
PHPB Attenuated Cognitive Impairment in Type 2 Diabetic KK-Ay Mice by Modulating SIRT1/Insulin Signaling Pathway and Inhibiting Generation of AGEs

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
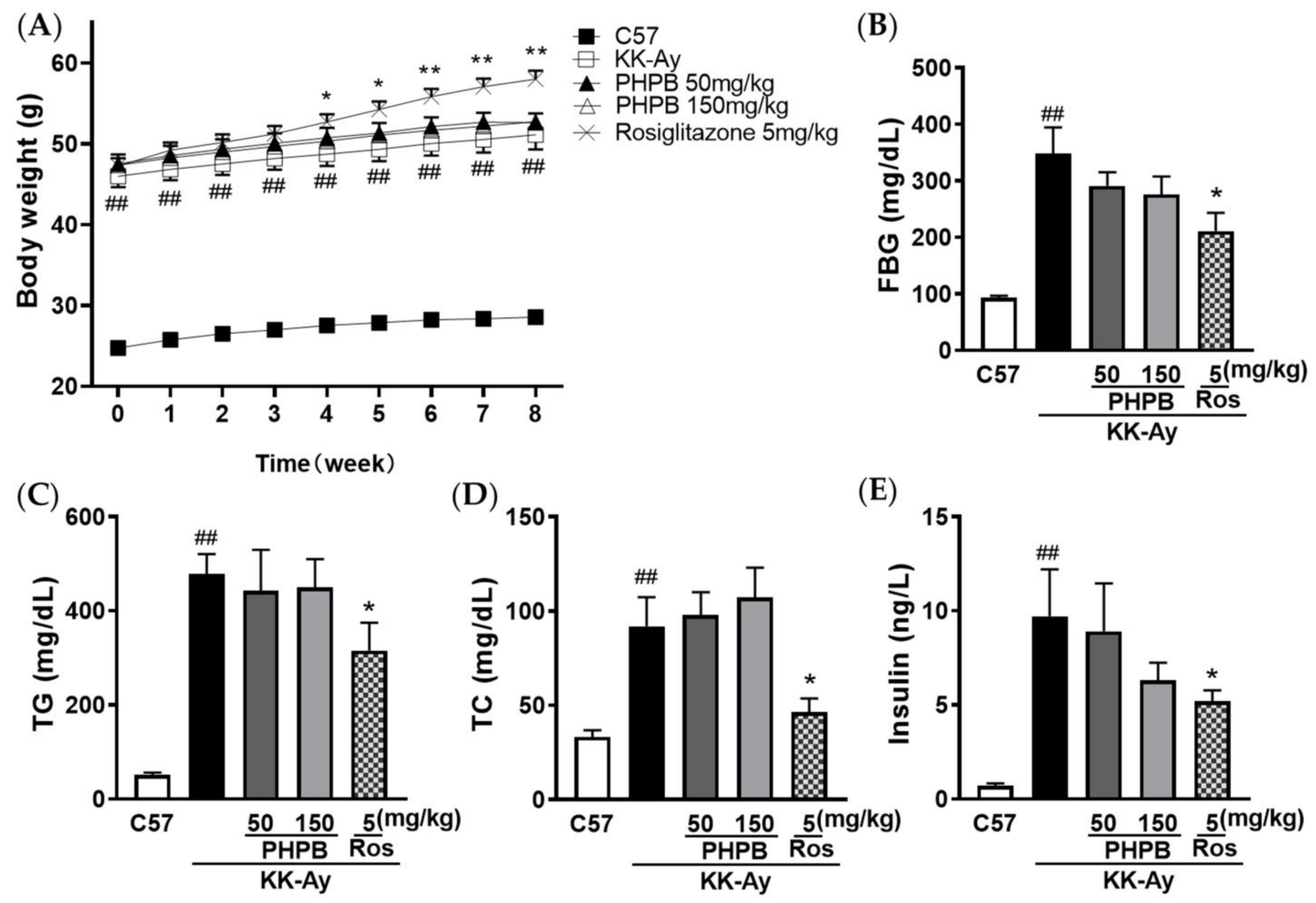
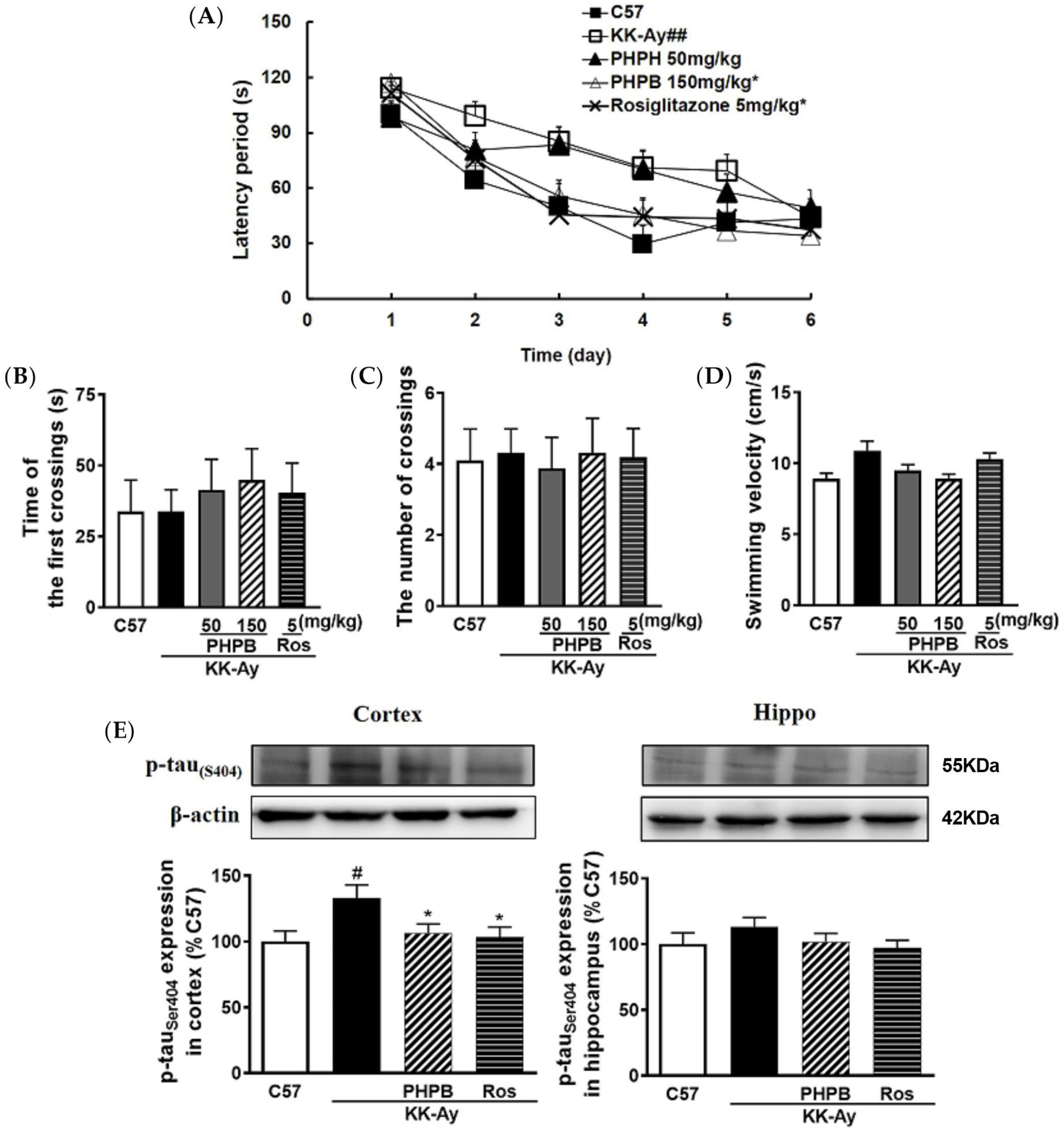
2.1. The Effects of PHPB on the Metabolic Features and Cognitive Function in KK-Ay Mice
