
Recent Advances in the Development of Adenovirus-Vectored Vaccines for Parasitic Infections

Abstract
:1. Introduction
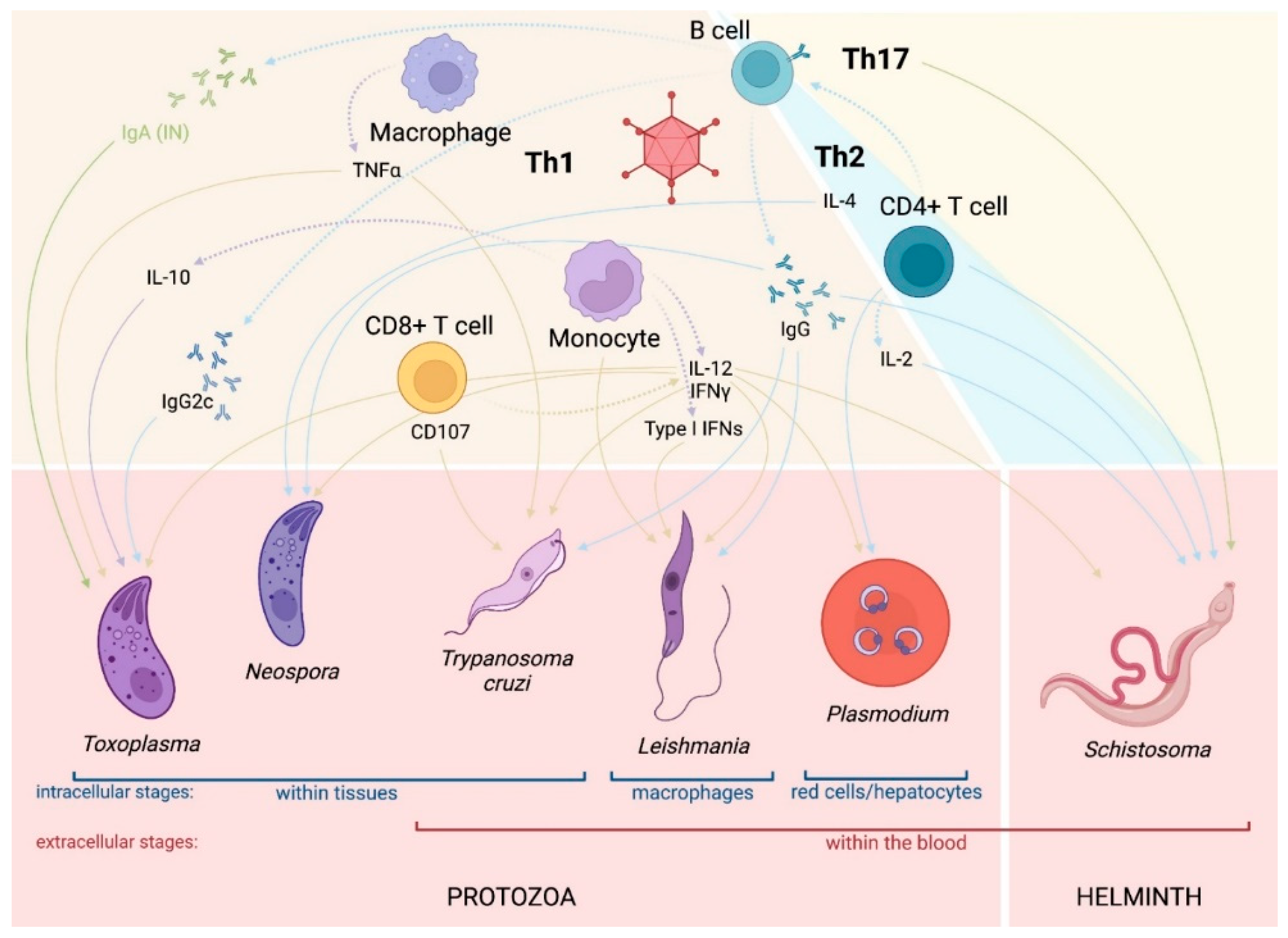
2. Parasite Vaccines
3. Adenoviruses as a Vaccine Vector
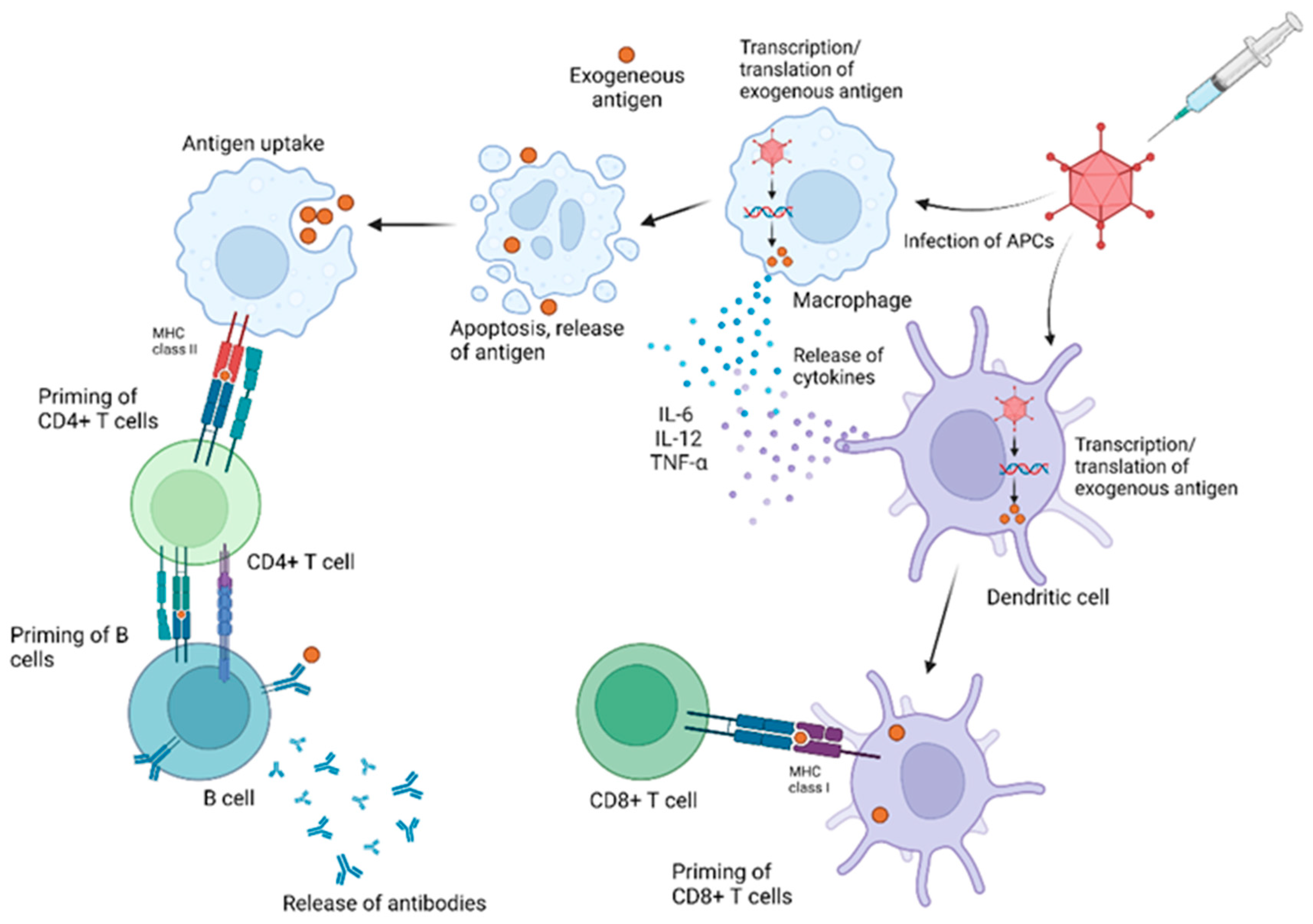
3.1. Immune Responses to AdV Vectors
3.2. Limitations of AdV Vectors: Concerns and Considerations
3.3. AdV Vaccines Approved and in Development
4. Adenovirus-Vectored Vaccines against Malaria
5. Adenovirus-Vectored Vaccines against Chagas Disease

{kind=link}
{kind=link}
| Immune skew | Adjuvant | Composition | Sensor/Receptor | Use in Approved Vaccines | Refs. |
|---|---|---|---|---|---|
| Th1 | IL12 | Recombinant IL-12 | IL12Rβ1 and IL12Rβ2 | [124] | |
| Freund’s complete adjuvant | Mineral Oil-in-water + heat-killed mycobacteria | NOD2 | Not for human use | [125] | |
| CpG | Synthetic oligodeoxynucleotides | TLR-9 | Hepatitis B (Heplisav-B) | [126] | |
| MPL-A | Detoxified LPS | TLR-4 | Component of AS04 | [127] | |
| LPS | Lipopolysaccharide | TLR-4 | [128] | ||
| Flagellin | Flagellin | TLR-5 | [129] | ||
| AS01 | Adjuvant system (MPL-A and QS-21) | TLR-4 | Shingles (Shingrix), malaria (RTS,S/AS01) | [130] | |
| Th2 | Alum | Aluminum Salts | NLPR3, likely additional mechanisms | Diphtheria-Pertussis- Tetanus (Tdap), Hepatitis A & B (Twinrix), etc. | [131] |
| Freund’s incomplete | Mineral Oil-in-water | Unknown | Human use discontinued in the 1950s | [125] | |
| Various helminth antigens | Various | [29,132] | |||
| Mixed | MF59 | Oil-in-water | Unknown | Influenza (Fluad Quadrivalent) | [133] |
| AS04 | Adjuvant system (MPL-A and Alum) | TLR-4 | HPV (Cervarix) | [127] | |
| AS03 | Adjuvant system (α- tocopherol and squalene) | Unknown | Influenza (Pandemrix, Q-pan) | [134] | |
| Matrix M | Saponins | Unknown | COVID-19 (Nuvaxovid) | [135] | |
| Mucosal | Cholera toxin B | Non-toxic subunit B of Cholera toxin | GM1 | Cholera (Dukoral) | [136] |
| dmLT | Modified heat-labile enterotoxin | GM1 | [137] |
6. Adenovirus-Vectored Vaccines against Schistosomiasis
7. Adenovirus-Vectored Vaccines against Leishmaniasis
8. Adenovirus-Vectored Vaccines against Toxoplasmosis
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riedel, S. Edward Jenner and the History of Smallpox and Vaccination. In Baylor University Medical Center Proceedings; Taylor & Francis: Abingdon-on-Thames, UK, 2005; Volume 18, pp. 21–25. [Google Scholar]
- Brimnes, N. Variolation, vaccination and popular resistance in early colonial south India. Med. Hist. 2004, 48, 199–228. [Google Scholar] [CrossRef] [Green Version]
- Mohebali, M.; Nadim, A.; Khamesipour, A. An overview of leishmanization experience: A successful control measure and a tool to evaluate candidate vaccines. Acta Trop. 2019, 200, 105173. [Google Scholar] [CrossRef]
- Plotkin, S. History of vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 12283–12287. [Google Scholar] [CrossRef] [Green Version]
- Kallerup, R.S.; Foged, C. Classification of Vaccines. In Subunit Vaccine Delivery; Foged, C., Rades, T., Perrie, Y., Hook, S., Eds.; Springer: New York, NY, USA, 2015; pp. 15–29. [Google Scholar]
- Moss, B.; Smith, G.L.; Gerin, J.L.; Purcell, R.H. Live recombinant vaccinia virus protects chimpanzees against hepatitis B. Nature 1984, 311, 67–69. [Google Scholar] [CrossRef]
- Draper, S.J.; Heeney, J.L. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol. 2010, 8, 62–73. [Google Scholar] [CrossRef]
- Travieso, T.; Li, J.; Mahesh, S.; Mello, J.D.F.R.E.; Blasi, M. The use of viral vectors in vaccine development. NPJ Vaccines 2022, 7, 75. [Google Scholar] [CrossRef]
- Afkhami, S.; Yao, Y.; Xing, Z. Methods and clinical development of adenovirus-vectored vaccines against mucosal pathogens. Mol. Ther. Methods Clin. Dev. 2016, 3, 16030. [Google Scholar] [CrossRef] [Green Version]
- Mendonça, S.A.; Lorincz, R.; Boucher, P.; Curiel, D.T. Adenoviral vector vaccine platforms in the SARS-CoV-2 pandemic. NPJ Vaccines 2021, 6, 97. [Google Scholar] [CrossRef]
- WHO. The Oxford/AstraZeneca (ChAdOx1-S [Recombinant] Vaccine) COVID-19 Vaccine: What You Need to Know. Available online: https://www.who.int/news-room/feature-stories/detail/the-oxford-astrazeneca-covid-19-vaccine-what-you-need-to-know (accessed on 26 January 2023).
