
In Vitro and In Vivo Characterization of the Transdermal Gel Formulation of Desloratadine for Prevention of Obesity and Metabolic Syndrome
 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Preliminary Data
2.2. Gel Characterization
2.3. Cohesiveness and Adhesiveness
2.4. Lag Time
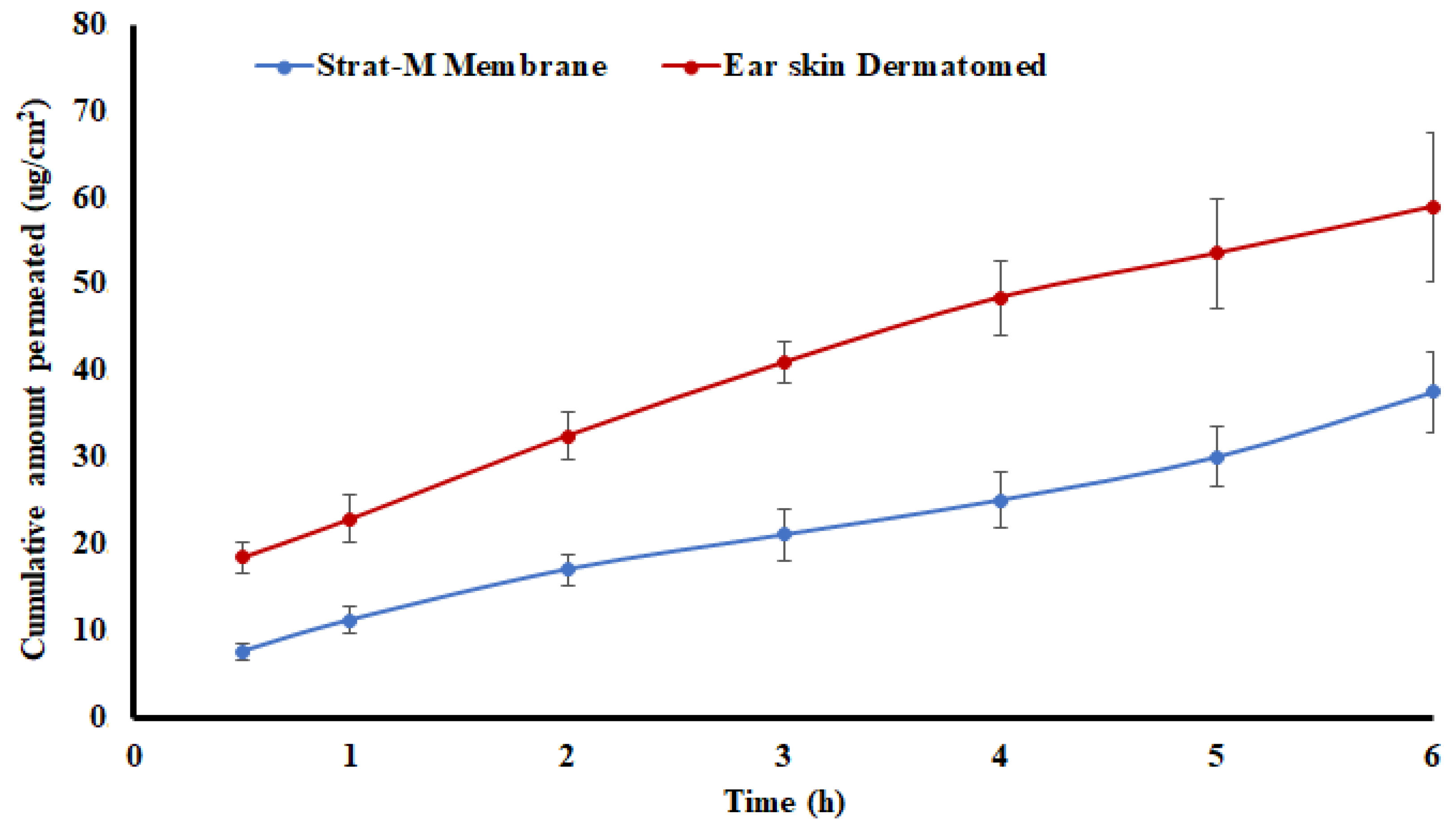
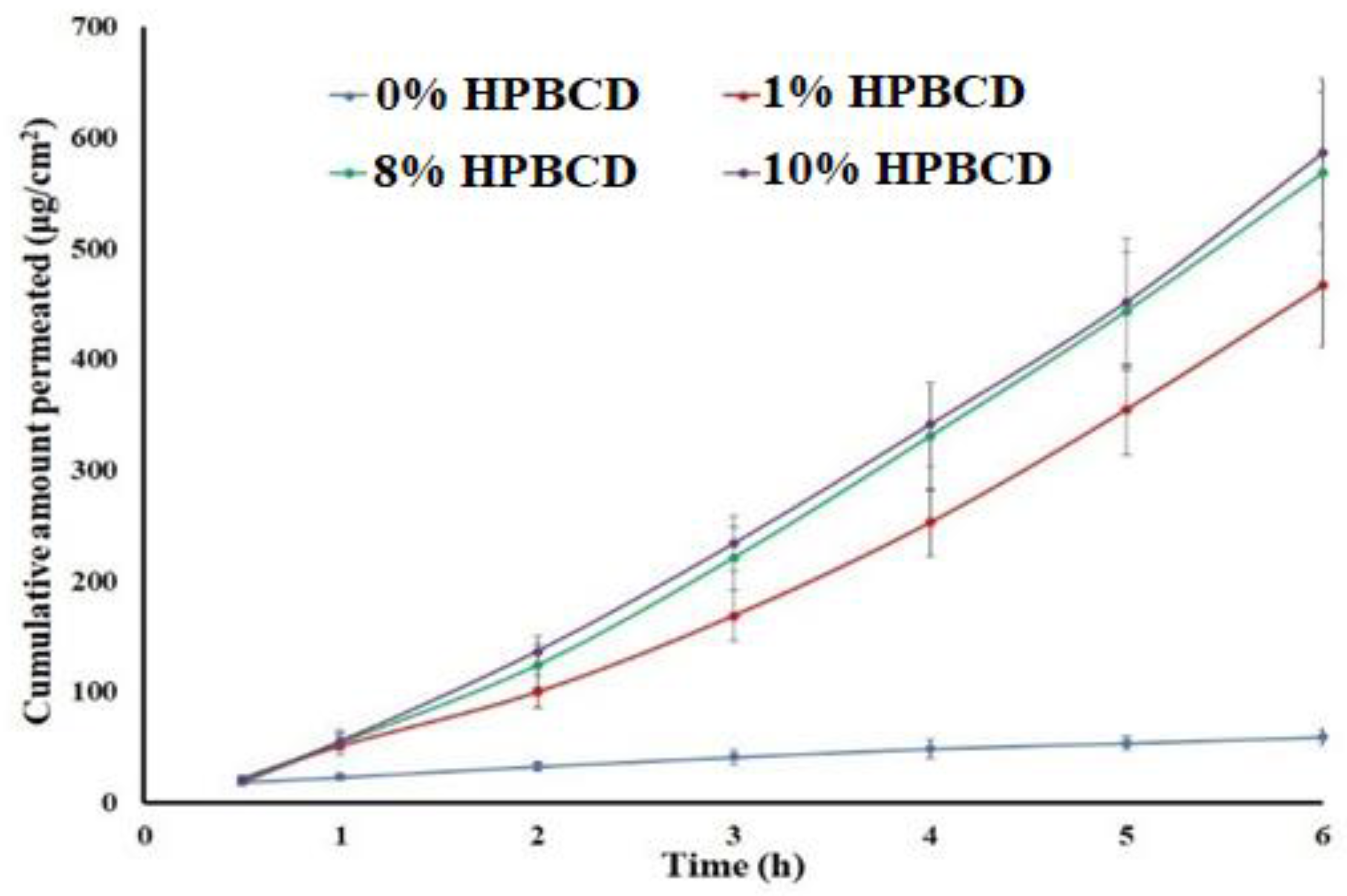
2.5. Flux and Apparent Permeation
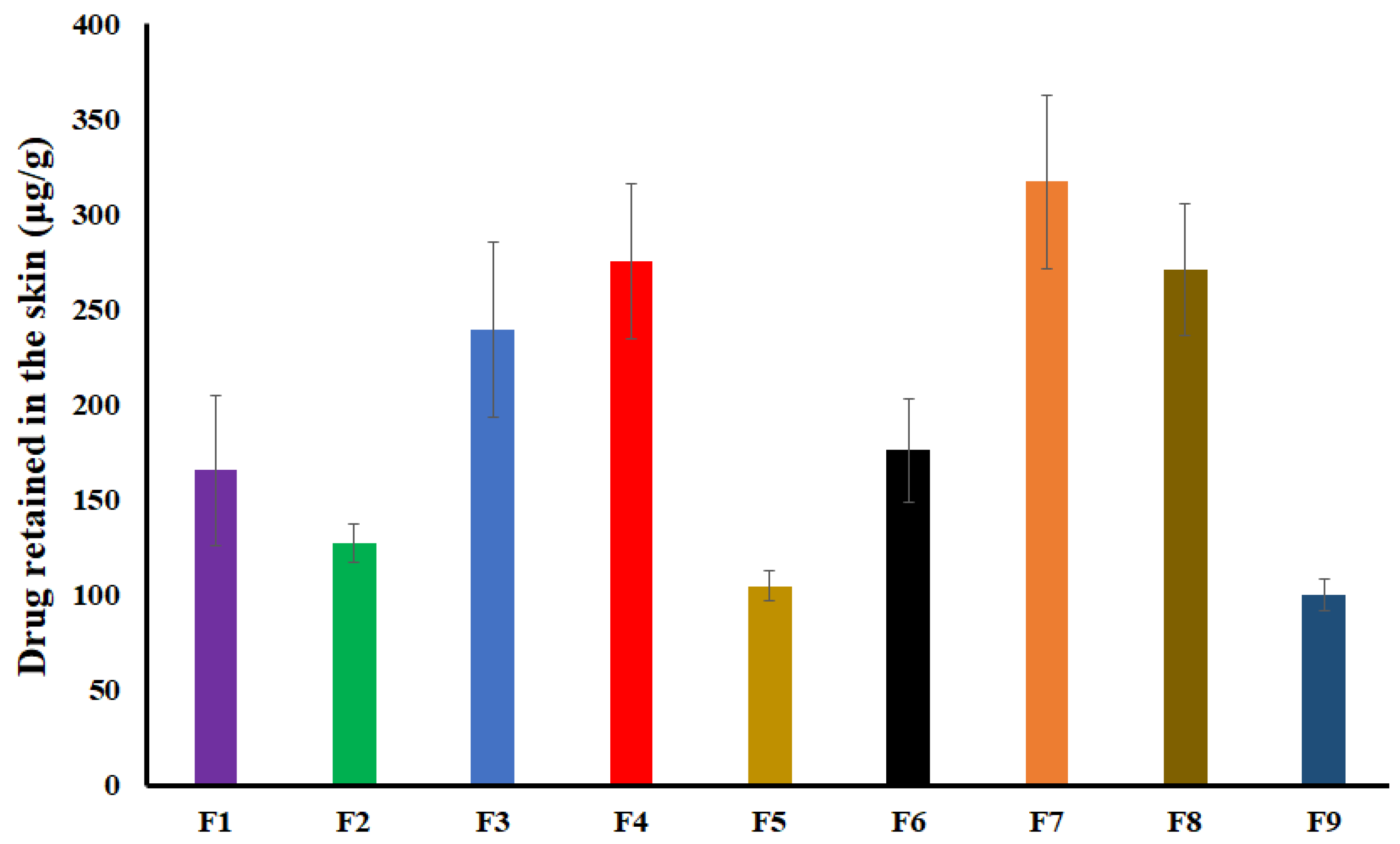
2.6. Drug Retained in the Skin
2.7. Pharmacokinetic Studies
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Drug Solubility
3.2.2. High Performance Liquid Chromatography Analysis
3.2.3. Pig Skin and Synthetic Membranes Preparation
