improves brain and cognitive function of anemic dialysis patients. Kidney Int. 1991, 39, 155–163. [Google Scholar] [CrossRef]
- Genc, S.; Koroglu, T.F.; Genc, K. Erythropoietin and the nervous system. Brain Res. 2004, 1000, 19–31. [Google Scholar] [CrossRef]
- Marti, H.H. Erythropoietin and the hypoxic brain. J. Exp. Biol. 2004, 207, 3233–3242. [Google Scholar] [CrossRef]
- Sirén, A.-L.; Knerlich, F.; Poser, W.; Gleiter, C.H.; Brück, W.; Ehrenreich, H. Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol. 2001, 101, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shacka, J.J.; Eells, J.B.; Suarez-Quian, C.; Przygodzki, R.M.; Beleslin-Cokic, B.; Lin, C.-S.; Nikodem, V.M.; Hempstead, B.; Flanders, K.C.; et al. Erythropoietin receptor signalling is required for normal brain development. Development 2002, 129, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.T.; Ohab, J.J.; Kertesz, N.; Groszer, M.; Matter, C.; Gao, J.; Liu, X.; Wu, H.; Carmichael, S.T. A Critical Role of Erythropoietin Receptor in Neurogenesis and Post-Stroke Recovery. J. Neurosci. 2006, 26, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Sadamoto, Y.; Igase, K.; Sakanaka, M.; Sato, K.; Otsuka, H.; Sakaki, S.; Masuda, S.; Sasaki, R. Erythropoietin Prevents Place Navigation Disability and Cortical Infarction in Rats with Permanent Occlusion of the Middle Cerebral Artery. Biochem. Biophys. Res. Commun. 1998, 253, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, M.; Marti, H.H.; Roussel, S.; Divoux, D.; Nouvelot, A.; MacKenzie, E.T.; Petit, E. A Potential Role for Erythropoietin in Focal Permanent Cerebral Ischemia in Mice. J. Cereb. Blood Flow Metab. 1999, 19, 643–651. [Google Scholar] [CrossRef]
- Aluclu, M.U.; Acar, A.; Guzel, A.; Bahceci, S.; Yaldiz, M. Evaluation of erythropoietin effects on cerebral ischemia in rats. Neuro Endocrinol. Lett. 2007, 28, 170–174. [Google Scholar]
- Villa, P.; Bigini, P.; Mennini, T.; Agnello, D.; Laragione, T.; Cagnotto, A.; Viviani, B.; Marinovich, M.; Cerami, A.; Coleman, T.R.; et al. Erythropoietin Selectively Attenuates Cytokine Production and Inflammation in Cerebral Ischemia by Targeting Neuronal Apoptosis. J. Exp. Med. 2003, 198, 971–975. [Google Scholar] [CrossRef]
- Gunnarson, E.; Song, Y.; Kowalewski, J.M.; Brismar, H.; Brines, M.; Cerami, A.; Andersson, U.; Zelenina, M.; Aperia, A. Erythropoietin modulation of astrocyte water permeability as a component of neuroprotection. Proc. Natl. Acad. Sci. USA 2009, 106, 1602–1607. [Google Scholar] [CrossRef]
- Chang, Y.S.; Mu, D.; Wendland, M.; Sheldon, R.A.; Vexler, Z.S.; McQuillen, P.S.; Ferriero, D.M. Erythropoietin Improves Functional and Histological Outcome in Neonatal Stroke. Pediatr. Res. 2005, 58, 106–111. [Google Scholar] [CrossRef]
- Spandou, E.; Papadopoulou, Z.; Soubasi, V.; Karkavelas, G.; Simeonidou, C.; Pazaiti, A.; Guiba-Tziampiri, O. Erythropoietin prevents long-term sensorimotor deficits and brain injury following neonatal hypoxia–ischemia in rats. Brain Res. 2005, 1045, 22–30. [Google Scholar] [CrossRef]