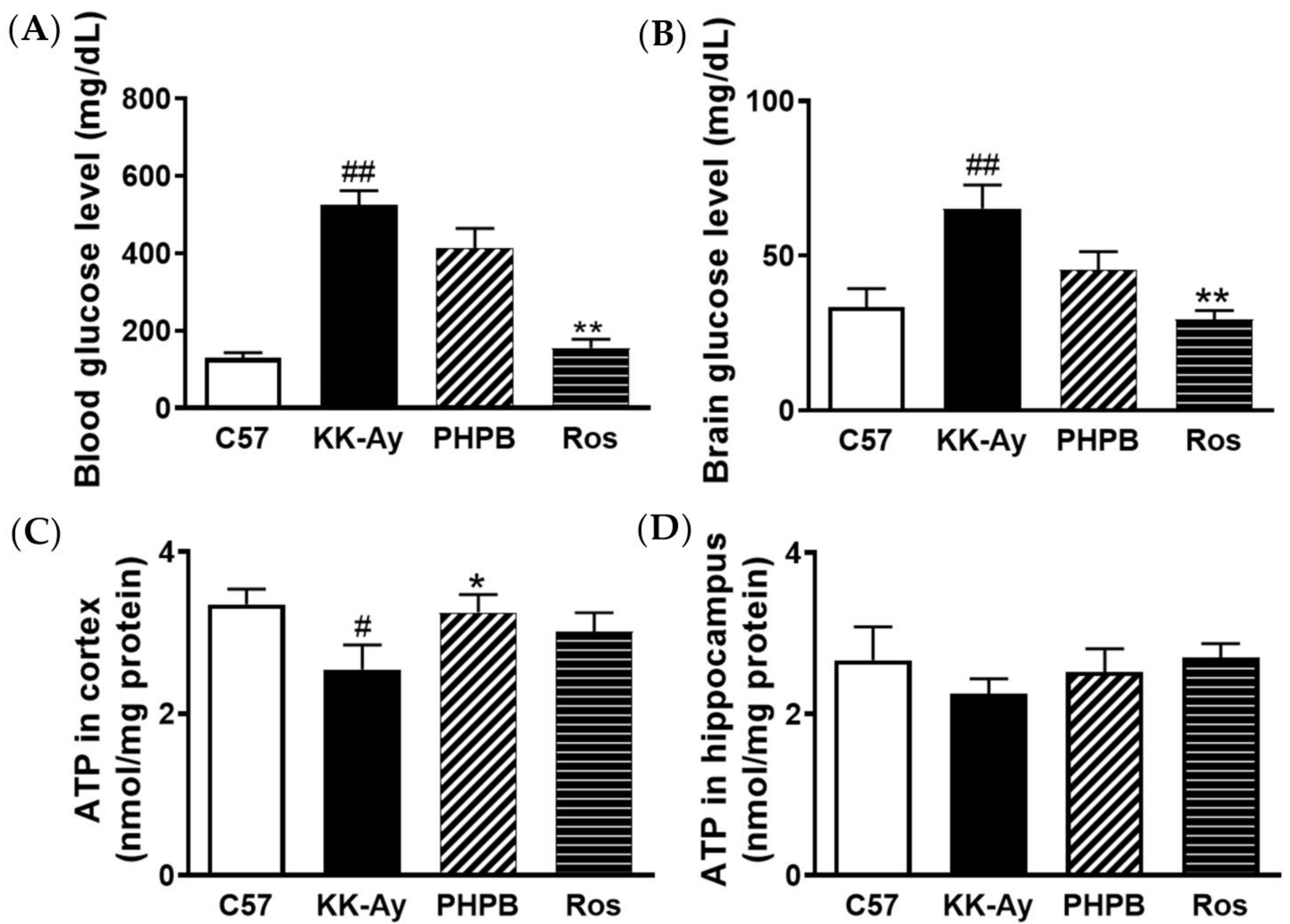
2.2. Effects of PHPB on Glucose Levels and ATP Contents in Brains of KK-Ay Mice
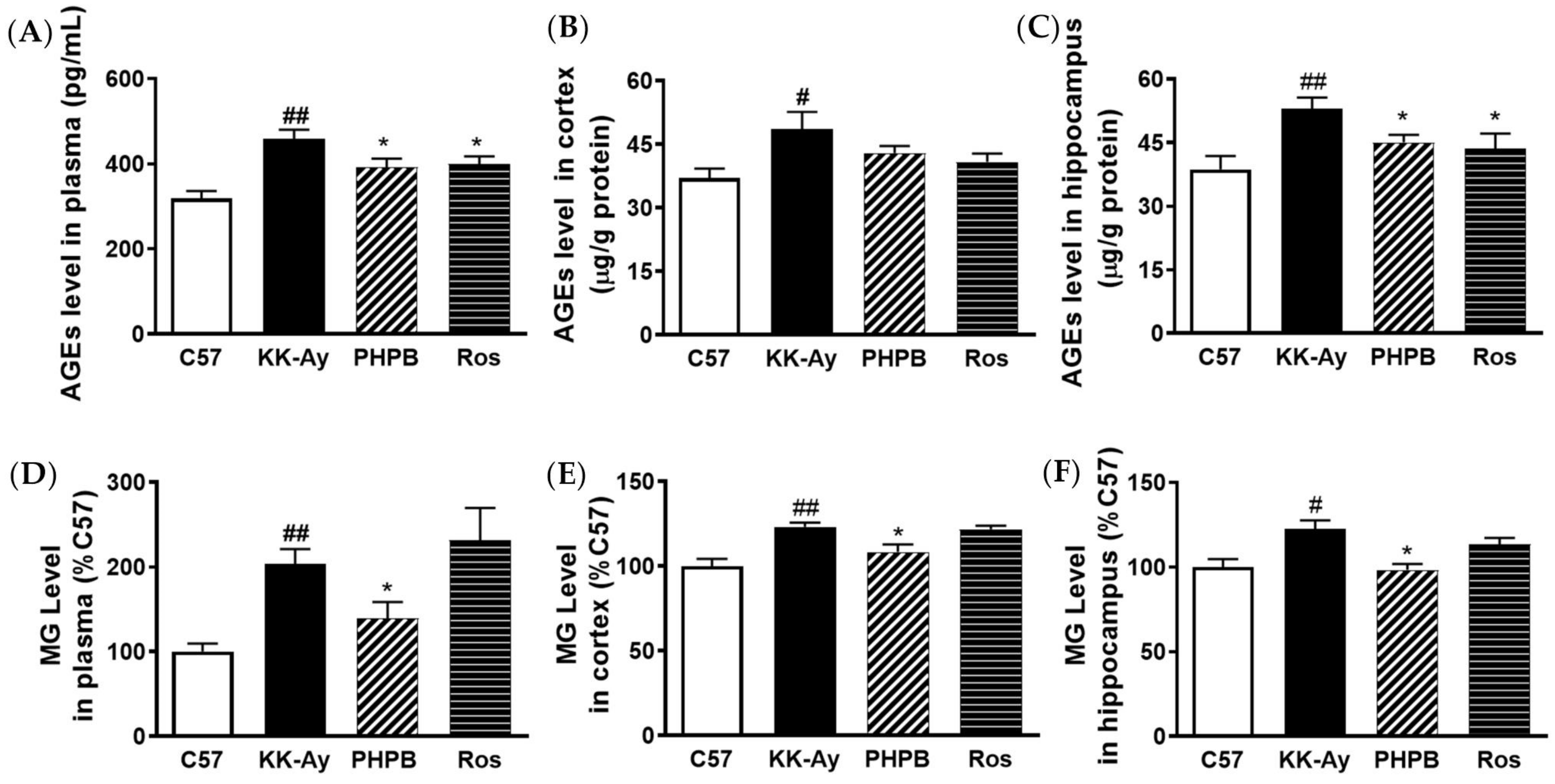