3.2.4. Transdermal Gel Preparation
3.2.5. Texture Analysis
3.2.6. In Vitro Permeation Study
3.2.7. Skin Retention Study
3.2.8. Pharmacokinetic Study
3.2.9. Ultra-Performance Liquid Chromatography Mass Spectrometry
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Qerem, W.; Ling, J.; Pullen, R.; Mcgarry, K. Reported prevalence of allergy and asthma in children from urban and rural Egypt. Air Qual. Atmos. Hleath 2016, 9, 613–620. [Google Scholar] [CrossRef]
- Gupta, R.S.; Warren, C.M.; Smith, B.M.; Jiang, J.; Blumenstock, J.A.; Davis, M.M.; Schleimer, R.P.; Nadeau, K.C. Prevalence and Severity of Food Allergies Among US Adults. JAMA Netw. Open 2019, 2, e185630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seité, S.; Kuo, A.M.; Taieb, C.; Strugar, T.L.; Lio, P. Self-Reported Prevalence of Allergies in the USA and Impact on Skin-An Epidemiological Study on a Representative Sample of American Adults. Int. J. Environ. Res. Public Health 2020, 17, 3360. [Google Scholar] [CrossRef]
- Gupta, R.; Holdford, D.; Bilaver, L.; Dyer, A.; Holl, J.L.; Meltzer, D. The economic impact of childhood food allergy in the United States. JAMA Pediatr. 2013, 167, 1026–1031. [Google Scholar] [CrossRef] [Green Version]
- Morgan Griffin, R. Allergy Statistics and Facts. 2021. Available online: https://www.webmd.com/allergies/allergy-statistics (accessed on 4 October 2022).