- Grasso, G.; Buemi, M.; Alafaci, C.; Sfacteria, A.; Passalacqua, M.; Sturiale, A.; Calapai, G.; De Vico, G.; Piedimonte, G.; Salpietro, F.M.; et al. Beneficial effects of systemic administration of recombinant human erythropoietin in rabbits subjected to subarachnoid hemorrhage. Proc. Natl. Acad. Sci. USA 2002, 99, 5627–5631. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Mahmood, A.; Qu, C.; Goussev, A.; Schallert, T.; Chopp, M. Erythropoietin Enhances Neurogenesis and Restores Spatial Memory in Rats after Traumatic Brain Injury. J. Neurotrauma 2005, 22, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Lu, D.; Qu, C.; Goussev, A.; Schallert, T.; Mahmood, A.; Chopp, M. Effects of erythropoietin on reducing brain damage and improving functional outcome after traumatic brain injury in mice. J. Neurosurg. 2008, 109, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Jia, R.; Tu, Y.; Xiao, S.; Ding, G. Erythropoietin prevents reactive oxygen species generation and renal tubular cell apoptosis at high glucose level. Biomed. Pharmacother. 2010, 64, 681–685. [Google Scholar] [CrossRef]
- Wang, R.; Wu, X.; Liang, J.; Qi, Z.; Liu, X.; Min, L.; Ji, X.; Luo, Y.; Zhao, H. Intra-artery infusion of recombinant human erythropoietin reduces blood–brain barrier disruption in rats following cerebral ischemia and reperfusion. Int. J. Neurosci. 2014, 125, 693–702. [Google Scholar] [CrossRef]
- Thériault, P.; Le Béhot, A.; ElAli, A.; Rivest, S. Sub-acute systemic erythropoietin administration reduces ischemic brain injury in an age-dependent manner. Oncotarget 2016, 7, 35552–35561. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.-Q.; Maiese, K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br. J. Pharmacol. 2003, 138, 1107–1118. [Google Scholar] [CrossRef]
- Shang, Y.C.; Chong, Z.Z.; Wang, S.; Maiese, K. Erythropoietin and Wnt1 govern pathways of mTOR, Apaf-1, and XIAP in inflammatory microglia. Curr. Neurovascular Res. 2011, 8, 270–285. [Google Scholar] [CrossRef]
- Morishita, E.; Masuda, S.; Nagao, M.; Yasuda, Y.; Sasaki, R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 1996, 76, 105–116. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.-H.; Kang, J.-Q.; Maiese, K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J. Neurosci. Res. 2002, 71, 659–669. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.-Q.; Maiese, K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J. Cereb. Blood Flow Metab. 2003, 23, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Kook, Y.-H.; Ka, M.; Um, M. Neuroprotective cytokines repress PUMA induction in the 1-methyl-4-phenylpyridinium (MPP+) model of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2011, 411, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Digicaylioglu, M.; Lipton, S.A. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-κB signalling cascades. Nature 2001, 412, 641–647. [Google Scholar] [CrossRef]
- Bond, W.S.; Rex, T.S. Evidence That Erythropoietin Modulates Neuroinflammation through Differential Action on Neurons, Astrocytes, and Microglia. Front. Immunol. 2014, 5, 523. [Google Scholar] [CrossRef] [PubMed]
- Beleslin-Cokic, B.B.; Cokic, V.P.; Yu, X.; Weksler, B.B.; Schechter, A.N.; Noguchi, C.T. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 2004, 104, 2073–2080. [Google Scholar] [CrossRef]
- Navarro, J.C.; Pillai, S.; Ponce, L.L.; Van, M.; Goodman, J.C.; Robertson, C.S. Endothelial Nitric Oxide Synthase Mediates the Cerebrovascular Effects of Erythropoietin in Traumatic Brain Injury. Front. Immunol. 2014, 5, 494. [Google Scholar] [CrossRef]
- Kertesz, N.; Wu, J.; Chen, T.H.-P.; Sucov, H.M.; Wu, H. The role of erythropoietin in regulating angiogenesis. Dev. Biol. 2004, 276, 101–110. [Google Scholar] [CrossRef]
- Chen, G.-H.; Li, X.-L.; Deng, Y.-Q.; Zhou, F.-M.; Zou, W.-Q.; Jiang, W.-X.; Shangguan, S.-Q.; Lu, Z.-N. The Molecular Mechanism of EPO Regulates the Angiogenesis after Cerebral Ischemia through AMPK-KLF2 Signaling Pathway. Crit. Rev. Eukaryot. Gene Expr. 2019, 29, 105–112. [Google Scholar] [CrossRef]