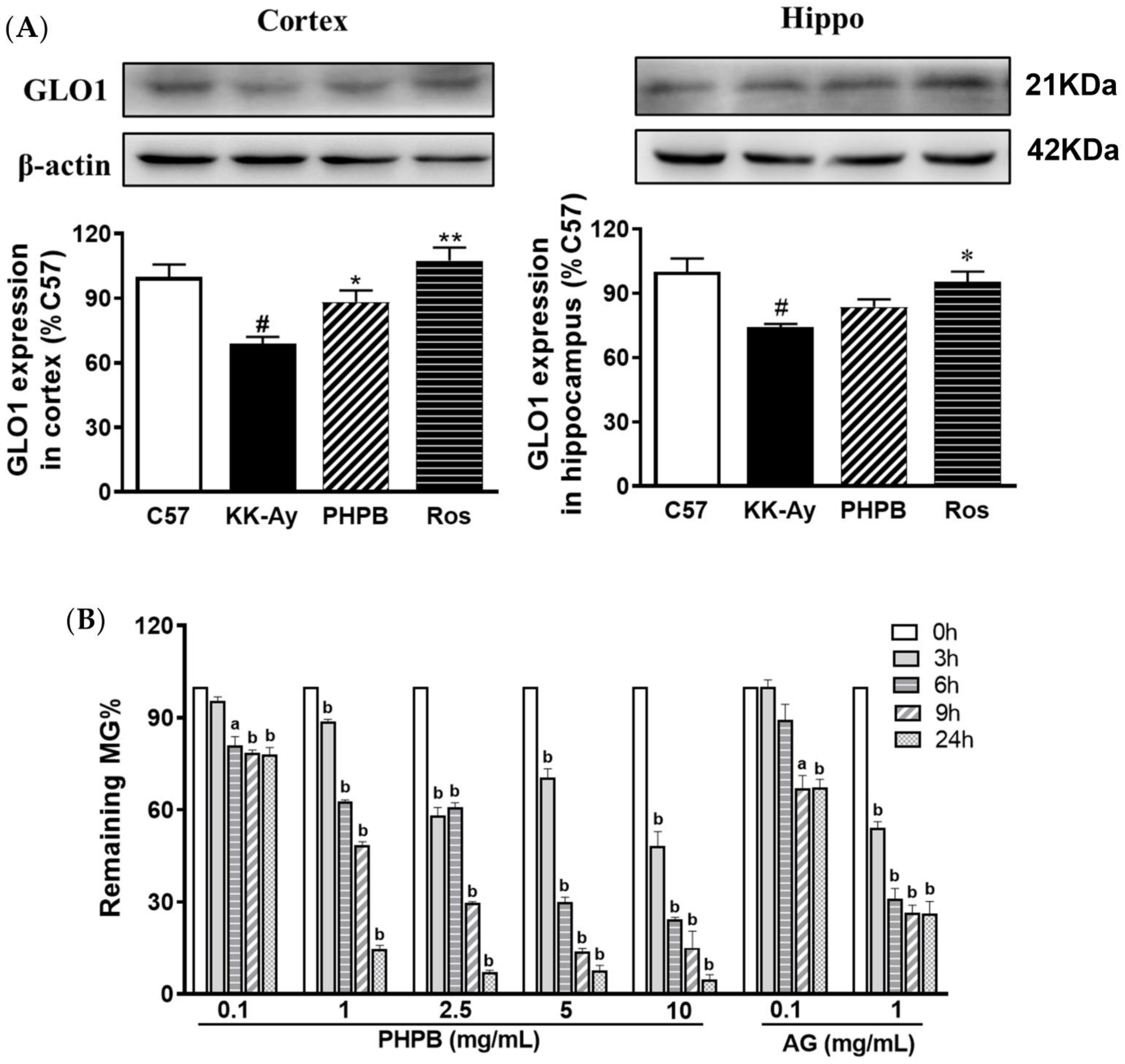
2.3. Effects of PHPB on Accumulation of AGEs and MG and Expression of GLO1 in KK-Ay Mice
2.4. Effect of PHPB on MG Clearance by MG Trapping In Vitro