- Arnold, J.J.; Williams, P.M. Anaphylaxis: Recognition and management. Am. Fam. Physician 2011, 84, 1111–1118. [Google Scholar] [PubMed]
- Zeke, A.; Sudhir, A. Management of allergic reactions and anaphylaxis in the emergency department. Emerg. Med. Pract. 2022, 24, 1–24. [Google Scholar] [PubMed]
- Chu, J.T. Histamine H1 receptor gene polymorphism acts as a biological indicator of the prediction of therapeutic efficacy in patients with allergic rhinitis in the Chinese Han population. J. Cell. Biochem. 2019, 120, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, H.; Yanai, K.; Wang, D.Y.; Itahashi, K.; Okubo, K. Antihistamines for Allergic Rhinitis Treatment from the Viewpoint of Nonsedative Properties. Int. J. Mol. Sci. 2019, 20, 213. [Google Scholar] [CrossRef] [Green Version]
- Matterne, U.; Böhmer, M.M.; Weisshaar, E.; Jupiter, A.; Carter, B.; Apfelbacher, C.J. Oral H1 antihistamines as ‘add-on’ therapy to topical treatment for eczema. Cochrane Database Syst. Rev. 2019, 1, Cd012167. [Google Scholar] [CrossRef]
- Gracious, B.; Abe, N.; Sundberg, J. The importance of taking a history of over-the-counter medication use: A brief review and case illustration of “PRN” antihistamine dependence in a hospitalized adolescent. J. Child Adolesc. Psychopharmacol. 2010, 20, 521–524. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; Miszta, P.; Filipek, S. Molecular Modeling of Histamine Receptors-Recent Advances in Drug Discovery. Molecules 2021, 26, 1778. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, M.E.; Wolff, A.H.; Bielory, L. Efficacy and tolerability of second- and third-generation antihistamines in the treatment of acquired cold urticaria: A meta-analysis. Ann. Allergy Asthma Immunol. 2010, 104, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, J.C.; Barber, J.A.; Palmese, L.B.; Reutenauer, E.L.; Tek, C. Association of prescription H1 antihistamine use with obesity: Results from the National Health and Nutrition Examination Survey. Obesity 2010, 18, 2398–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, M.; Syed, S.; Ilyas, M.; Gashev, A.A. Antihistamines Increase Body Mass Index Percentiles and Z-Scores in Hispanic Children. Children 2020, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Nizamutdinova, I.T.; Maejima, D.; Nagai, T.; Meininger, C.J.; Gashev, A.A. Histamine as an Endothelium-Derived Relaxing Factor in Aged Mesenteric Lymphatic Vessels. Lymphat. Res. Biol. 2017, 15, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Gasheva, O.Y.; Zawieja, D.C.; Meininger, C.J.; Gashev, A.A. Histamine-mediated autocrine signaling in mesenteric perilymphatic mast cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R590–R604. [Google Scholar] [CrossRef]
- Pal, S.; Meininger, C.J.; Gashev, A.A. Aged Lymphatic Vessels and Mast Cells in Perilymphatic Tissues. Int. J. Mol. Sci. 2017, 18, 965. [Google Scholar] [CrossRef] [Green Version]
- Gasheva, O.Y.; Tsoy Nizamutdinova, I.; Hargrove, L.; Gobbell, C.; Troyanova-Wood, M.; Alpini, S.F.; Pal, S.; Du, C.; Hitt, A.R.; Yakovlev, V.V.; et al. Prolonged intake of desloratadine: Mesenteric lymphatic vessel dysfunction and development of obesity/metabolic syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G217–G227. [Google Scholar] [CrossRef]
- Troyanova-Wood, M.; Gobbell, C.; Meng, Z.; Gasheva, O.; Gashev, A.; Yakovlev, V.V. Assessing the effect of prolonged use of desloratadine on adipose Brillouin shift and composition in rats. J. Biophotonics 2021, 14, e202000269. [Google Scholar] [CrossRef]