- Studer, L.; Csete, M.; Lee, S.-H.; Kabbani, N.; Walikonis, J.; Wold, B.; McKay, R. Enhanced Proliferation, Survival, and Dopaminergic Differentiation of CNS Precursors in Lowered Oxygen. J. Neurosci. 2000, 20, 7377–7383. [Google Scholar] [CrossRef]
- Shingo, T.; Sorokan, S.T.; Shimazaki, T.; Weiss, S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J. Neurosci. 2001, 21, 9733–9743. [Google Scholar] [CrossRef]
- Digicaylioglu, M.; Garden, G.; Timberlake, S.; Fletcher, L.; Lipton, S.A. Acute neuroprotective synergy of erythropoietin and insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 2004, 101, 9855–9860. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Kilic, U.; Soliz, J.; Bassetti, C.L.; Gassmann, M.; Hermann, D.M. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005, 19, 2026–2028. [Google Scholar] [CrossRef]
- Um, M.; Lodish, H.F. Antiapoptotic Effects of Erythropoietin in Differentiated Neuroblastoma SH-SY5Y Cells Require Activation of Both the STAT5 and AKT Signaling Pathways. J. Biol. Chem. 2006, 281, 5648–5656. [Google Scholar] [CrossRef]
- Sergio, C.-M.; Rolando, C.-A. Erythropoietin regulates signaling pathways associated with neuroprotective events. Exp. Brain Res. 2022, 240, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.-C.; Sadamoto, Y.; Tanaka, J.; Zhu, P.; Nakata, K.; Ma, Y.-J.; Hata, R.; Sakanaka, M. Erythropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by up-regulating Bcl-xL expression. J. Neurosci. Res. 2002, 67, 795–803. [Google Scholar] [CrossRef]
- Yamamoto, M.; Koshimura, K.; Sohmiya, M.; Murakami, Y.; Kato, Y. Effect of erythropoietin on nitric oxide production in the rat hippocampus using in vivo brain microdialysis. Neuroscience 2004, 128, 163–168. [Google Scholar] [CrossRef]
- Yip, H.-K.; Tsai, T.-H.; Lin, H.-S.; Chen, S.-F.; Sun, C.-K.; Leu, S.; Yuen, C.-M.; Tan, T.-Y.; Lan, M.-Y.; Liou, C.-W.; et al. Effect of erythropoietin on level of circulating endothelial progenitor cells and outcome in patients after acute ischemic stroke. Crit. Care 2011, 15, R40. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Jumbe, N.L.; Farrell, C.L.; Niehoff, M.L.; Heatherington, A.C. Passage of erythropoietic agents across the blood–brain barrier: A comparison of human and murine erythropoietin and the analog darbepoetin alfa. Eur. J. Pharmacol. 2004, 505, 93–101. [Google Scholar] [CrossRef]
- Ábrahám, C.S.; Harada, N.; Deli, M.A.; Niwa, M. Transient forebrain ischemia increases the blood-brain barrier permeability for albumin in stroke-prone spontaneously hypertensive rats. Cell. Mol. Neurobiol. 2002, 22, 455–462. [Google Scholar] [CrossRef]
- Xenocostas, A.; Cheung, W.K.; Farrell, F.; Zakszewski, C.; Kelley, M.; Lutynski, A.; Crump, M.; Lipton, J.H.; Kiss, T.L.; Lau, C.Y.; et al. The pharmacokinetics of erythropoietin in the cerebrospinal fluid after intravenous administration of recombinant human erythropoietin. Eur. J. Clin. Pharmacol. 2005, 61, 189–195. [Google Scholar] [CrossRef]
- Ishii, T.; Asai, T.; Urakami, T.; Oku, N. Accumulation of macromolecules in brain parenchyma in acute phase of cerebral infarction/reperfusion. Brain Res. 2010, 1321, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.; Krawczyk, M.; Ahn, D.; Ahmet, I.; Paik, D.; Lakatta, E.G.; Talan, M.I. Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc. Natl. Acad. Sci. USA 2003, 100, 11612–11617. [Google Scholar] [CrossRef] [PubMed]