- WHO. The Janssen Ad26.COV2.S COVID-19 Vaccine: What You Need to Know. Available online: https://www.who.int/news-room/feature-stories/detail/the-j-j-covid-19-vaccine-what-you-need-to-know (accessed on 26 January 2023).
- Rappuoli, R. Bridging the knowledge gaps in vaccine design. Nat. Biotechnol. 2007, 25, 1361–1366. [Google Scholar] [CrossRef]
- Anisuzzaman; Tsuji, N. Schistosomiasis and hookworm infection in humans: Disease burden, pathobiology and anthelmintic vaccines. Parasitol. Int. 2020, 75, 102051. [Google Scholar] [CrossRef]
- Liew, F.Y.; Vickerman, K.; Hill, A.V.S.; Jepson, A.; Plebanski, M.; Gilbert, S.C. Genetic analysis of host–parasite coevolution in human malaria. Philos. Trans. R. Soc. B Biol. Sci. 1997, 352, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Cox, F.E. History of human parasitology. Clin. Microbiol. Rev. 2002, 15, 595–612. [Google Scholar] [CrossRef] [Green Version]
- Limbach, K.J.; Richie, T.L. Viral vectors in malaria vaccine development. Parasite Immunol. 2009, 31, 501–519. [Google Scholar] [CrossRef]
- Sacks, D.L.; Peters, N.C.; Bethony, J.M. Chapter 17—Vaccines Against Parasites. In The Vaccine Book, 2nd ed.; Bloom, B.R., Lambert, P.-H., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 331–360. [Google Scholar]
- WHO. Neglected Tropical Diseases. Available online: https://www.who.int/health-topics/neglected-tropical-diseases (accessed on 11 December 2022).
- WHO. WHO Recommends Groundbreaking Malaria Vaccine for Children at Risk. Available online: https://www.who.int/news/item/06-10-2021-who-recommends-groundbreaking-malaria-vaccine-for-children-at-risk (accessed on 12 December 2022).
- Gordon, D.M.; McGovern, T.W.; Krzych, U.; Cohen, J.C.; Schneider, I.; LaChance, R.; Heppner, D.G.; Yuan, G.; Hollingdale, M.; Slaoui, M.; et al. Safety, immunogenicity, and efficacy of a recombinantly produced Plasmodium falciparum circumsporozoite protein-hepatitis B surface antigen subunit vaccine. J. Infect. Dis. 1995, 171, 1576–1585. [Google Scholar] [CrossRef]
- RTS,S Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 2015, 386, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Maxmen, A. Scientists Hail Historic Malaria Vaccine Approval—But Point to Challenges Ahead. Available online: https://www.nature.com/articles/d41586-021-02755-5#ref-CR1 (accessed on 13 December 2022).
- Datoo, M.S.; Natama, H.M.; Somé, A.; Bellamy, D.; Traoré, O.; Rouamba, T.; Tahita, M.C.; Ido, N.F.A.; Yameogo, P.; Valia, D.; et al. Efficacy and immunogenicity of R21/Matrix-M vaccine against clinical malaria after 2 years’ follow-up in children in Burkina Faso: A phase 1/2b randomised controlled trial. Lancet Infect. Dis. 2022, 22, 1728–1736. [Google Scholar] [CrossRef]
- Collins, K.A.; Snaith, R.; Cottingham, M.G.; Gilbert, S.C.; Hill, A.V.S. Enhancing protective immunity to malaria with a highly immunogenic virus-like particle vaccine. Sci. Rep. 2017, 7, 46621. [Google Scholar] [CrossRef] [Green Version]
- Palatnik-de-Sousa, C.B.; Nico, D. The Delay in the Licensing of Protozoal Vaccines: A Comparative History. Front. Immunol. 2020, 11, 204. [Google Scholar] [CrossRef]
- McAllister, M.M. Successful vaccines for naturally occurring protozoal diseases of animals should guide human vaccine research. A review of protozoal vaccines and their designs. Parasitology 2014, 141, 624–640. [Google Scholar] [CrossRef] [Green Version]
- Stutzer, C.; Richards, S.A.; Ferreira, M.; Baron, S.; Maritz-Olivier, C. Metazoan Parasite Vaccines: Present Status and Future Prospects. Front. Cell. Infect. Microbiol. 2018, 8, 67. [Google Scholar] [CrossRef] [Green Version]
- Perera, D.J.; Ndao, M. Promising Technologies in the Field of Helminth Vaccines. Front. Immunol. 2021, 12, 711650. [Google Scholar] [CrossRef]
- Benkő, M.; Aoki, K.; Arnberg, N.; Davison, A.J.; Echavarría, M.; Hess, M.; Jones, M.S.; Kaján, G.L.; Kajon, A.E.; Mittal, S.K.; et al. ICTV Virus Taxonomy Profile: Adenoviridae 2022. J. Gen. Virol. 2022, 103, 001721. [Google Scholar] [CrossRef]
- Tatsis, N.; Ertl, H.C.J. Adenoviruses as vaccine vectors. Mol. Ther. 2004, 10, 616–629. [Google Scholar] [CrossRef]
- Lynch, J.P., 3rd; Kajon, A.E. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chirmule, N.; Gao, G.-P.; Qian, R.; Croyle, M.; Joshi, B.; Tazelaar, J.; Wilson, J.M. Acute Cytokine Response to Systemic Adenoviral Vectors in Mice Is Mediated by Dendritic Cells and Macrophages. Mol. Ther. 2001, 3, 697–707. [Google Scholar] [CrossRef]
- Reyes-Sandoval, A.; Harty, J.T.; Todryk, S.M. Viral vector vaccines make memory T cells against malaria. Immunology 2007, 121, 158–165. [Google Scholar] [CrossRef]
- Barouch, D.H.; Kik, S.V.; Weverling, G.J.; Dilan, R.; King, S.L.; Maxfield, L.F.; Clark, S.; Ng’ang’a, D.; Brandariz, K.L.; Abbink, P.; et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine 2011, 29, 5203–5209. [Google Scholar] [CrossRef] [Green Version]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors. Hum. Vaccine Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [Green Version]
- Van Winkel, C.A.J.; Moreno, A.; Curiel, D.T. Capsid-Incorporation Strategy To Display Antigens for an Alternative Adenoviral Vector Vaccine Approach. Mol. Pharm. 2018, 15, 5446–5453. [Google Scholar] [CrossRef]
- Baker, A.T.; Boyd, R.J.; Sarkar, D.; Teijeira-Crespo, A.; Chan, C.K.; Bates, E.; Waraich, K.; Vant, J.; Wilson, E.; Truong, C.D.; et al. ChAdOx1 interacts with CAR and PF4 with implications for thrombosis with thrombocytopenia syndrome. Sci. Adv. 2021, 7, eabl8213. [Google Scholar] [CrossRef]
- Buoninfante, A.; Andeweg, A.; Baker, A.T.; Borad, M.; Crawford, N.; Dogné, J.-M.; Garcia-Azorin, D.; Greinacher, A.; Helfand, R.; Hviid, A.; et al. Understanding thrombosis with thrombocytopenia syndrome after COVID-19 vaccination. NPJ Vaccines 2022, 7, 141. [Google Scholar] [CrossRef]
- WHO. Status of COVID-19 Vaccines within WHO EUL/PQ Evaluation Process. Available online: https://www.who.int/teams/regulation-prequalification/eul/covid-19 (accessed on 11 December 2022).
- Dai, Y.; Wang, X.; Zhao, S.; Tang, J.; Zhang, L.; Dai, J.; Zeng, M.; Lu, S.; Zhu, Y.; Su, C. Construction and evaluation of replication-defective recombinant optimized triosephosphate isomerase adenoviral vaccination in Schistosoma japonicum challenged mice. Vaccine 2014, 32, 771–778. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, X.; Tang, J.; Zhao, S.; Xing, Y.; Dai, J.; Jin, X.; Zhu, Y. Enhancement of protective efficacy through adenoviral vectored vaccine priming and protein boosting strategy encoding triosephosphate isomerase (SjTPI) against Schistosoma japonicum in mice. PLoS ONE 2015, 10, e0120792. [Google Scholar] [CrossRef]
- Dai, Y.; Zhao, S.; Tang, J.; Xing, Y.; Qu, G.; Dai, J.; Jin, X.; Wang, X. Evaluation of protective efficacy induced by different heterologous prime-boost strategies encoding triosephosphate isomerase against Schistosoma japonicum in mice. Parasites Vectors 2017, 10, 111. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Zhu, L.; Luo, R.; Dao, J.; Zhao, J.; Shi, Y.; Li, H.; Lu, K.; Feng, X.; Lin, J.; et al. Evaluation of protective immune response in mice by vaccination the recombinant adenovirus for expressing Schistosoma japonicum inhibitor apoptosis protein. Parasitol. Res. 2014, 113, 4261–4269. [Google Scholar] [CrossRef]
- Perera, D.J.; Hassan, A.S.; Liu, S.S.; Elahi, S.M.; Gadoury, C.; Weeratna, R.D.; Gilbert, R.; Ndao, M. A low dose adenovirus vectored vaccine expressing Schistosoma mansoni Cathepsin B protects from intestinal schistosomiasis in mice. EBioMedicine 2022, 80, 104036. [Google Scholar] [CrossRef]
- WHO. World Malaria Report 2022; World Health Organization: Geneva, Switzerland, 2022.