- Berginc, K.; Sibinovska, N.; Žakelj, S.; Trontelj, J.; Legen, I. Biopharmaceutical classification of desloratadine—Not all drugs are classified the easy way. Acta Pharm. 2020, 70, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Minhua, F.; Shuangyu, J.; Hua, L.; Liming, F.; Xulei, Z. Desloratadine External Preparation and Preparation Method Thereof. China Pat. Appl. 2009. CN101991574A. Hainan Pulin Pharmaceutical Co Ltd., Zhejiang Ridae Pharmaceutical Co Ltd., Hangzhou Saili Medicine Inst Co Ltd. Available online: https://patents.google.com/patent/CN101991574A/en#legalEvents (accessed on 4 October 2022).
- Minhua, F.; Jun, S.; Hua, L.; Hui, L. Desloratadine Gel and Its Preparation Method. 2012, CN103877006A. Zhejiang Poly Pharmaceutical Co., Ltd.; Hangzhou Saili Medicine Inst Co., Ltd.; Hainan Pulin Pharmaceutical Co Ltd.; China. Available online: https://patents.google.com/patent/CN103877006A/en (accessed on 4 October 2022).
- Trivedi, D.G.; Patel, H.J.; Bhandari, A.; Shah, D. Preparation and evaluation of transdermal patch of desloratadine. Int. J. Biomed. Res. 2011, 6, 359–373. [Google Scholar] [CrossRef] [Green Version]
- Yurtdaş-Kırımlıoğlu, G. A promising approach to design thermosensitive in situ gel based on solid dispersions of desloratadine with Kolliphor® 188 and Pluronic® F127. J. Therm. Anal Calorim. 2022, 147, 1307–1327. [Google Scholar] [CrossRef]
- Mura, P.; Faucci, M.T.; Bramanti, G.; Corti, P. Evaluation of transcutol as a clonazepam transdermal permeation enhancer from hydrophilic gel formulations. Eur. J. Pharm. Sci. 2000, 9, 365–372. [Google Scholar] [CrossRef]
- Neupane, R.; Boddu, S.H.S.; Renukuntla, J.; Babu, R.J.; Tiwari, A.K. Alternatives to Biological Skin in Permeation Studies: Current Trends and Possibilities. Pharmaceutics 2020, 12, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todo, H. Transdermal Permeation of Drugs in Various Animal Species. Pharmaceutics 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, P.; Picchini, U.; Beck, B.; van Gelder, J.; Delbar, N.; DeGaetano, A. A general approach to the apparent permeability index. J. Pharmacokinet. Pharmacodyn. 2008, 35, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Vogensen, S.B.; Brewster, M.E.; Konrádsdóttir, F. Effects of cyclodextrins on drug delivery through biological membranes. J. Pharm. Sci. 2007, 96, 2532–2546. [Google Scholar] [CrossRef]
- Másson, M.; Loftsson, T.; Másson, G.; Stefánsson, E. Cyclodextrins as permeation enhancers: Some theoretical evaluations and in vitro testing. J. Control Release 1999, 59, 107–118. [Google Scholar] [CrossRef]
- Hamed, R.; Al Baraghthi, T.; Alkilani, A.Z.; Abu-Huwaij, R. Correlation Between Rheological Properties and In Vitro Drug Release from Penetration Enhancer-Loaded Carbopol® Gels. J. Pharm. Innov. 2016, 11, 339–351. [Google Scholar] [CrossRef]
- Suhail, M.; Fang, C.-W.; Khan, A.; Minhas, M.U.; Wu, P.-C. Fabrication and In Vitro Evaluation of pH-Sensitive Polymeric Hydrogels as Controlled Release Carriers. Gels 2021, 7, 110. [Google Scholar] [CrossRef]
- Robnik, B.; Naumoska, K.; Časar, Z. A Novel Testing Approach for Oxidative Degradation Dependent Incompatibility of Amine Moiety Containing Drugs with PEGs in Solid-State. Pharmaceutics 2020, 12, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, M.; Joginapalli, S.; Billa, P.R. Desloratadine-Containing Formulation Stabilized with Cyclodextrin. 2007, EP1988893A2. Orchid Chemicals and Pharmaceuticals Ltd. Available online: https://patents.google.com/patent/EP1988893A2/en (accessed on 4 October 2022).