- Parsa, C.J.; Matsumoto, A.; Kim, J.; Riel, R.U.; Pascal, L.S.; Walton, G.B.; Thompson, R.B.; Petrofski, J.A.; Annex, B.H.; Stamler, J.S.; et al. A novel protective effect of erythropoietin in the infarcted heart. J. Clin. Investig. 2003, 112, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Ning, N.; Niu, X.; Dang, Y.; Dong, X.; Wei, J.; Zhu, C. Erythropoietin treatment in patients with acute myocardial infarction: A meta-analysis of randomized controlled trials. Am. Heart J. 2012, 164, 715–727. [Google Scholar] [CrossRef]
- Wen, Y.; Xu, J.; Ma, X.; Gao, Q. High-Dose Erythropoietin in Acute ST-Segment Elevation Myocardial Infarction: A Meta-Analysis of Randomized Controlled Trials. Am. J. Cardiovasc. Drugs 2013, 13, 435–442. [Google Scholar] [CrossRef]
- Ali-Hassan-Sayegh, S.; Mirhosseini, S.J.; Tahernejad, M.; Mahdavi, P.; Haddad, F.; Shahidzadeh, A.; Lotfaliani, M.R.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Weymann, A.; et al. Administration of erythropoietin in patients with myocardial infarction: Does it make sense? An updated and comprehensive meta-analysis and systematic review. Cardiovasc. Revascularization Med. 2015, 16, 179–189. [Google Scholar] [CrossRef]
- Jean-Baptiste, W.; Ali, A.Y.; Inyang, B.; Koshy, F.S.; George, K.; Poudel, P.; Chalasani, R.; Goonathilake, M.R.; Waqar, S.; George, S.; et al. Are There Any Cardioprotective Effects or Safety Concerns of Erythropoietin in Patients With Myocardial Infarction? A Systematic Review. Cureus 2022, 14, e25671. [Google Scholar] [CrossRef]
- Bennett, C.L.; Silver, S.M.; Djulbegovic, B.; Samaras, A.T.; Blau, C.A.; Gleason, K.J.; Barnato, S.E.; Elverman, K.M.; Courtney, D.M.; McKoy, J.M.; et al. Venous Thromboembolism and Mortality Associated With Recombinant Erythropoietin and Darbepoetin Administration for the Treatment of Cancer-Associated Anemia. JAMA 2008, 299, 914–924. [Google Scholar] [CrossRef]
- Steppich, B.; Groha, P.; Ibrahim, T.; Schunkert, H.; Laugwitz, K.-L.; Hadamitzky, M.; Kastrati, A.; Ott, I. Effect of Erythropoietin in patients with acute myocardial infarction: Five-year results of the REVIVAL-3 trial. BMC Cardiovasc. Disord. 2017, 17, 1–8. [Google Scholar] [CrossRef]
- Ruschitzka, F.T.; Wenger, R.H.; Stallmach, T.; Quaschning, T.; de Wit, C.; Wagner, K.; Labugger, R.; Kelm, M.; Noll, G.; Rülicke, T.; et al. Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proc. Natl. Acad. Sci. USA 2000, 97, 11609–11613. [Google Scholar] [CrossRef]
- Quaschning, T.; Ruschitzka, F.; Stallmach, T.; Shaw, S.; Morawietz, H.; Goettsch, W.; Hermann, M.; Slowinski, T.; Theuring, F.; Hocher, B.; et al. Erythropoietin-induced excessive erythrocytosis activates the tissue endothelin system in mice. FASEB J. 2002, 17, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Lim, V.S. Recombinant human erythropoietin in predialysis patients. Am. J. Kidney Dis. 1991, 18, 34–37. [Google Scholar] [PubMed]
- Smith, K.J.; Bleyer, A.J.; Little, W.C.; Sane, D.C. The cardiovascular effects of erythropoietin. Cardiovasc. Res. 2003, 59, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.M.R.; Apaiajai, N.; Chattipakorn, N.; Chattipakorn, S.C. The Protective and Reparative Role of Colony-Stimulating Factors in the Brain with Cerebral Ischemia/Reperfusion Injury. Neuroendocrinology 2020, 111, 1029–1065. [Google Scholar] [CrossRef] [PubMed]
- Talan, M.I.; Ahmet, I.; Lakatta, E. Did Clinical Trials in Which Erythropoietin Failed to Reduce Acute Myocardial Infarct Size Miss a Narrow Therapeutic Window? PLoS ONE 2012, 7, e34819. [Google Scholar] [CrossRef]