- WHO. Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 11 December 2022).
- Abuga, K.M.; Jones-Warner, W.; Hafalla, J.C.R. Immune responses to malaria pre-erythrocytic stages: Implications for vaccine development. Parasite Immunol. 2021, 43, e12795. [Google Scholar] [CrossRef]
- Hafalla, J.C.; Silvie, O.; Matuschewski, K. Cell biology and immunology of malaria. Immunol. Rev. 2011, 240, 297–316. [Google Scholar] [CrossRef]
- Hoffman, S.L.; Goh, L.M.L.; Luke, T.C.; Schneider, I.; Le, T.P.; Doolan, D.L.; Sacci, J.; de la Vega, P.; Dowler, M.; Paul, C.; et al. Protection of Humans against Malaria by Immunization with Radiation-Attenuated Plasmodium falciparum Sporozoites. J. Infect. Dis. 2002, 185, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Silvie, O.; Mota, M.M.; Matuschewski, K.; Prudêncio, M. Interactions of the malaria parasite and its mammalian host. Curr. Opin. Microbiol. 2008, 11, 352–359. [Google Scholar] [CrossRef]
- Shears, M.J.; Sekhar Nirujogi, R.; Swearingen, K.E.; Renuse, S.; Mishra, S.; Jaipal Reddy, P.; Moritz, R.L.; Pandey, A.; Sinnis, P. Proteomic Analysis of Plasmodium Merosomes: The Link between Liver and Blood Stages in Malaria. J. Proteome Res. 2019, 18, 3404–3418. [Google Scholar] [CrossRef]
- Hoffman, S.L.; Richie, T.L. 6—Disease States and Vaccines: Selected Cases: Part I. Malaria. In The Vaccine Book; Bloom, B.R., Lambert, P.-H., Eds.; Academic Press: San Diego, CA, USA, 2003; pp. 291–310. [Google Scholar]
- Ogwang, C.; Kimani, D.; Edwards, N.J.; Roberts, R.; Mwacharo, J.; Bowyer, G.; Bliss, C.; Hodgson, S.H.; Njuguna, P.; Viebig, N.K.; et al. Prime-boost vaccination with chimpanzee adenovirus and modified vaccinia Ankara encoding TRAP provides partial protection against Plasmodium falciparum infection in Kenyan adults. Sci. Transl. Med. 2015, 7, 286re5. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, S.H.; Ewer, K.J.; Bliss, C.M.; Edwards, N.J.; Rampling, T.; Anagnostou, N.A.; de Barra, E.; Havelock, T.; Bowyer, G.; Poulton, I.D.; et al. Evaluation of the efficacy of ChAd63-MVA vectored vaccines expressing circumsporozoite protein and ME-TRAP against controlled human malaria infection in malaria-naive individuals. J. Infect. Dis. 2015, 211, 1076–1086. [Google Scholar] [CrossRef]
- Mensah, V.A.; Gueye, A.; Ndiaye, M.; Edwards, N.J.; Wright, D.; Anagnostou, N.A.; Syll, M.; Ndaw, A.; Abiola, A.; Bliss, C.; et al. Safety, Immunogenicity and Efficacy of Prime-Boost Vaccination with ChAd63 and MVA Encoding ME-TRAP against Plasmodium falciparum Infection in Adults in Senegal. PLoS ONE 2016, 11, e0167951. [Google Scholar] [CrossRef] [Green Version]
- Rampling, T.; Ewer, K.J.; Bowyer, G.; Bliss, C.M.; Edwards, N.J.; Wright, D.; Payne, R.O.; Venkatraman, N.; de Barra, E.; Snudden, C.M.; et al. Safety and High Level Efficacy of the Combination Malaria Vaccine Regimen of RTS,S/AS01B with Chimpanzee Adenovirus 63 and Modified Vaccinia Ankara Vectored Vaccines Expressing ME-TRAP. J. Infect. Dis. 2016, 214, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Afolabi, M.O.; Tiono, A.B.; Adetifa, U.J.; Yaro, J.B.; Drammeh, A.; Nébié, I.; Bliss, C.; Hodgson, S.H.; Anagnostou, N.A.; Sanou, G.S.; et al. Safety and Immunogenicity of ChAd63 and MVA ME-TRAP in West African Children and Infants. Mol. Ther. 2016, 24, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Mensah, V.A.; Roetynck, S.; Kanteh, E.K.; Bowyer, G.; Ndaw, A.; Oko, F.; Bliss, C.M.; Jagne, Y.J.; Cortese, R.; Nicosia, A.; et al. Safety and Immunogenicity of Malaria Vectored Vaccines Given with Routine Expanded Program on Immunization Vaccines in Gambian Infants and Neonates: A Randomized Controlled Trial. Front. Immunol. 2017, 8, 1551. [Google Scholar] [CrossRef] [Green Version]
- Venkatraman, N.; Anagnostou, N.; Bliss, C.; Bowyer, G.; Wright, D.; Lövgren-Bengtsson, K.; Roberts, R.; Poulton, I.; Lawrie, A.; Ewer, K.; et al. Safety and immunogenicity of heterologous prime-boost immunization with viral-vectored malaria vaccines adjuvanted with Matrix-M™. Vaccine 2017, 35, 6208–6217. [Google Scholar] [CrossRef]
- Tiono, A.B.; Nébié, I.; Anagnostou, N.; Coulibaly, A.S.; Bowyer, G.; Lam, E.; Bougouma, E.C.; Ouedraogo, A.; Yaro, J.B.B.; Barry, A.; et al. First field efficacy trial of the ChAd63 MVA ME-TRAP vectored malaria vaccine candidate in 5-17 months old infants and children. PLoS ONE 2018, 13, e0208328. [Google Scholar] [CrossRef]
- Rampling, T.; Ewer, K.J.; Bowyer, G.; Edwards, N.J.; Wright, D.; Sridhar, S.; Payne, R.; Powlson, J.; Bliss, C.; Venkatraman, N.; et al. Safety and efficacy of novel malaria vaccine regimens of RTS,S/AS01B alone, or with concomitant ChAd63-MVA-vectored vaccines expressing ME-TRAP. NPJ Vaccines 2018, 3, 49. [Google Scholar] [CrossRef] [Green Version]
- Bliss, C.M.; Bowyer, G.; Anagnostou, N.A.; Havelock, T.; Snudden, C.M.; Davies, H.; de Cassan, S.C.; Grobbelaar, A.; Lawrie, A.M.; Venkatraman, N.; et al. Assessment of novel vaccination regimens using viral vectored liver stage malaria vaccines encoding ME-TRAP. Sci. Rep. 2018, 8, 3390. [Google Scholar] [CrossRef] [Green Version]
- Ewer, K.J.; O’Hara, G.A.; Duncan, C.J.; Collins, K.A.; Sheehy, S.H.; Reyes-Sandoval, A.; Goodman, A.L.; Edwards, N.J.; Elias, S.C.; Halstead, F.D.; et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nat. Commun. 2013, 4, 2836. [Google Scholar] [CrossRef] [Green Version]
- Elias, S.C.; Collins, K.A.; Halstead, F.D.; Choudhary, P.; Bliss, C.M.; Ewer, K.J.; Sheehy, S.H.; Duncan, C.J.; Biswas, S.; Hill, A.V.; et al. Assessment of immune interference, antagonism, and diversion following human immunization with biallelic blood-stage malaria viral-vectored vaccines and controlled malaria infection. J. Immunol. 2013, 190, 1135–1147. [Google Scholar] [CrossRef] [Green Version]
- Ogwang, C.; Afolabi, M.; Kimani, D.; Jagne, Y.J.; Sheehy, S.H.; Bliss, C.M.; Duncan, C.J.; Collins, K.A.; Garcia Knight, M.A.; Kimani, E.; et al. Safety and immunogenicity of heterologous prime-boost immunisation with Plasmodium falciparum malaria candidate vaccines, ChAd63 ME-TRAP and MVA ME-TRAP, in healthy Gambian and Kenyan adults. PLoS ONE 2013, 8, e57726. [Google Scholar] [CrossRef] [Green Version]
- Bauza, K.; Malinauskas, T.; Pfander, C.; Anar, B.; Jones, E.Y.; Billker, O.; Hill, A.V.; Reyes-Sandoval, A. Efficacy of a Plasmodium vivax malaria vaccine using ChAd63 and modified vaccinia Ankara expressing thrombospondin-related anonymous protein as assessed with transgenic Plasmodium berghei parasites. Infect. Immun. 2014, 82, 1277–1286. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Choudhary, P.; Elias, S.C.; Miura, K.; Milne, K.H.; de Cassan, S.C.; Collins, K.A.; Halstead, F.D.; Bliss, C.M.; Ewer, K.J.; et al. Assessment of humoral immune responses to blood-stage malaria antigens following ChAd63-MVA immunization, controlled human malaria infection and natural exposure. PLoS ONE 2014, 9, e107903. [Google Scholar] [CrossRef]
- de Barra, E.; Hodgson, S.H.; Ewer, K.J.; Bliss, C.M.; Hennigan, K.; Collins, A.; Berrie, E.; Lawrie, A.M.; Gilbert, S.C.; Nicosia, A.; et al. A phase Ia study to assess the safety and immunogenicity of new malaria vaccine candidates ChAd63 CS administered alone and with MVA CS. PLoS ONE 2014, 9, e115161. [Google Scholar] [CrossRef]
- Elias, S.C.; Choudhary, P.; de Cassan, S.C.; Biswas, S.; Collins, K.A.; Halstead, F.D.; Bliss, C.M.; Ewer, K.J.; Hodgson, S.H.; Duncan, C.J.; et al. Analysis of human B-cell responses following ChAd63-MVA MSP1 and AMA1 immunization and controlled malaria infection. Immunology 2014, 141, 628–644. [Google Scholar] [CrossRef]