- Ahmed, S.; Mahmood, S.; Danish Ansari, M.; Gull, A.; Sharma, N.; Sultana, Y. Nanostructured lipid carrier to overcome stratum corneum barrier for the delivery of agomelatine in rat brain; formula optimization, characterization and brain distribution study. Int. J. Pharm. 2021, 607, 121006. [Google Scholar] [CrossRef] [PubMed]
- Binder, L.; Mazál, J.; Petz, R.; Klang, V.; Valenta, C. The role of viscosity on skin penetration from cellulose ether-based hydrogels. Skin Res. Technol. 2019, 25, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolla, P.K.; Clark, B.A.; Juluri, A.; Cheruvu, H.S.; Renukuntla, J. Evaluation of Formulation Parameters on Permeation of Ibuprofen from Topical Formulations Using Strat-M(®) Membrane. Pharmaceutics 2020, 12, 151. [Google Scholar] [CrossRef] [Green Version]
- Mustapha, R.B.; Lafforgue, C.; Fenina, N.; Marty, J.P. Influence of drug concentration on the diffusion parameters of caffeine. Indian. J. Pharmacol. 2011, 43, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, D.W.; Musakhanian, J. Skin Penetration and Permeation Properties of Transcutol®-Neat or Diluted Mixtures. AAPS PharmSciTech 2018, 19, 3512–3533. [Google Scholar] [CrossRef] [PubMed]
- Health, P. Regulatory, Authority. Desloratadine 0.5 mg/ml Oral Solution, Public Assessment Report for a Medicinal Product for Human Use. 2020. Available online: https://www.hpra.ie/img/uploaded/swedocuments/Public_AR_PA2315-012-001_19112020104445.pdf (accessed on 4 October 2022).
- EMEA, S. Discussion. EMEA Scientific Discussion—Desloratadine. 2004. Available online: https://www.ema.europa.eu/en/documents/scientific-discussion/aerius-epar-scientific-discussion_en.pdf (accessed on 4 October 2022).
- Gupta, S.; Banfield, C.; Affrime, M.; Marbury, T.; Padhi, D.; Glue, P. Oral bioavailability of desloratadine is unaffected by food. Clin. Pharmacokinet. 2002, 41 (Suppl. S1), 7–12. [Google Scholar] [CrossRef]
- Molimard, M.; Diquet, B.; Benedetti, M.S. Comparison of pharmacokinetics and metabolism of desloratadine, fexofenadine, levocetirizine and mizolastine in humans. Fundam. Clin. Pharmacol. 2004, 18, 399–411. [Google Scholar] [CrossRef]
- Kapoor, Y.; Kumar, K. Structural and clinical impact of anti-allergy agents: An overview. Bioorg. Chem. 2020, 94, 103351. [Google Scholar] [CrossRef]
- Cho, C.W.; Choi, J.S.; Kim, S.J.; Shin, S.C. Enhanced bioavailability and antihistamine effects of loratadine from the transdermal EVA matrix system in rats. J. Drug Deliv. Sci. Technol. 2010, 20, 107–110. [Google Scholar] [CrossRef]