- Talan, M.I.; Latini, R. Myocardial infarction: Cardioprotection by erythropoietin. Breast Cancer 2013, 982, 265–302. [Google Scholar] [CrossRef]
- Pearl, R.G. Erythropoietin and organ protection: Lessons from negative clinical trials. Crit. Care 2014, 18, 1–3. [Google Scholar] [CrossRef]
- Brines, M.; Cerami, A. The Receptor That Tames the Innate Immune Response. Mol. Med. 2011, 18, 486–496. [Google Scholar] [CrossRef]
- Pradeep, S.; Huang, J.; Mora, E.M.; Nick, A.M.; Cho, M.S.; Wu, S.Y.; Noh, K.; Pecot, C.V.; Rupaimoole, R.; Stein, M.A.; et al. Erythropoietin Stimulates Tumor Growth via EphB4. Cancer Cell 2015, 28, 610–622. [Google Scholar] [CrossRef]
- Zhang, F.; Xing, J.; Liou, A.K.-F.; Wang, S.; Gan, Y.; Luo, Y.; Ji, X.; Stetler, R.A.; Chen, J.; Cao, G. Enhanced Delivery of Erythropoietin Across the Blood–Brain Barrier for Neuroprotection Against Ischemic Neuronal Injury. Transl. Stroke Res. 2010, 1, 113–121. [Google Scholar] [CrossRef]
- Najjar, S.S.; Rao, S.V.; Melloni, C.; Raman, S.V.; Povsic, T.J.; Melton, L.; Barsness, G.W.; Prather, K.; Heitner, J.F.; Kilaru, R.; et al. Intravenous Erythropoietin in Patients With ST-Segment Elevation Myocardial Infarction. JAMA 2011, 305, 1863–1872. [Google Scholar] [CrossRef]
- Imai, N.; Higuchi, M.; Kawamura, A.; Tomonoh, K.; Oh-Eda, M.; Fujiwara, M.; Shimonaka, Y.; Ochi, N. Physicochemical and biological characterization of asialoerythropoietin. Suppressive effects of sialic acid in the expression of biological activity of human erythropoietin in vitro. JBIC J. Biol. Inorg. Chem. 1990, 194, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Mennini, T.; De Paola, M.; Bigini, P.; Mastrotto, C.; Fumagalli, E.; Barbera, S.; Mengozzi, M.; Viviani, B.; Corsini, E.; Marinovich, M.; et al. Nonhematopoietic Erythropoietin Derivatives Prevent Motoneuron Degeneration In Vitro and In Vivo. Mol. Med. 2006, 12, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.; Altmann, F.; Arrenberg, C.K.; Koprivova, A.; Beike, A.K.; Stemmer, C.; Gorr, G.; Reski, R.; Decker, E.L. Moss-based production of asialo-erythropoietin devoid of Lewis A and other plant-typical carbohydrate determinants. Plant Biotechnol. J. 2012, 10, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Kittur, F.S.; Hung, C.-Y.; Darlington, D.E.; Sane, D.C.; Xie, J. N-Glycosylation engineering of tobacco plants to produce asialoerythropoietin. Plant Cell Rep. 2012, 31, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Kittur, F.S.; Bah, M.; Archer-Hartmann, S.; Hung, C.-Y.; Azadi, P.; Ishihara, M.; Sane, D.C.; Xie, J. Cytoprotective Effect of Recombinant Human Erythropoietin Produced in Transgenic Tobacco Plants. PLoS ONE 2013, 8, e76468. [Google Scholar] [CrossRef]
- Kittur, F.S.; Hung, C.-Y.; Zhu, C.; Shajahan, A.; Azadi, P.; Thomas, M.D.; Pearce, J.L.; Gruber, C.; Kallolimath, S.; Xie, J. Glycoengineering tobacco plants to stably express recombinant human erythropoietin with different N-glycan profiles. Int. J. Biol. Macromol. 2020, 157, 158–169. [Google Scholar] [CrossRef]
- Weise, A.; Altmann, F.; Rodriguez-Franco, M.; Sjoberg, E.R.; Bäumer, W.; Launhardt, H.; Kietzmann, M.; Gorr, G. High-level expression of secreted complex glycosylated recombinant human erythropoietin in the Physcomitrella Delta-fuc-t Delta-xyl-t mutant. Plant Biotechnol. J. 2007, 5, 389–401. [Google Scholar] [CrossRef]