- Kimani, D.; Jagne, Y.J.; Cox, M.; Kimani, E.; Bliss, C.M.; Gitau, E.; Ogwang, C.; Afolabi, M.O.; Bowyer, G.; Collins, K.A.; et al. Translating the immunogenicity of prime-boost immunization with ChAd63 and MVA ME-TRAP from malaria naive to malaria-endemic populations. Mol. Ther. 2014, 22, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, S.H.; Choudhary, P.; Elias, S.C.; Milne, K.H.; Rampling, T.W.; Biswas, S.; Poulton, I.D.; Miura, K.; Douglas, A.D.; Alanine, D.G.; et al. Combining viral vectored and protein-in-adjuvant vaccines against the blood-stage malaria antigen AMA1: Report on a phase 1a clinical trial. Mol. Ther. 2014, 22, 2142–2154. [Google Scholar] [CrossRef] [Green Version]
- Spencer, A.J.; Cottingham, M.G.; Jenks, J.A.; Longley, R.J.; Capone, S.; Colloca, S.; Folgori, A.; Cortese, R.; Nicosia, A.; Bregu, M.; et al. Enhanced vaccine-induced CD8+ T cell responses to malaria antigen ME-TRAP by fusion to MHC class II invariant chain. PLoS ONE 2014, 9, e100538. [Google Scholar] [CrossRef] [Green Version]
- de Cassan, S.C.; Shakri, A.R.; Llewellyn, D.; Elias, S.C.; Cho, J.S.; Goodman, A.L.; Jin, J.; Douglas, A.D.; Suwanarusk, R.; Nosten, F.H.; et al. Preclinical Assessment of Viral Vectored and Protein Vaccines Targeting the Duffy-Binding Protein Region II of Plasmodium Vivax. Front. Immunol. 2015, 6, 348. [Google Scholar] [CrossRef] [Green Version]
- Bauza, K.; Atcheson, E.; Malinauskas, T.; Blagborough, A.M.; Reyes-Sandoval, A. Tailoring a Combination Preerythrocytic Malaria Vaccine. Infect. Immun. 2015, 84, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Douglas, A.D.; Baldeviano, G.C.; Lucas, C.M.; Lugo-Roman, L.A.; Crosnier, C.; Bartholdson, S.J.; Diouf, A.; Miura, K.; Lambert, L.E.; Ventocilla, J.A.; et al. A PfRH5-based vaccine is efficacious against heterologous strain blood-stage Plasmodium falciparum infection in aotus monkeys. Cell Host Microbe 2015, 17, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Ewer, K.J.; Sierra-Davidson, K.; Salman, A.M.; Illingworth, J.J.; Draper, S.J.; Biswas, S.; Hill, A.V. Progress with viral vectored malaria vaccines: A multi-stage approach involving “unnatural immunity”. Vaccine 2015, 33, 7444–7451. [Google Scholar] [CrossRef] [Green Version]
- Longley, R.J.; Halbroth, B.R.; Ewer, K.J.; Hill, A.V.; Spencer, A.J. Identification of Immunodominant Responses to the Plasmodium falciparum Antigens PfUIS3, PfLSA1 and PfLSAP2 in Multiple Strains of Mice. PLoS ONE 2015, 10, e0144515. [Google Scholar] [CrossRef]
- Pearson, F.E.; O’Mahony, C.; Moore, A.C.; Hill, A.V. Induction of CD8(+) T cell responses and protective efficacy following microneedle-mediated delivery of a live adenovirus-vectored malaria vaccine. Vaccine 2015, 33, 3248–3255. [Google Scholar] [CrossRef]
- Ewer, K.J.; Lambe, T.; Rollier, C.S.; Spencer, A.J.; Hill, A.V.; Dorrell, L. Viral vectors as vaccine platforms: From immunogenicity to impact. Curr. Opin. Immunol. 2016, 41, 47–54. [Google Scholar] [CrossRef]
- Li, Y.; Leneghan, D.B.; Miura, K.; Nikolaeva, D.; Brian, I.J.; Dicks, M.D.J.; Fyfe, A.J.; Zakutansky, S.E.; de Cassan, S.; Long, C.A.; et al. Enhancing immunogenicity and transmission-blocking activity of malaria vaccines by fusing Pfs25 to IMX313 multimerization technology. Sci. Rep. 2016, 6, 18848. [Google Scholar] [CrossRef] [Green Version]
- Bliss, C.M.; Drammeh, A.; Bowyer, G.; Sanou, G.S.; Jagne, Y.J.; Ouedraogo, O.; Edwards, N.J.; Tarama, C.; Ouedraogo, N.; Ouedraogo, M.; et al. Viral Vector Malaria Vaccines Induce High-Level T Cell and Antibody Responses in West African Children and Infants. Mol. Ther. 2017, 25, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Milicic, A.; Rollier, C.S.; Tang, C.K.; Longley, R.; Hill, A.V.S.; Reyes-Sandoval, A. Adjuvanting a viral vectored vaccine against pre-erythrocytic malaria. Sci. Rep. 2017, 7, 7284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, R.J.; Halbroth, B.R.; Salman, A.M.; Ewer, K.J.; Hodgson, S.H.; Janse, C.J.; Khan, S.M.; Hill, A.V.S.; Spencer, A.J. Assessment of the Plasmodium falciparum Preerythrocytic Antigen UIS3 as a Potential Candidate for a Malaria Vaccine. Infect. Immun. 2017, 85, e00641-16. [Google Scholar] [CrossRef] [Green Version]
- Payne, R.O.; Silk, S.E.; Elias, S.C.; Milne, K.H.; Rawlinson, T.A.; Llewellyn, D.; Shakri, A.R.; Jin, J.; Labbé, G.M.; Edwards, N.J.; et al. Human vaccination against Plasmodium vivax Duffy-binding protein induces strain-transcending antibodies. JCI Insight 2017, 2, e93683. [Google Scholar] [CrossRef] [Green Version]
- Payne, R.O.; Silk, S.E.; Elias, S.C.; Miura, K.; Diouf, A.; Galaway, F.; de Graaf, H.; Brendish, N.J.; Poulton, I.D.; Griffiths, O.J.; et al. Human vaccination against RH5 induces neutralizing antimalarial antibodies that inhibit RH5 invasion complex interactions. JCI Insight 2017, 2, e96381. [Google Scholar] [CrossRef]
- Bowyer, G.; Grobbelaar, A.; Rampling, T.; Venkatraman, N.; Morelle, D.; Ballou, R.W.; Hill, A.V.S.; Ewer, K.J. CXCR3(+) T Follicular Helper Cells Induced by Co-Administration of RTS,S/AS01B and Viral-Vectored Vaccines Are Associated with Reduced Immunogenicity and Efficacy Against Malaria. Front. Immunol. 2018, 9, 1660. [Google Scholar] [CrossRef] [Green Version]
- Halbroth, B.R.; Sebastian, S.; Salman, A.M.; Ulaszewska, M.; Gola, A.; Longley, R.J.; Janse, C.J.; Khan, S.M.; Hill, A.V.S.; Spencer, A.J. Preclinical Development and Assessment of Viral Vectors Expressing a Fusion Antigen of Plasmodium falciparum LSA1 and LSAP2 for Efficacy against Liver-Stage Malaria. Infect. Immun. 2020, 88, e00573-19. [Google Scholar] [CrossRef] [Green Version]
- de Graaf, H.; Payne, R.O.; Taylor, I.; Miura, K.; Long, C.A.; Elias, S.C.; Zaric, M.; Minassian, A.M.; Silk, S.E.; Li, L.; et al. Safety and Immunogenicity of ChAd63/MVA Pfs25-IMX313 in a Phase I First-in-Human Trial. Front. Immunol. 2021, 12, 694759. [Google Scholar] [CrossRef]
- Longley, R.J.; Salman, A.M.; Cottingham, M.G.; Ewer, K.; Janse, C.J.; Khan, S.M.; Spencer, A.J.; Hill, A.V.S. Comparative assessment of vaccine vectors encoding ten malaria antigens identifies two protective liver-stage candidates. Sci. Rep. 2015, 5, 11820. [Google Scholar] [CrossRef] [Green Version]
- Shahnaij, M.; Iyori, M.; Mizukami, H.; Kajino, M.; Yamagoshi, I.; Syafira, I.; Yusuf, Y.; Fujiwara, K.; Yamamoto, D.S.; Kato, H.; et al. Liver-Directed AAV8 Booster Vaccine Expressing Plasmodium falciparum Antigen Following Adenovirus Vaccine Priming Elicits Sterile Protection in a Murine Model. Front. Immunol. 2021, 12, 612910. [Google Scholar] [CrossRef]
- Shiratsuchi, T.; Rai, U.; Kaneko, I.; Zhang, M.; Iwanaga, S.; Yuda, M.; Tsuji, M. A potent malaria vaccine based on adenovirus with dual modifications at Hexon and pVII. Vaccine 2017, 35, 6990–7000. [Google Scholar] [CrossRef]
- McGuire, K.A.; Miura, K.; Wiethoff, C.M.; Williamson, K.C. New adenovirus-based vaccine vectors targeting Pfs25 elicit antibodies that inhibit Plasmodium falciparum transmission. Malar. J. 2017, 16, 254. [Google Scholar] [CrossRef] [Green Version]
- PAHO. Chagas Disease. Available online: https://www.paho.org/en/topics/chagas-disease (accessed on 11 December 2022).
- WHO. Chagas Disease (American Trypanosomiasis). Available online: https://www.who.int/health-topics/chagas-disease (accessed on 11 December 2022).