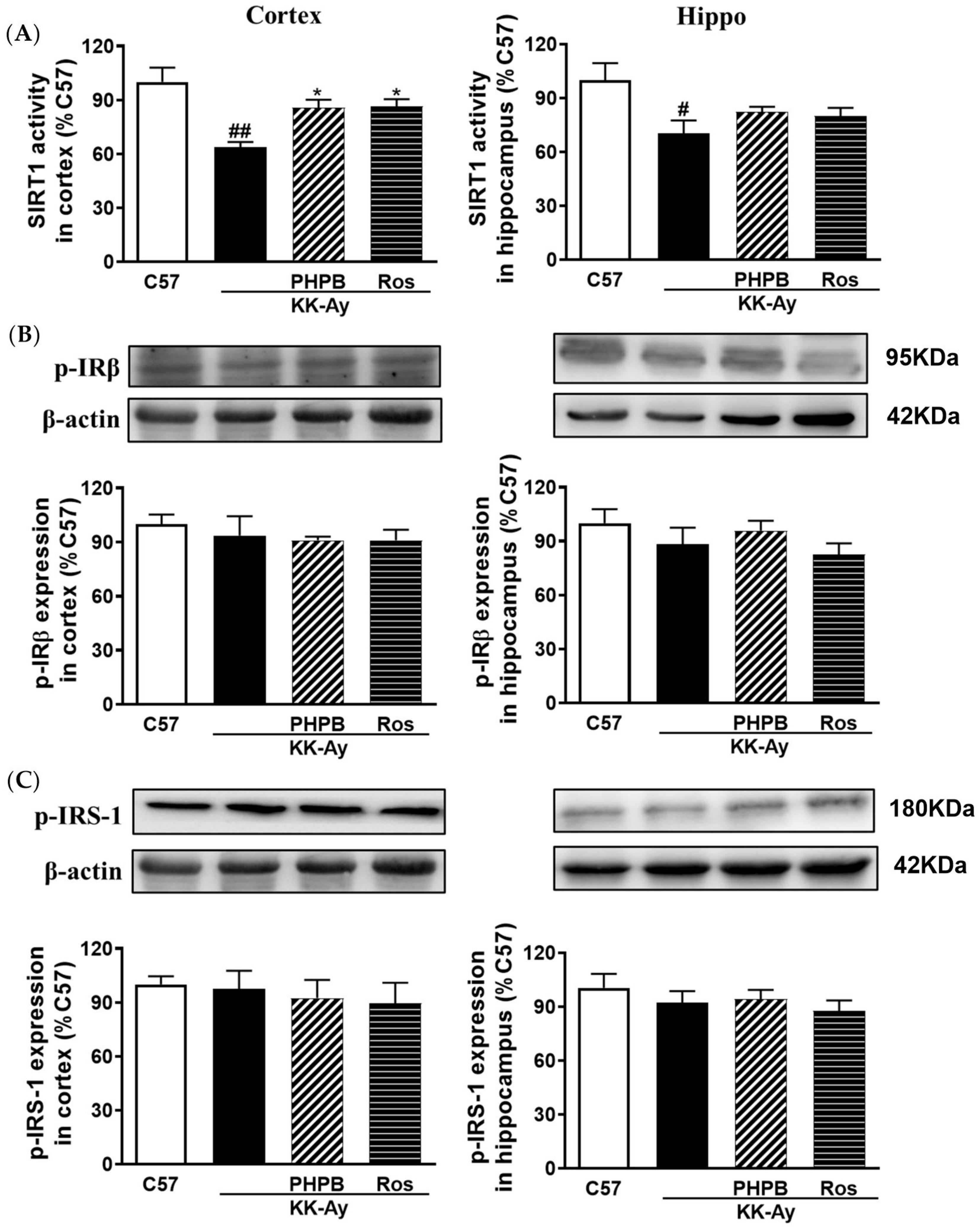
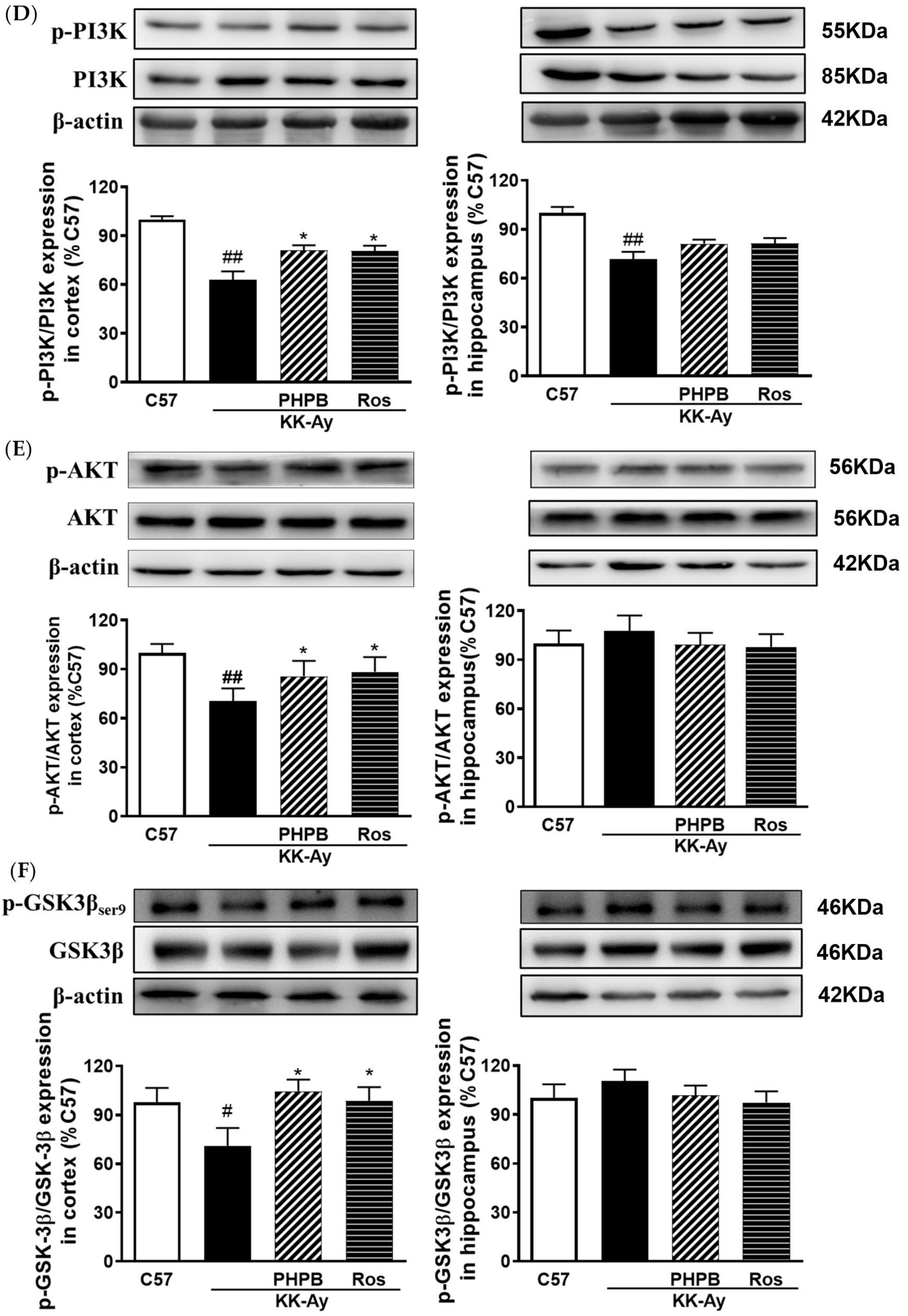
2.5. Effects of PHPB on the SIRT1 Deacetylase Activity and Insulin Signaling Pathway in the KK-Ay Mice