- Wee, E.G.-T.; Sherrier, D.J.; Prime, T.A.; DuPree, P. Targeting of Active Sialyltransferase to the Plant Golgi Apparatus. Plant Cell 1998, 10, 1759–1768. [Google Scholar] [CrossRef]
- Ma, J.K.-C.; Drake, P.M.W.; Christou, P. The production of recombinant pharmaceutical proteins in plants. Nat. Rev. Genet. 2003, 4, 794–805. [Google Scholar] [CrossRef]
- Gomord, V.; Faye, L. Posttranslational modification of therapeutic proteins in plants. Curr. Opin. Plant Biol. 2004, 7, 171–181. [Google Scholar] [CrossRef]
- Palacpac, N.Q.; Yoshida, S.; Sakai, H.; Kimura, Y.; Fujiyama, K.; Yoshida, T.; Seki, T. Stable expression of human β1,4-galactosyltransferase in plant cells modifies N-linked glycosylation patterns. Proc. Natl. Acad. Sci. USA 1999, 96, 4692–4697. [Google Scholar] [CrossRef] [PubMed]
- Bakker, H.; Rouwendal, G.J.A.; Karnoup, A.S.; Florack, D.E.A.; Stoopen, G.M.; Helsper, J.P.F.G.; van Ree, R.; van Die, I.; Bosch, D. An antibody produced in tobacco expressing a hybrid β-1,4-galactosyltransferase is essentially devoid of plant carbohydrate epitopes. Proc. Natl. Acad. Sci. USA 2006, 103, 7577–7582. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Streatfield, S.J.; Wycoff, K. Medical molecular farming: Production of antibodies, biopharmaceuticals and edible vaccines in plants. Trends Plant Sci. 2001, 6, 219–226. [Google Scholar] [CrossRef]
- Kittur, F.S.; Lalgondar, M.; Hung, C.-Y.; Sane, D.C.; Xie, J. C-Terminally fused affinity Strep-tag II is removed by proteolysis from recombinant human erythropoietin expressed in transgenic tobacco plants. Plant Cell Rep. 2014, 34, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Boissel, J.; Showers, M.; Ruch, B.; Bunn, H. Erythropoietin structure-function relationships. Identification of functionally important domains. J. Biol. Chem. 1994, 269, 22839–22846. [Google Scholar] [CrossRef]
- Chargelegue, D.; Vine, N.D.; Van Dolleweerd, C.J.; Drake, P.M.; Ma, J.K.-C. A murine monoclonal antibody produced in transgenic plants with plant-specific glycans is not immunogenic in mice. Transgenic Res. 2000, 9, 187–194. [Google Scholar] [CrossRef]
- Jin, C.; Altmann, F.; Strasser, R.; Mach, L.; Schähs, M.; Kunert, R.; Rademacher, T.; Glössl, J.; Steinkellner, H. A plant-derived human monoclonal antibody induces an anti-carbohydrate immune response in rabbits. Glycobiology 2007, 18, 235–241. [Google Scholar] [CrossRef]
- Ward, B.J.; Landry, N.; Trépanier, S.; Mercier, G.; Dargis, M.; Couture, M.; D’aoust, M.-A.; Vézina, L.-P. Human antibody response to N-glycans present on plant-made influenza virus-like particle (VLP) vaccines. Vaccine 2014, 32, 6098–6106. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, D.; Shi, J.; Yang, D. Expression of α-1,6-fucosyltransferase (FUT8) in rice grain and immunogenicity evaluation of plant-specific glycans. J. Biotechnol. 2017, 242, 111–121. [Google Scholar] [CrossRef]
- Cheung, E.C.C.; McBride, H.M.; Slack, R.S. Mitochondrial dynamics in the regulation of neuronal cell death. Apoptosis 2007, 12, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Balog, J.; Mehta, S.L.; Vemuganti, R. Mitochondrial fission and fusion in secondary brain damage after CNS insults. J. Cereb. Blood Flow Metab. 2016, 36, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Mihara, K. Mitochondrial Dynamics: Functional Link with Apoptosis. Int. J. Cell Biol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Markus, T.M.; Tsai, S.-Y.; Bs, M.R.B.; Farrer, R.G.; O’Brien, T.E.; Kindler-Baumann, D.R.; Rausch, M.; Rudin, M.; Wiessner, C.; Mir, A.K.; et al. Recovery and brain reorganization after stroke in adult and aged rats. Ann. Neurol. 2005, 58, 950–953. [Google Scholar] [CrossRef]