- Melo, R.C.; Brener, Z. Tissue Tropism of Different Trypanosoma cruzi Strains. J. Parasitol. 1978, 64, 475–482. [Google Scholar] [CrossRef]
- Zhang, L.; Tarleton, R.L. Parasite Persistence Correlates with Disease Severity and Localization in Chronic Chagas’ Disease. J. Infect. Dis. 1999, 180, 480–486. [Google Scholar] [CrossRef]
- Guarner, J.; Bartlett, J.; Zaki, S.R.; Grijalva, M.J.; Colley, D.G.; Powell, M.R. Mouse model for Chagas disease: Immunohistochemical distribution of different stages of Trypanosoma cruzi in tissues throughout infection. Am. J. Trop. Med. Hyg. 2001, 65, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Camandaroba, E.; Thé, T.S.; Pessina, D.H.; Andrade, S.G. Trypanosoma cruzi: Clones isolated from the Colombian strain, reproduce the parental strain characteristics, with ubiquitous histotropism. Int. J. Exp. Pathol. 2006, 87, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Mazliah, D.; Ward, A.I.; Lewis, M.D. Host-parasite dynamics in Chagas disease from systemic to hyper-local scales. Parasite Immunol. 2021, 43, e12786. [Google Scholar] [CrossRef]
- Bafica, A.; Santiago, H.C.; Goldszmid, R.; Ropert, C.; Gazzinelli, R.T.; Sher, A. Cutting Edge: TLR9 and TLR2 Signaling Together Account for MyD88-Dependent Control of Parasitemia in Trypanosoma cruzi Infection. J. Immunol. 2006, 177, 3515–3519. [Google Scholar] [CrossRef] [Green Version]
- Chessler, A.-D.C.; Unnikrishnan, M.; Bei, A.K.; Daily, J.P.; Burleigh, B.A. Trypanosoma cruzi Triggers an Early Type I IFN Response In Vivo at the Site of Intradermal Infection. J. Immunol. 2009, 182, 2288–2296. [Google Scholar] [CrossRef] [Green Version]
- Mesías, A.C.; Garg, N.J.; Zago, M.P. Redox Balance Keepers and Possible Cell Functions Managed by Redox Homeostasis in Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 2019, 9, 435. [Google Scholar] [CrossRef]
- Padilla, A.M.; Simpson, L.J.; Tarleton, R.L. Insufficient TLR Activation Contributes to the Slow Development of CD8+ T Cell Responses in Trypanosoma cruzi Infection. J. Immunol. 2009, 183, 1245–1252. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.A.; Ellis-Neyes, L.A.; Slifer, T.; Kanaly, S.; Grünig, G.; Fort, M.; Rennick, D.; Araujo, F.G. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J. Immunol. 1997, 158, 3311–3316. [Google Scholar] [CrossRef]
- Bivona, A.E.; Alberti, A.S.; Cerny, N.; Trinitario, S.N.; Malchiodi, E.L. Chagas disease vaccine design: The search for an efficient Trypanosoma cruzi immune-mediated control. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165658. [Google Scholar] [CrossRef]
- Machado, A.V.; Cardoso, J.E.; Claser, C.; Rodrigues, M.M.; Gazzinelli, R.T.; Bruna-Romero, O. Long-term protective immunity induced against Trypanosoma cruzi infection after vaccination with recombinant adenoviruses encoding amastigote surface protein-2 and trans-sialidase. Hum. Gene Ther. 2006, 17, 898–908. [Google Scholar] [CrossRef] [Green Version]
- de Alencar, B.C.; Persechini, P.M.; Haolla, F.A.; de Oliveira, G.; Silverio, J.C.; Lannes-Vieira, J.; Machado, A.V.; Gazzinelli, R.T.; Bruna-Romero, O.; Rodrigues, M.M. Perforin and gamma interferon expression are required for CD4+ and CD8+ T-cell-dependent protective immunity against a human parasite, Trypanosoma cruzi, elicited by heterologous plasmid DNA prime-recombinant adenovirus 5 boost vaccination. Infect. Immun. 2009, 77, 4383–4395. [Google Scholar] [CrossRef] [Green Version]
- Haolla, F.A.; Claser, C.; de Alencar, B.C.; Tzelepis, F.; de Vasconcelos, J.R.; de Oliveira, G.; Silvério, J.C.; Machado, A.V.; Lannes-Vieira, J.; Bruna-Romero, O.; et al. Strain-specific protective immunity following vaccination against experimental Trypanosoma cruzi infection. Vaccine 2009, 27, 5644–5653. [Google Scholar] [CrossRef]
- Rigato, P.O.; de Alencar, B.C.; de Vasconcelos, J.R.; Dominguez, M.R.; Araújo, A.F.; Machado, A.V.; Gazzinelli, R.T.; Bruna-Romero, O.; Rodrigues, M.M. Heterologous plasmid DNA prime-recombinant human adenovirus 5 boost vaccination generates a stable pool of protective long-lived CD8(+) T effector memory cells specific for a human parasite, Trypanosoma cruzi. Infect. Immun. 2011, 79, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, M.R.; Silveira, E.L.; de Vasconcelos, J.R.; de Alencar, B.C.; Machado, A.V.; Bruna-Romero, O.; Gazzinelli, R.T.; Rodrigues, M.M. Subdominant/cryptic CD8 T cell epitopes contribute to resistance against experimental infection with a human protozoan parasite. PLoS ONE 2011, 6, e22011. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, M.R.; Ersching, J.; Lemos, R.; Machado, A.V.; Bruna-Romero, O.; Rodrigues, M.M.; de Vasconcelos, J.R. Re-circulation of lymphocytes mediated by sphingosine-1-phosphate receptor-1 contributes to resistance against experimental infection with the protozoan parasite Trypanosoma cruzi. Vaccine 2012, 30, 2882–2891. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, J.R.; Bruña-Romero, O.; Araújo, A.F.; Dominguez, M.R.; Ersching, J.; de Alencar, B.C.; Machado, A.V.; Gazzinelli, R.T.; Bortoluci, K.R.; Amarante-Mendes, G.P.; et al. Pathogen-induced proapoptotic phenotype and high CD95 (Fas) expression accompany a suboptimal CD8+ T-cell response: Reversal by adenoviral vaccine. PLoS Pathog. 2012, 8, e1002699. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, J.R.; Dominguez, M.R.; Neves, R.L.; Ersching, J.; Araújo, A.; Santos, L.I.; Virgilio, F.S.; Machado, A.V.; Bruna-Romero, O.; Gazzinelli, R.T.; et al. Adenovirus vector-induced CD8⁺ T effector memory cell differentiation and recirculation, but not proliferation, are important for protective immunity against experimental Trypanosoma cruzi Infection. Hum. Gene Ther. 2014, 25, 350–363. [Google Scholar] [CrossRef] [Green Version]
- Araújo, A.F.; de Oliveira, G.; Vasconcelos, J.F.; Ersching, J.; Dominguez, M.R.; Vasconcelos, J.R.; Machado, A.V.; Gazzinelli, R.T.; Bruna-Romero, O.; Soares, M.B.; et al. Genetic vaccination against experimental infection with myotropic parasite strains of Trypanosoma cruzi. Mediators Inflamm. 2014, 2014, 605023. [Google Scholar] [CrossRef] [Green Version]
- Pereira, I.R.; Vilar-Pereira, G.; Marques, V.; da Silva, A.A.; Caetano, B.; Moreira, O.C.; Machado, A.V.; Bruna-Romero, O.; Rodrigues, M.M.; Gazzinelli, R.T.; et al. A human type 5 adenovirus-based Trypanosoma cruzi therapeutic vaccine re-programs immune response and reverses chronic cardiomyopathy. PLoS Pathog. 2015, 11, e1004594. [Google Scholar] [CrossRef]
- Ferreira, C.P.; Cariste, L.M.; Santos Virgílio, F.D.; Moraschi, B.F.; Monteiro, C.B.; Vieira Machado, A.M.; Gazzinelli, R.T.; Bruna-Romero, O.; Menin Ruiz, P.L.; Ribeiro, D.A.; et al. LFA-1 Mediates Cytotoxicity and Tissue Migration of Specific CD8(+) T Cells after Heterologous Prime-Boost Vaccination against Trypanosoma cruzi Infection. Front. Immunol. 2017, 8, 1291. [Google Scholar] [CrossRef] [Green Version]
- Pontes Ferreira, C.; Cariste, L.M.; Ferri Moraschi, B.; Ferrarini Zanetti, B.; Won Han, S.; Araki Ribeiro, D.; Vieira Machado, A.; Lannes-Vieira, J.; Gazzinelli, R.T.; Vasconcelos, J.R.C. CXCR3 chemokine receptor guides Trypanosoma cruzi-specific T-cells triggered by DNA/adenovirus ASP2 vaccine to heart tissue after challenge. PLoS Negl. Trop. Dis. 2019, 13, e0007597. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, F.A.P.; Pontes, C.; Gazzinelli, R.T.; Romero, O.B.; Lazzarin, M.C.; Dos Santos, J.F.; de Oliveira, F.; Pisani, L.P.; de Vasconcelos, J.R.C.; Ribeiro, D.A. Therapeutic effects of vaccine derived from amastigote surface protein-2 (ASP-2) against Chagas disease in mouse liver. Cytokine 2019, 113, 285–290. [Google Scholar] [CrossRef]
- Barbosa, R.P.; Filho, B.G.; Dos Santos, L.I.; Junior, P.A.; Marques, P.E.; Pereira, R.V.; Cara, D.C.; Bruña-Romero, O.; Rodrigues, M.M.; Gazzinelli, R.T.; et al. Vaccination using recombinants influenza and adenoviruses encoding amastigote surface protein-2 are highly effective on protection against Trypanosoma cruzi infection. PLoS ONE 2013, 8, e61795. [Google Scholar] [CrossRef] [Green Version]
- Jovicic, N.; Jeftic, I.; Jovanovic, I.; Radosavljevic, G.; Arsenijevic, N.; Lukic, M.L.; Pejnovic, N. Differential Immunometabolic Phenotype in Th1 and Th2 Dominant Mouse Strains in Response to High-Fat Feeding. PLoS ONE 2015, 10, e0134089. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, F.A.P.; Pontes, C.; Machado, A.M.V.; Bruna-Romero, O.; Quintana, H.T.; De Oliveira, F.; De Vasconcelos, J.R.C.; Ribeiro, D.A. Therapeutical effects of vaccine from Trypanosoma cruzi amastigote surface protein 2 by simultaneous inoculation with live parasites. J. Cell. Biochem. 2019, 120, 3373–3383. [Google Scholar] [CrossRef]
- Pulendran, B.; Arunachalam, P.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- Metzger, D.W. Interleukin-12 as an adjuvant for induction of protective antibody responses. Cytokine 2010, 52, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Lindblad, E.B. Freund’s Adjuvants. In Vaccine Adjuvants: Preparation Methods and Research Protocols; O’Hagan, D.T., Ed.; Springer: Totowa, NJ, USA, 2000; pp. 49–63. [Google Scholar]
- Klinman, D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 2004, 4, 249–259. [Google Scholar] [CrossRef]
- Garçon, N.; Wettendorff, M.; Van Mechelen, M. Role of AS04 in human papillomavirus vaccine: Mode of action and clinical profile. Expert Opin. Biol. Ther. 2011, 11, 667–677. [Google Scholar] [CrossRef]
- McAleer, J.P.; Vella, A.T. Educating CD4 T cells with vaccine adjuvants: Lessons from lipopolysaccharide. Trends Immunol. 2010, 31, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Hajam, I.A.; Dar, P.A.; Shahnawaz, I.; Jaume, J.C.; Lee, J.H. Bacterial flagellin—A potent immunomodulatory agent. Exp. Mol. Med. 2017, 49, e373. [Google Scholar] [CrossRef] [Green Version]
- Didierlaurent, A.M.; Laupèze, B.; Di Pasquale, A.; Hergli, N.; Collignon, C.; Garçon, N. Adjuvant system AS01: Helping to overcome the challenges of modern vaccines. Expert Rev. Vaccines 2017, 16, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Marrack, P.; McKee, A.S.; Munks, M.W. Towards an understanding of the adjuvant action of aluminium. Nat. Rev. Immunol. 2009, 9, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; George, P.J.; Deng, W.; Koussa, J.; Parkhouse, K.; Hensley, S.E.; Jiang, J.; Lu, J.; Liu, Z.; Wei, J.; et al. The parasite-derived rOv-ASP-1 is an effective antigen-sparing CD4+ T cell-dependent adjuvant for the trivalent inactivated influenza vaccine, and functions in the absence of MyD88 pathway. Vaccine 2018, 36, 3650–3665. [Google Scholar] [CrossRef]
- O’Hagan, D.T.; Ott, G.S.; Nest, G.V.; Rappuoli, R.; Giudice, G.D. The history of MF59® adjuvant: A phoenix that arose from the ashes. Expert Rev. Vaccines 2013, 12, 13–30. [Google Scholar] [CrossRef]
- Morel, S.; Didierlaurent, A.; Bourguignon, P.; Delhaye, S.; Baras, B.; Jacob, V.; Planty, C.; Elouahabi, A.; Harvengt, P.; Carlsen, H.; et al. Adjuvant System AS03 containing α-tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine 2011, 29, 2461–2473. [Google Scholar] [CrossRef]
- Keech, C.; Albert, G.; Cho, I.; Robertson, A.; Reed, P.; Neal, S.; Plested, J.S.; Zhu, M.; Cloney-Clark, S.; Zhou, H.; et al. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N. Engl. J. Med. 2020, 383, 2320–2332. [Google Scholar] [CrossRef]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant––An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef] [Green Version]
- Clements, J.D.; Norton, E.B. The Mucosal Vaccine Adjuvant LT(R192G/L211A) or dmLT. mSphere 2018, 3, e00215–e00218. [Google Scholar] [CrossRef] [Green Version]
- Farrow, A.L.; Rachakonda, G.; Gu, L.; Krendelchtchikova, V.; Nde, P.N.; Pratap, S.; Lima, M.F.; Villalta, F.; Matthews, Q.L. Immunization with Hexon modified adenoviral vectors integrated with gp83 epitope provides protection against Trypanosoma cruzi infection. PLoS Negl. Trop. Dis. 2014, 8, e3089. [Google Scholar] [CrossRef] [Green Version]
- Farrow, A.L.; Peng, B.J.; Gu, L.; Krendelchtchikov, A.; Matthews, Q.L. A Novel Vaccine Approach for Chagas Disease Using Rare Adenovirus Serotype 48 Vectors. Viruses 2016, 8, 78. [Google Scholar] [CrossRef] [Green Version]
- Matthews, Q.L.; Farrow, A.L.; Rachakonda, G.; Gu, L.; Nde, P.; Krendelchtchikov, A.; Pratap, S.; Sakhare, S.S.; Sabbaj, S.; Lima, M.F.; et al. Epitope Capsid-Incorporation: New Effective Approach for Vaccine Development for Chagas Disease. Pathog. Immun. 2016, 1, 214–233. [Google Scholar] [CrossRef] [Green Version]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- WHO. Schistosomiasis (Bilharzia). Available online: https://www.who.int/health-topics/schistosomiasis (accessed on 5 December 2022).