- Davies, D.J.; Heylings, J.R.; McCarthy, T.J.; Correa, C.M. Development of an in vitro model for studying the penetration of chemicals through compromised skin. Toxicol. In Vitr. 2015, 29, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirilov, P.; Tran, V.H.; Ducrotté-Tassel, A.; Salvi, J.P.; Perrot, S.; Haftek, M.; Boulieu, R.; Pirot, F. Ex-Vivo percutaneous absorption of enrofloxacin: Comparison of LMOG organogel vs. pentravan cream. Int. J. Pharm. 2016, 498, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Sintov, A.C. Cumulative evidence of the low reliability of frozen/thawed pig skin as a model for in vitro percutaneous permeation testing. Eur. J. Pharm. Sci. 2017, 102, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, T.; Mohamed, E.M.; Dharani, S.; Kayalar, C.; Ozkan, T.; Kuttolamadom, M.A.; Rahman, Z.; Khan, M.A. Very-Rapidly Dissolving Printlets of Isoniazid Manufactured by SLS 3D Printing: In Vitro and In Vivo Characterization. Mol. Pharm. 2022, 19, 2937–2949. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Polymer (%) | Drug (%) | Transcutol® (%) | Viscosity (Pas) | Flux- Ear Skin (μg/cm2.h) | Papp- Ear Skin (cm/h) | Flux- Strat-M® (μg/cm2.h) | Papp- Strat-M® (cm/h) |
|---|---|---|---|---|---|---|---|---|
| F1 | 2.0 | 2.50 | 20.0 | 31.9 ± 2.7 | 248.6 ± 31.4 | 1.24 × 10−2 | 12.6 ± 1.5 | 6.28 × 10−4 |
| F2 | 3.0 | 2.50 | 20.0 | 243.9 ± 21.5 | 269.3 ± 35.8 | 1.35 × 10−2 | 34.7 ± 3.7 | 1.74 × 10−3 |
| F3 | 2.5 | 3.75 | 17.5 | 180.9 ± 16.3 | 256.7 ± 27.4 | 8.56 × 10−3 | 36.4 ± 3.9 | 1.21 × 10−3 |
| F4 | 3.0 | 5.00 | 20.0 | 374.5 ± 25.4 | 302.5 ± 38.2 | 7.56 × 10−3 | 38.2 ± 4.1 | 9.55 × 10−3 |
| F5 | 3.0 | 2.50 | 15.0 | 493.4 ± 35.9 | 149.7 ± 17.9 | 7.49 × 10−3 | 24.5 ± 2.7 | 1.23 × 10−3 |
| F6 | 3.0 | 5.00 | 15.0 | 519.9 ± 48.3 | 291.5 ± 30.5 | 7.29 × 10−3 | 36.5 ± 3.8 | 9.11 × 10−3 |
| F7 | 2.0 | 5.00 | 15.0 | 104.6 ± 9.0 | 287.9 ± 31.6 | 7.20 × 10−3 | 46.9 ± 4.8 | 1.17 × 10−3 |
| F8 | 2.0 | 5.00 | 20.0 | 65.5 ± 5.5 | 436.8 ± 44.2 | 1.09 × 10−3 | 97.4 ± 9.9 | 2.43 × 10−3 |
| F9 | 2.0 | 2.50 | 15.0 | 58.2 ± 4.5 | 347.0 ± 36.9 | 8.68 × 10−3 | 51.7 ± 5.1 | 1.29 × 10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, E.M.; Dharani, S.; Khuroo, T.; Hamed, R.; Khan, M.A.; Rahman, Z. In Vitro and In Vivo Characterization of the Transdermal Gel Formulation of Desloratadine for Prevention of Obesity and Metabolic Syndrome. Pharmaceuticals 2023, 16, 578. https://doi.org/10.3390/ph16040578
Mohamed EM, Dharani S, Khuroo T, Hamed R, Khan MA, Rahman Z. In Vitro and In Vivo Characterization of the Transdermal Gel Formulation of Desloratadine for Prevention of Obesity and Metabolic Syndrome. Pharmaceuticals. 2023; 16(4):578. https://doi.org/10.3390/ph16040578
Chicago/Turabian StyleMohamed, Eman M., Sathish Dharani, Tahir Khuroo, Rania Hamed, Mansoor A. Khan, and Ziyaur Rahman. 2023. "In Vitro and In Vivo Characterization of the Transdermal Gel Formulation of Desloratadine for Prevention of Obesity and Metabolic Syndrome" Pharmaceuticals 16, no. 4: 578. https://doi.org/10.3390/ph16040578