- Yang, W.; Paschen, W. Is age a key factor contributing to the disparity between success of neuroprotective strategies in young animals and limited success in elderly stroke patients? Focus on protein homeostasis. J. Cereb. Blood Flow Metab. 2017, 37, 3318–3324. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Buga, A.-M.; Turner, R.C.; Rosen, C.L.; Toescu, E. Cerebrovascular Disorders: Role of Aging. J. Aging Res. 2012, 2012, 1–4. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Glavan, D.-G.; Olaru, A.; Olaru, D.-G.; Margaritescu, O.; Tica, O.; Surugiu, R.; Sandu, R.E. Present Status and Future Challenges of New Therapeutic Targets in Preclinical Models of Stroke in Aged Animals with/without Comorbidities. Int. J. Mol. Sci. 2018, 19, 356. [Google Scholar] [CrossRef]
- Chen, R.-L.; Balami, J.S.; Esiri, M.M.; Chen, L.-K.; Buchan, A.M. Ischemic stroke in the elderly: An overview of evidence. Nat. Rev. Neurol. 2010, 6, 256–265. [Google Scholar] [CrossRef]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef]
- Hankey, G. Population Impact of Potentially Modifiable Risk Factors for Stroke. Stroke 2020, 51, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Floros, N.; Ibing, W.; Schelzig, H.; Knapsis, A. Neurotherapeutic potential of erythropoietin after ischemic injury of the central nervous system. Neural Regen. Res. 2019, 14, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Buga, A.-M.; Doeppner, T.R.; Hermann, D.M. Stem cell therapies in preclinical models of stroke associated with aging. Front. Cell. Neurosci. 2014, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-K.; Yang, M.-T.; Kang, K.-H.; Liou, H.-C.; Lu, D.-H.; Fu, W.-M.; Lin, W.-L. Targeted Delivery of Erythropoietin by Transcranial Focused Ultrasound for Neuroprotection against Ischemia/Reperfusion-Induced Neuronal Injury: A Long-Term and Short-Term Study. PLoS ONE 2014, 9, e90107. [Google Scholar] [CrossRef]
- Ostrowski, D.; Heinrich, R. Alternative Erythropoietin Receptors in the Nervous System. J. Clin. Med. 2018, 7, 24. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type | Source | Terminal Sugar | Properties | Activities |
|---|---|---|---|---|
| Endogenous EPO | The major amount is produced by the kidneys in adults and small amounts by other organs | Sialic acid | Molecular weight: ~30.4 kDa; pI: 3.92–4.42; half-life in the circulation system: ~5 h | Erythropoiesis; tissue protection |
| RhuEPOM | Overexpressing human EPO gene in mammalian cells | Sialic acid | Molecular weight: 26–36 kDa; pI: 4.42–5.11; half-life in the circulation system: 4–8 h | Erythropoiesis; tissue protection |
| Asialo-rhuEPOE | Enzymatic removal of sialic acids from rhuEPOM | β1,4-Galactose | Molecular weight: undetermined; pI: ~8.5; half-life in the circulation system: 2–3 min | Tissue protection |
| Asialo-rhuEPOP | Overexpressing human EPO and GalT genes in plants | β1,4-Galactose | Molecular weight: 28–30 kDa; pI: ~8.75; half-life in the circulation system: undetermined. | Tissue protection |
| Number of Patients | Dosage | Route | Time of Evaluation | Results | Ref. |
|---|---|---|---|---|---|
| rhuEPOM (n = 21); placebo (n = 19) | 99,000 IU (33,000 IU each within 8 h of symptom onset, and followed up at 24 and 48 h) | IV | 30 days | Reduced infarct size, improved recovery of neurocognitive function and the neurological deficit, and ameliorated stroke-related disability at 30 days | [31] |