- Centers for Disease Control and Prevention. Schistosomiasis. Available online: https://www.cdc.gov/parasites/schistosomiasis/ (accessed on 5 December 2022).
- McManus, D.P.; Bergquist, R.; Cai, P.; Ranasinghe, S.; Tebeje, B.M.; You, H. Schistosomiasis—From immunopathology to vaccines. Semin. Immunopathol. 2020, 42, 355–371. [Google Scholar] [CrossRef]
- Hay, S.I.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; et al. Global, regional, and national disability-adjusted life-years (DALYs) for 333 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1260–1344. [Google Scholar] [CrossRef] [Green Version]
- Toor, J.; Alsallaq, R.; Truscott, J.E.; Turner, H.C.; Werkman, M.; Gurarie, D.; King, C.H.; Anderson, R.M. Are We on Our Way to Achieving the 2020 Goals for Schistosomiasis Morbidity Control Using Current World Health Organization Guidelines? Clin. Infect. Dis. 2018, 66, S245–S252. [Google Scholar] [CrossRef]
- Colley, D.G.; Secor, W.E. Immunology of human schistosomiasis. Parasite Immunol. 2014, 36, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Frahm, S.; Anisuzzaman, A.; Prodjinotho, U.F.; Vejzagić, N.; Verschoor, A.; Prazeres da Costa, C. A novel cell-free method to culture Schistosoma mansoni from cercariae to juvenile worm stages for in vitro drug testing. PLoS Negl. Trop. Dis. 2019, 13, e0006590. [Google Scholar] [CrossRef] [Green Version]
- Anisuzzaman; Frahm, S.; Prodjinotho, U.F.; Bhattacharjee, S.; Verschoor, A.; Prazeres da Costa, C. Host-Specific Serum Factors Control the Development and Survival of Schistosoma mansoni. Front. Immunol. 2021, 12, 635622. [Google Scholar] [CrossRef]
- Molehin, A.J. Current Understanding of Immunity against Schistosomiasis: Impact on Vaccine and Drug Development. Res. Rep. Trop. Med. 2020, 11, 119–128. [Google Scholar] [CrossRef]
- Costain, A.H.; MacDonald, A.S.; Smits, H.H. Schistosome Egg Migration: Mechanisms, Pathogenesis and Host Immune Responses. Front. Immunol. 2018, 9, 3042. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Mwinzi, P.N.; Black, C.L.; Muok, E.M.; Karanja, D.M.; Secor, W.E.; Colley, D.G. T regulatory cell levels decrease in people infected with Schistosoma mansoni on effective treatment. Am. J. Trop. Med. Hyg. 2007, 77, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.A.; Coulson, P.S. Strategies for a schistosome vaccine: Can we manipulate the immune response effectively? Microbes Infect. 1999, 1, 535–543. [Google Scholar] [CrossRef]
- Hoffmann, K.F.; Cheever, A.W.; Wynn, T.A. IL-10 and the Dangers of Immune Polarization: Excessive Type 1 and Type 2 Cytokine Responses Induce Distinct Forms of Lethal Immunopathology in Murine Schistosomiasis. J. Immunol. 2000, 164, 6406–6416. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zhu, J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int. J. Mol. Sci. 2020, 21, 8011. [Google Scholar] [CrossRef]
- Eveland, L.K.; Morse, S.I. Schistosoma mansoni: Infectivity and immunizing effects of in vitro derived schistosomula attenuated by X irradiation. Exp. Parasitol. 1978, 45, 19–25. [Google Scholar] [CrossRef]
- Minard, P.; Dean, D.A.; Jacobson, R.H.; Vannier, W.E.; Murrell, K.D. Immunization of Mice with Cobalt-60 Irradiated Schistosoma Mansoni Cercariae. Am. J. Trop. Med. Hyg. 1978, 27, 76–86. [Google Scholar] [CrossRef]
- Driciru, E.; Koopman, J.P.R.; Cose, S.; Siddiqui, A.A.; Yazdanbakhsh, M.; Elliott, A.M.; Roestenberg, M. Immunological Considerations for Schistosoma Vaccine Development: Transitioning to Endemic Settings. Front. Immunol. 2021, 12, 635985. [Google Scholar] [CrossRef]
- El Ridi, R.; Tallima, H. Why the radiation-attenuated cercarial immunization studies failed to guide the road for an effective schistosomiasis vaccine: A review. J. Adv. Res. 2015, 6, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, K.F.; James, S.L.; Cheever, A.W.; Wynn, T.A. Studies with double cytokine-deficient mice reveal that highly polarized Th1- and Th2-type cytokine and antibody responses contribute equally to vaccine-induced immunity to Schistosoma mansoni. J. Immunol. 1999, 163, 927–938. [Google Scholar] [CrossRef]
- Gray, D.J.; Williams, G.M.; Li, Y.; McManus, D.P. Transmission Dynamics of Schistosoma japonicum in the Lakes and Marshlands of China. PLoS ONE 2009, 3, e4058. [Google Scholar] [CrossRef]
- Zhu, Y.; Si, J.; Harn, D.A.; Yu, C.; Liang, Y.; Ren, J.; Yin, X.; He, W.; Hua, W. The protective immunity of a DNA vaccine encoding Schistosoma japonicum Chinese strain triose-phosphate isomerase in infected BALB/C mice. Southeast Asian J. Trop. Med. Public Health 2004, 35, 518–522. [Google Scholar]
- Choudhry, A.; Mathena, J.; Albano, J.D.; Yacovone, M.; Collins, L. Safety evaluation of adenovirus type 4 and type 7 vaccine live, oral in military recruits. Vaccine 2016, 34, 4558–4564. [Google Scholar] [CrossRef] [Green Version]
- Mo, A.X.; Colley, D.G. Workshop report: Schistosomiasis vaccine clinical development and product characteristics. Vaccine 2016, 34, 995–1001. [Google Scholar] [CrossRef]
- WHO. Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 6 December 2022).
- Zijlstra, E.E.; Musa, A.M.; Khalil, E.A.G.; El Hassan, I.M.; El-Hassan, A.M. Post-kala-azar dermal leishmaniasis. Lancet Infect. Dis. 2003, 3, 87–98. [Google Scholar] [CrossRef]
- WHO. Leishmaniasis. Available online: https://www.who.int/data/gho/data/themes/topics/indicator-groups/indicator-group-details/GHO/leishmaniasis (accessed on 6 December 2022).
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votýpka, J.; Marty, P.; Delaunay, P.; Sereno, D. A Historical Overview of the Classification, Evolution, and Dispersion of Leishmania Parasites and Sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef]
- Müller, I.; Pedrazzini, T.; Farrell, J.P.; Louis, J. T-cell Responses and Immunity to Experimental Infection with Leishmania major. Annu. Rev. Immunol. 1989, 7, 561–578. [Google Scholar] [CrossRef]
- Handman, E.; Ceredig, R.; Mitchell, G.F. Murine cutaneous leishmaniasis: Disease patterns in intact and nude mice of various genotypes and examination of some differences between normal and infected macrophages. Aust. J. Exp. Biol. Med. Sci. 1979, 57, 9–29. [Google Scholar] [CrossRef]
- Sacks, D.; Noben-Trauth, N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2002, 2, 845–858. [Google Scholar] [CrossRef]
- Costa-da-Silva, A.C.; Nascimento, D.O.; Ferreira, J.R.M.; Guimarães-Pinto, K.; Freire-de-Lima, L.; Morrot, A.; Decote-Ricardo, D.; Filardy, A.A.; Freire-de-Lima, C.G. Immune Responses in Leishmaniasis: An Overview. Trop. Med. Infect. Dis. 2022, 7, 54. [Google Scholar] [CrossRef]
- Nasseri, M.; Modabber, F.Z. Generalized infection and lack of delayed hypersensitivity in BALB/c mice infected with Leishmania tropica major. Infect. Immun. 1979, 26, 611–614. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.; Singh, V.; Naik, S. Immune response to leishmania: Paradox rather than paradigm. FEMS Immunol. Med. Microbiol. 2007, 51, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Samant, M.; Sahu, U.; Pandey, S.C.; Khare, P. Role of Cytokines in Experimental and Human Visceral Leishmaniasis. Front. Cell. Infect. Microbiol. 2021, 11, 624009. [Google Scholar] [CrossRef]
- Stager, S.; Rafati, S. CD8+ T Cells in Leishmania Infections: Friends or Foes? Front. Immunol. 2012, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Tsagozis, P.; Karagouni, E.; Dotsika, E. CD8+ T cells with parasite-specific cytotoxic activity and a Tc1 profile of cytokine and chemokine secretion develop in experimental visceral leishmaniasis. Parasite Immunol. 2003, 25, 569–579. [Google Scholar] [CrossRef]
- Alexander, J.; Russell, D.G. The Interaction of Leishmania Species with Macrophages. In Advances in Parasitology; Baker, J.R., Muller, R., Eds.; Academic Press: San Diego, CA, USA, 1992; Volume 31, pp. 175–254. [Google Scholar]
- Wei, X.-q.; Charles, I.G.; Smith, A.; Ure, J.; Feng, G.-j.; Huang, F.-p.; Xu, D.; Mullers, W.; Moncada, S.; Liew, F.Y. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 1995, 375, 408–411. [Google Scholar] [CrossRef]
- Carneiro, P.P.; Conceição, J.; Macedo, M.; Magalhães, V.; Carvalho, E.M.; Bacellar, O. The Role of Nitric Oxide and Reactive Oxygen Species in the Killing of Leishmania braziliensis by Monocytes from Patients with Cutaneous Leishmaniasis. PLoS ONE 2016, 11, e0148084. [Google Scholar] [CrossRef] [Green Version]
- Carstens-Kass, J.; Paulini, K.; Lypaczewski, P.; Matlashewski, G. A review of the leishmanin skin test: A neglected test for a neglected disease. PLoS Negl. Trop. Dis. 2021, 15, e0009531. [Google Scholar] [CrossRef]
- Pacheco-Fernandez, T.; Volpedo, G.; Gannavaram, S.; Bhattacharya, P.; Dey, R.; Satoskar, A.; Matlashewski, G.; Nakhasi, H.L. Revival of Leishmanization and Leishmanin. Front. Cell. Infect. Microbiol. 2021, 11, 639801. [Google Scholar] [CrossRef]
- Moafi, M.; Rezvan, H.; Sherkat, R.; Taleban, R. Leishmania Vaccines Entered in Clinical Trials: A Review of Literature. Int. J. Prev. Med. 2019, 10, 95. [Google Scholar] [CrossRef]
- Grimaldi, G., Jr.; Teva, A.; Porrozzi, R.; Pinto, M.A.; Marchevsky, R.S.; Rocha, M.G.; Dutra, M.S.; Bruña-Romero, O.; Fernandes, A.P.; Gazzinelli, R.T. Clinical and parasitological protection in a Leishmania infantum-macaque model vaccinated with adenovirus and the recombinant A2 antigen. PLoS Negl. Trop. Dis. 2014, 8, e2853. [Google Scholar] [CrossRef]
- Osman, M.; Mistry, A.; Keding, A.; Gabe, R.; Cook, E.; Forrester, S.; Wiggins, R.; Di Marco, S.; Colloca, S.; Siani, L.; et al. A third generation vaccine for human visceral leishmaniasis and post kala azar dermal leishmaniasis: First-in-human trial of ChAd63-KH. PLoS Negl. Trop. Dis. 2017, 11, e0005527. [Google Scholar] [CrossRef] [Green Version]
- Younis, B.M.; Osman, M.; Khalil, E.A.G.; Santoro, F.; Furini, S.; Wiggins, R.; Keding, A.; Carraro, M.; Musa, A.E.A.; Abdarahaman, M.A.A.; et al. Safety and immunogenicity of ChAd63-KH vaccine in post-kala-azar dermal leishmaniasis patients in Sudan. Mol. Ther. 2021, 29, 2366–2377. [Google Scholar] [CrossRef]
- Velez, R.; Gállego, M. Commercially approved vaccines for canine leishmaniosis: A review of available data on their safety and efficacy. Trop. Med. Int. Health 2020, 25, 540–557. [Google Scholar] [CrossRef] [Green Version]
- Maroof, A.; Brown, N.; Smith, B.; Hodgkinson, M.R.; Maxwell, A.; Losch, F.O.; Fritz, U.; Walden, P.; Lacey, C.N.; Smith, D.F.; et al. Therapeutic vaccination with recombinant adenovirus reduces splenic parasite burden in experimental visceral leishmaniasis. J. Infect. Dis. 2012, 205, 853–863. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Flegr, J.; Prandota, J.; Sovičková, M.; Israili, Z.H. Toxoplasmosis—A global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS ONE 2014, 9, e90203. [Google Scholar] [CrossRef] [Green Version]
- Molan, A.; Nosaka, K.; Hunter, M.; Wang, W. Global status of Toxoplasma gondii infection: Systematic review and prevalence snapshots. Trop. Biomed. 2019, 36, 898–925. [Google Scholar]
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. Toxoplasmosis (Toxoplasma infection). Available online: https://www.cdc.gov/parasites/toxoplasmosis/ (accessed on 10 December 2022).
- McAuley, J.B. Congenital Toxoplasmosis. J. Pediatr. Infect. Dis. Soc. 2014, 3 (Suppl. S1), S30–S35. [Google Scholar] [CrossRef]
- Robert-Gangneux, F.; Dardé, M.L. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin. Microbiol. Rev. 2012, 25, 264–296. [Google Scholar] [CrossRef] [Green Version]
- Milne, G.; Webster, J.P.; Walker, M. Toxoplasma gondii: An Underestimated Threat? Trends Parasitol. 2020, 36, 959–969. [Google Scholar] [CrossRef]
- Huang, J.; Zheng, J.; Liu, B.; Lu, L.; Wu, H.; Lin, S.; Li, D. The association between Toxoplasma infection and mortality: The NHANES epidemiologic follow-up study. Parasites Vectors 2022, 15, 284. [Google Scholar] [CrossRef]
- Peng, H.-J.; Chen, X.-G.; Lindsay, D.S. A Review: Competence, Compromise, and Concomitance—Reaction of the Host Cell To Toxoplasma gondii Infection and Development. J. Parasitol. 2011, 97, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Sasai, M.; Yamamoto, M. Innate, adaptive, and cell-autonomous immunity against Toxoplasma gondii infection. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.A.; Moretto, M. Immune responses to Toxoplasma gondii. Curr. Opin. Immunol. 2022, 77, 102226. [Google Scholar] [CrossRef]
- Safronova, A.; Araujo, A.; Camanzo, E.T.; Moon, T.J.; Elliott, M.R.; Beiting, D.P.; Yarovinsky, F. Alarmin S100A11 initiates a chemokine response to the human pathogen Toxoplasma gondii. Nat. Immunol. 2019, 20, 64–72. [Google Scholar] [CrossRef]
- Andrade, W.A.; do Carmo Souza, M.; Ramos-Martinez, E.; Nagpal, K.; Dutra, M.S.; Melo, M.B.; Bartholomeu, D.C.; Ghosh, S.; Golenbock, D.T.; Gazzinelli, R.T. Combined Action of Nucleic Acid-Sensing Toll-like Receptors and TLR11/TLR12 Heterodimers Imparts Resistance to Toxoplasma gondii in Mice. Cell Host Microbe 2013, 13, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.A.; Subauste, C.S.; Van Cleave, V.H.; Remington, J.S. Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: Regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect. Immun. 1994, 62, 2818–2824. [Google Scholar] [CrossRef] [Green Version]
- Sasai, M.; Pradipta, A.; Yamamoto, M. Host immune responses to Toxoplasma gondii. Int. Immunol. 2018, 30, 113–119. [Google Scholar] [CrossRef]
- Suzuki, Y. The immune system utilizes two distinct effector mechanisms of T cells depending on two different life cycle stages of a single pathogen, Toxoplasma gondii, to control its cerebral infection. Parasitol. Int. 2020, 76, 102030. [Google Scholar] [CrossRef]
- Bhadra, R.; Gigley, J.P.; Weiss, L.M.; Khan, I.A. Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1–PDL-1 blockade. Proc. Natl. Acad. Sci. USA 2011, 108, 9196–9201. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Cobb, D.A.; Bhadra, R.; Youngblood, B.; Khan, I.A. Blimp-1–mediated CD4 T cell exhaustion causes CD8 T cell dysfunction during chronic toxoplasmosis. J. Exp. Med. 2016, 213, 1799–1818. [Google Scholar] [CrossRef] [Green Version]
- Chu, K.B.; Quan, F.S. Advances in Toxoplasma gondii Vaccines: Current Strategies and Challenges for Vaccine Development. Vaccines 2021, 9, 413. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Lu, S.; Zheng, B. Toxoplasmosis vaccines: What we have and where to go? NPJ Vaccines 2022, 7, 131. [Google Scholar] [CrossRef]
- Buxton, D.; Innes, E.A. A commercial vaccine for ovine toxoplasmosis. Parasitology 1995, 110, S11–S16. [Google Scholar] [CrossRef]
- Dubey, J.P. Toxoplasmosis in sheep—The last 20 years. Vet. Parasitol. 2009, 163, 1–14. [Google Scholar] [CrossRef]
- Caetano, B.C.; Bruña-Romero, O.; Fux, B.; Mendes, E.A.; Penido, M.L.; Gazzinelli, R.T. Vaccination with replication-deficient recombinant adenoviruses encoding the main surface antigens of toxoplasma gondii induces immune response and protection against infection in mice. Hum. Gene Ther. 2006, 17, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Machado, A.V.; Caetano, B.C.; Barbosa, R.P.; Salgado, A.P.; Rabelo, R.H.; Garcia, C.C.; Bruna-Romero, O.; Escriou, N.; Gazzinelli, R.T. Prime and boost immunization with influenza and adenovirus encoding the Toxoplasma gondii surface antigen 2 (SAG2) induces strong protective immunity. Vaccine 2010, 28, 3247–3256. [Google Scholar] [CrossRef]