| rhuEPOM (n = 37); placebo (n = 43) | 56,000 IU (16,000 IU as a bolus dose followed by 8000 IU each at 12 h intervals for remaining 5 doses) | IV | 28 days | Effective reduction of ischemic stroke complication | [33] |
| rhuEPOM and placebo each (n = 71) | 10,000 IU (5000 IU each at 48 and 72 h after stroke) | SC | 90 days and 5 years | Reduced the scale of Barthel index but did not affect long-term recurrent stroke and mortality; significantly improved long-term neurological outcomes | [34] |
| rhuEPOM (n = 256); placebo (n = 266) | 120,000 IU (40,000 IU each within 6 h of symptom onset, and followed up at 24 and 48 h) | IV | 90 days | The treatment of rhuEPOM or combined with rtPA did not show any improvement in clinical outcomes but had a higher overall death rate | [32] * |
| Author/Year | Cell Line/Animal | Treatment/Model | Outcome |
|---|---|---|---|
| Erbayraktar et al. [35]/2003 | PC-12 cells | Nerve growth factor (NGF) absence-triggered cell death | 34% protection |
| Erbayraktar et al. [35]/2003 | P-19 cells | Hypoxia for 15 h | 43% protection |
| Mennini et al. [133]/2006 | Motoneuron culture | Kainate-induced cell death | Increased survival rate by 57% |
| Ishii et al. [40]/2012 | PC-12 cells | Nerve growth factor (NGF) absence-triggered cell death | No observed protection |
| Kittur et al. [136]/2013 | N2A cells | Staurosporine-induced cell death | 44% protection |
| Erbayraktar et al. [35]/2003 | Sprague Dawley male rats | MCAO model | Reduced infarct volume by ~50% |
| Erbayraktar et al. [35]/2003 | Sprague Dawley male rats | Spinal cord compression | Restricted injury with better neuron survival and motor score |
| Erbayraktar et al. [35]/2003 | Sprague Dawley male rats | Sciatic nerve crush model | Reduced functional loss and improved motor testing score |
| Wang et al. [36]/2004 | Wistar rat pups (7 days old) | Hypoxia–ischemia model | Reduced infarct volume by 52% |
| Grasso et al. [37]/2006 | Sprague Dawley rats | Spinal cord compression | Significantly recovered affected motor function |
| Mennini et al. [133]/2006 | Homozygous wobbler mice | Amyotrophic lateral sclerosis model carrying a mutation of Vps54 gene | Improved motor behavior and reduced inflammation |
| Price et al. [38]/2010 | Sprague Dawley male rats | MCAO model | Significantly reduced infarct volume with reduced cell death |
| Yamashita et al. [39]/2010 | Mongolian male gerbils | Occlusion of the common carotid arteries | Improved learning and memory function with better neuron survival |
| Ishii et al. [40]/2012 | Wistar male rats | MCAO model | Significantly reduced cerebral I/R injury |
| He et al. [41]/2022 | BALB/c male mice | MCAO model | Significant decreased neurological deficits, infarction volume, and edema volume with better neuron survival |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kittur, F.S.; Hung, C.-Y.; Li, P.A.; Sane, D.C.; Xie, J. Asialo-rhuEPO as a Potential Neuroprotectant for Ischemic Stroke Treatment. Pharmaceuticals 2023, 16, 610. https://doi.org/10.3390/ph16040610
Kittur FS, Hung C-Y, Li PA, Sane DC, Xie J. Asialo-rhuEPO as a Potential Neuroprotectant for Ischemic Stroke Treatment. Pharmaceuticals. 2023; 16(4):610. https://doi.org/10.3390/ph16040610
Chicago/Turabian StyleKittur, Farooqahmed S., Chiu-Yueh Hung, P. Andy Li, David C. Sane, and Jiahua Xie. 2023. "Asialo-rhuEPO as a Potential Neuroprotectant for Ischemic Stroke Treatment" Pharmaceuticals 16, no. 4: 610. https://doi.org/10.3390/ph16040610