- Mendes, E.A.; Caetano, B.C.; Penido, M.L.; Bruna-Romero, O.; Gazzinelli, R.T. MyD88-dependent protective immunity elicited by adenovirus 5 expressing the surface antigen 1 from Toxoplasma gondii is mediated by CD8+ T lymphocytes. Vaccine 2011, 29, 4476–4484. [Google Scholar] [CrossRef]
- Mendes, É.A.; Fonseca, F.G.; Casério, B.M.; Colina, J.P.; Gazzinelli, R.T.; Caetano, B.C. Recombinant vaccines against T. gondii: Comparison between homologous and heterologous vaccination protocols using two viral vectors expressing SAG1. PLoS ONE 2013, 8, e63201. [Google Scholar] [CrossRef]
- Li, X.Z.; Wang, X.H.; Xia, L.J.; Weng, Y.B.; Hernandez, J.A.; Tu, L.Q.; Li, L.T.; Li, S.J.; Yuan, Z.G. Protective efficacy of recombinant canine adenovirus type-2 expressing TgROP18 (CAV-2-ROP18) against acute and chronic Toxoplasma gondii infection in mice. BMC Infect. Dis. 2015, 15, 114. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Z.; Lv, L.; Zhang, X.; Anchang, K.Y.; Abdullahi, A.Y.; Tu, L.; Wang, X.; Xia, L.; Zhang, X.X.; Feng, W.; et al. Recombinant canine adenovirus type-2 expressing TgROP16 provides partial protection against acute Toxoplasma gondii infection in mice. Infect. Genet. Evol. 2016, 45, 447–453. [Google Scholar] [CrossRef]
- Sklar, M.J.; Maiolatesi, S.; Patterson, N.; Sedegah, M.; Limbach, K.; Teneza-Mora, N.; Chuang, I.; Hollis-Perry, K.M.; Banania, J.G.; Guzman, I.; et al. A three-antigen Plasmodium falciparum DNA prime-Adenovirus boost malaria vaccine regimen is superior to a two-antigen regimen and protects against controlled human malaria infection in healthy malaria-naïve adults. PLoS ONE 2021, 16, e0256980. [Google Scholar] [CrossRef]
- Yin, H.; Zhao, L.; Wang, T.; Zhou, H.; He, S.; Cong, H. A Toxoplasma gondii vaccine encoding multistage antigens in conjunction with ubiquitin confers protective immunity to BALB/c mice against parasite infection. Parasites Vectors 2015, 8, 498. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Yin, H.; Li, Y.; Zhao, L.; Sun, X.; Cong, H. Vaccination with recombinant adenovirus expressing multi-stage antigens of Toxoplasma gondii by the mucosal route induces higher systemic cellular and local mucosal immune responses than with other vaccination routes. Parasite 2017, 24, 12. [Google Scholar] [CrossRef] [Green Version]
- Lavelle, E.C.; Ward, R.W. Mucosal vaccines—Fortifying the frontiers. Nat. Rev. Immunol. 2022, 22, 236–250. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, N.; Chen, Q. Vaccination with recombinant adenoviruses expressing Toxoplasma gondii MIC3, ROP9, and SAG2 provide protective immunity against acute toxoplasmosis in mice. Vaccine 2019, 37, 1118–1125. [Google Scholar] [CrossRef]
- Zheng, B.; Ding, J.; Chen, X.; Yu, H.; Lou, D.; Tong, Q.; Kong, Q.; Lu, S. Immuno-Efficacy of a T. gondii Secreted Protein with an Altered Thrombospondin Repeat (TgSPATR) As a Novel DNA Vaccine Candidate against Acute Toxoplasmosis in BALB/c Mice. Front. Microbiol. 2017, 8, 216. [Google Scholar] [CrossRef] [Green Version]
- Dubey, J.P.; Carpenter, J.L.; Speer, C.A.; Topper, M.J.; Uggla, A. Newly recognized fatal protozoan disease of dogs. J. Am. Vet. Med. Assoc. 1988, 192, 1269–1285. [Google Scholar]
- Dubey, J.P.; Schares, G.; Ortega-Mora, L.M. Epidemiology and control of neosporosis and Neospora caninum. Clin. Microbiol. Rev. 2007, 20, 323–367. [Google Scholar] [CrossRef] [Green Version]
- Barr, B.C.; Conrad, P.A.; Sverlow, K.W.; Tarantal, A.F.; Hendrickx, A.G. Experimental fetal and transplacental Neospora infection in the nonhuman primate. Lab. Investig. 1994, 71, 236–242. [Google Scholar]
- Jia, L.; Guo, H.; Liu, M.; Gao, Y.; Zhang, L.; Li, H.; Xie, S.; Zhang, N. Construction of an Adenovirus Vaccine Expressing the Cross-reactive Antigen AMA1 for Neospora caninum and Toxoplasma gondii and Its Immune Response in an Animal Model. Iran. J. Parasitol. 2018, 13, 235–243. [Google Scholar]
- Jordan, K.A.; Hunter, C.A. Regulation of CD8+ T cell responses to infection with parasitic protozoa. Exp. Parasitol. 2010, 126, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Rothel, J.S.; Boyle, D.B.; Both, G.W.; Pye, A.D.; Waterkeyn, J.G.; Wood, P.R.; Lightowlers, M.W. Sequential nucleic acid and recombinant adenovirus vaccination induces host-protective immune responses against Taenia ovis infection in sheep. Parasite Immunol. 1997, 19, 221–227. [Google Scholar] [CrossRef]
- Terrazas, L.I.; Bojalil, R.; Govezensky, T.; Larralde, C. Shift from an early protective Th1-type immune response to a late permissive Th2-type response in murine cysticercosis (Taenia crassiceps). J. Parasitol. 1998, 84, 74–81. [Google Scholar] [CrossRef]
- Tharmalingam, J.; Prabhakar, A.T.; Gangadaran, P.; Dorny, P.; Vercruysse, J.; Geldhof, P.; Rajshekhar, V.; Alexander, M.; Oommen, A. Host Th1/Th2 immune response to Taenia solium cyst antigens in relation to cyst burden of neurocysticercosis. Parasite Immunol. 2016, 38, 628–634. [Google Scholar] [CrossRef]
- Hill, A.V.; Reyes-Sandoval, A.; O’Hara, G.; Ewer, K.; Lawrie, A.; Goodman, A.; Nicosia, A.; Folgori, A.; Colloca, S.; Cortese, R.; et al. Prime-boost vectored malaria vaccines: Progress and prospects. Hum. Vaccine 2010, 6, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Hollingdale, M.R.; Sedegah, M.; Limbach, K. Development of replication-deficient adenovirus malaria vaccines. Expert Rev. Vaccines 2017, 16, 261–271. [Google Scholar] [CrossRef]
- Humphreys, I.R.; Sebastian, S. Novel viral vectors in infectious diseases. Immunology 2018, 153, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lu, S. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 2009, 21, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Pance, A. Diversify and Conquer: The Vaccine Escapism of Plasmodium falciparum. Microorganisms 2020, 8, 1748. [Google Scholar] [CrossRef]


| Type of Pathogen or Condition | Disease | Vector | Phase | Status | ID Number |
|---|---|---|---|---|---|
| Virus | COVID-19 | ChAdOx1 | II/III | Active | NCT04400838 |
| HAdV-26 | III | Active | NCT04505722 | ||
| Respiratory syncytial virus | HAdV-26 | III | Recruiting | NCT04908683 | |
| Norovirus | HAdV-5 | IIb | Recruiting | NCT05212168 | |
| HIV/AIDS | HAdV-26 | III | Active | NCT03964415 | |
| Influenza | HAdV-5 | II | Completed, with results | NCT02918006 | |
| Ebola | HAdV-26 | III | Completed, with results | NCT02509494 | |
| Bacteria | Tuberculosis | ChAdOx1 | I/IIa | Completed | NCT03681860 |
| Parasites | P. falciparum Malaria | HAdV-35/HAdV-26 | I/IIa | Completed | NCT01397227 |
| ChAd63 | II | Completed | NCT01666925 | ||
| P. vivax Malaria | ChAd63 | II | Completed | NCT04009096 | |
| Leishmaniasis | ChAd63 | IIb | Active | NCT03969134 | |
| Cancer | Advanced/metastatic solid tumors | HAdV-5 | I/II | Active | NCT02285816 |
| Non-small cell lung cancer | ChAdOx1 | I/II | Recruiting | NCT04908111 | |
| Lynch syndrome cancer prevention | Gad20 | Ib/II | Recruiting | NCT05078866 | |
| Drug dependence | Cocaine dependence | HAdV-5 | I | Recruiting | NCT02455479 |
| T Helper Subset | Polarizing Cytokines | Master Transcription Factor | Key Effector Cytokines | General Targets | Related Cells/Effectors |
|---|---|---|---|---|---|
| Th1 | IFN-γ IL-12 | T-bet | IFN-γ | Intracellular pathogens | CD8+ T cells M1 macrophages Natural killer cells (NKs) IgG2 |
| Th2 | IL-4 IL-2 | GATA-3 | IL-4 IL-5 IL-13 | Helminths/large extracellular pathogens | M2 macrophages Eosinophils IgE Goblet cells IgG1 |
| Th17 | IL-1β IL-6 IL-23 | RORγt | IL-17A IL-17F IL-22 | Extracellular bacteria and fungi | Nitric oxide Antimicrobial peptides Neutrophils |
| Treg | IL-2 TGF-β | Foxp3 | IL-10 TGF-β IL-35 | Regulation of the immune response | IL-2 consumption Negative signal transmission (CTLA-4, CD39 etc.) Perforin/granzyme B killing of antigen-presenting cells |
| Tfh | IL-6 IL-21 | Bcl6 | IL-21 | Promotion of B cell maturation and antibody production | B cells CXCR5 Antibodies |
| Disease | Parasite Speices | Antigen | Vector | Current Stage of Development | Refs. |
|---|---|---|---|---|---|
| Chagas disease | T. cruzi | ASP2 and/or TS | HAdV-5 | Mice | [107,108,109,110,111,112,113,114,115,116,117,118,119,120,122] |
| ASP2 | HAdV-5 | Mice | [139,140] | ||
| gp83 | HAdV-5 | Mice | [138,139] | ||
| ASP2 | HAdV-48 | Mice | [139] | ||
| gp83 | HAdV-48 | Mice | |||
| Schistosomiasis | S. mansoni | SmCB | HAdV-5 | Mice | [45] |
| S. japonicum | SjTPI | HAdV-5 | Mice | [41,42,43] | |
| SjIAP | HAdV-5 | Mice | [44] | ||
| Leishmaniasis and PKDL | unspecific | KMP-11 and HASBP | ChAd63 | Clinical trials phase IIa | [185,186] |
| L. infantum | A2 | HAdV-5 | NHP(Rhesus macaque) | [184] | |
| Toxoplasmosis | T. gondii | TgMIC3 | HAdV-5 | Mice | [222] |
| TgSAG2 | |||||
| TgROP9 | |||||
| UMAS | HAdV-5 | Mice | [219,220] | ||
| ROP16 | CAV-2 | Mice | [217] | ||
| ROP18 | CAV-2 | Mice | [216] | ||
| SAG1 | HAdV-5 | Mice | [215] | ||
| Toxoplasmosis/Neosporosis | T. gondii and N. caninum | NcAMA1 | HAdV-5 | Mice | [225] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koger-Pease, C.; Perera, D.J.; Ndao, M. Recent Advances in the Development of Adenovirus-Vectored Vaccines for Parasitic Infections. Pharmaceuticals 2023, 16, 334. https://doi.org/10.3390/ph16030334
Koger-Pease C, Perera DJ, Ndao M. Recent Advances in the Development of Adenovirus-Vectored Vaccines for Parasitic Infections. Pharmaceuticals. 2023; 16(3):334. https://doi.org/10.3390/ph16030334
Chicago/Turabian StyleKoger-Pease, Cal, Dilhan J. Perera, and Momar Ndao. 2023. "Recent Advances in the Development of Adenovirus-Vectored Vaccines for Parasitic Infections" Pharmaceuticals 16, no. 3: 334. https://doi.org/10.3390/ph16030334