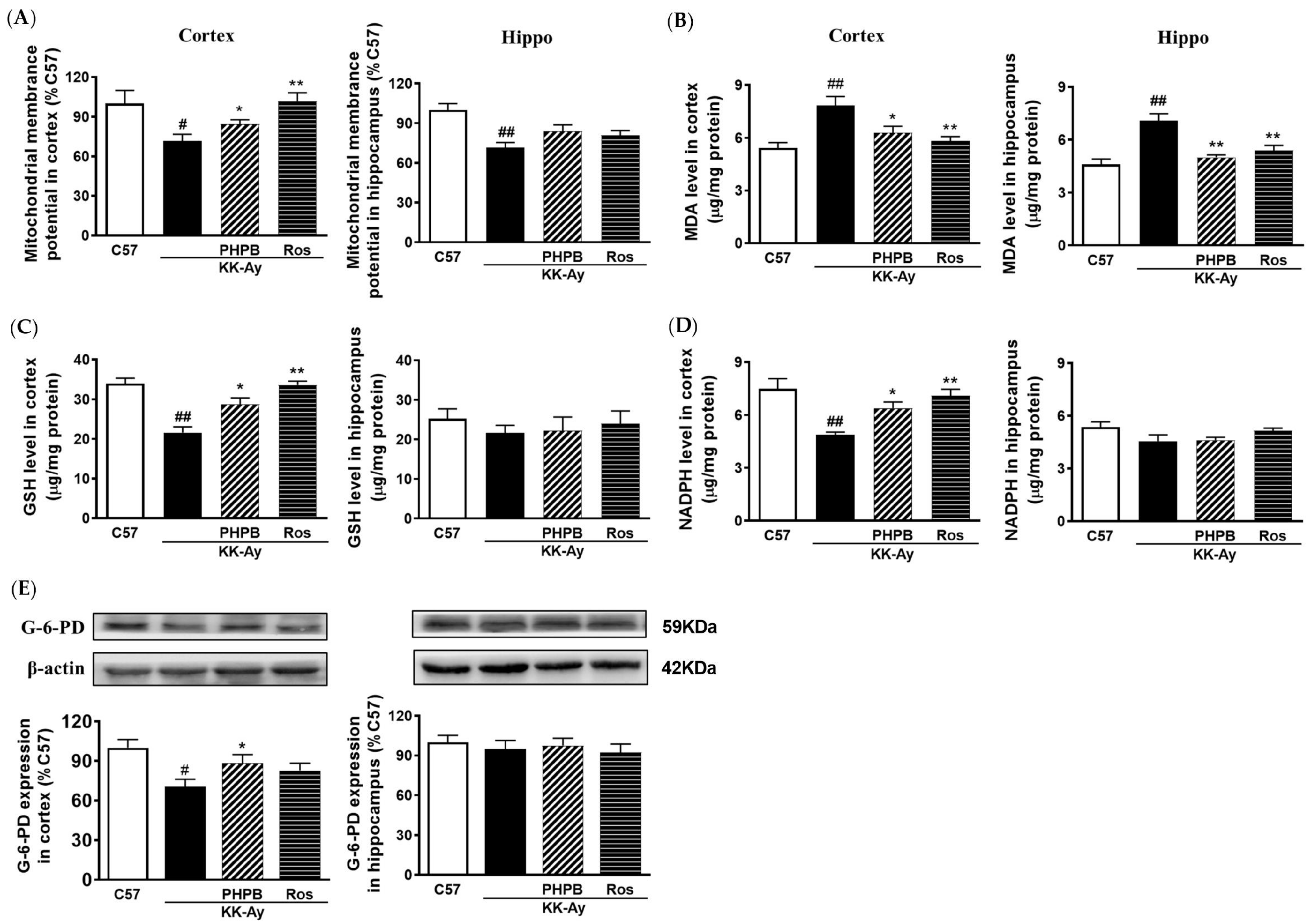
2.6. Effects of PHPB on Oxidative Stress and Mitochondrial Function in KK-Ay Mice
3. Discussion
4. Materials and Methods
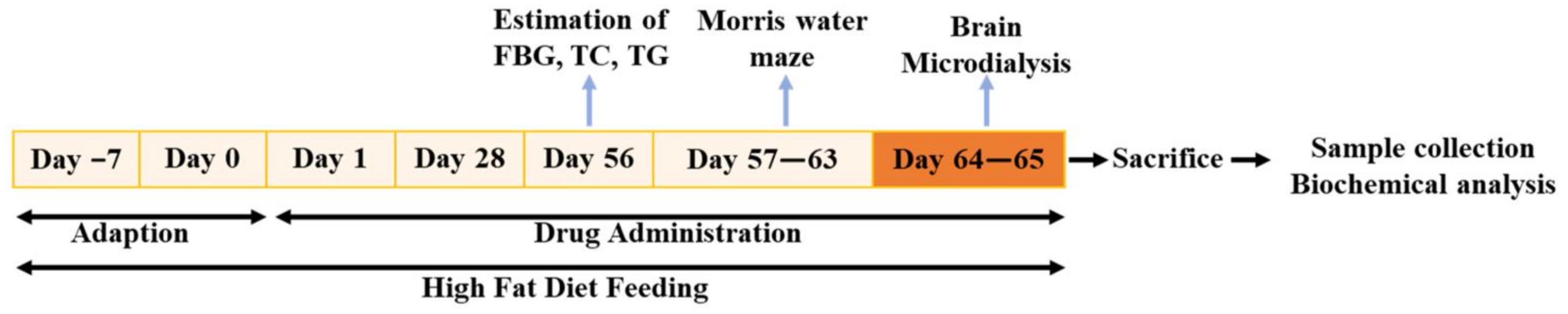
4.1. Animals and Drug Administration
4.2. Morris Water Maze Test
4.3. Metabolic Determination
4.4. Microdialysis
4.4.1. Surgery
4.4.2. Tissue Microdialysis
4.5. Tissue Processing
4.6. Biochemical Analysis
4.7. Methylglyoxal Measurement
4.8. Determination of Methylglyoxal (MG) Trapping of PHPB In Vitro
4.9. Western Blotting
4.10. Measurement of Mitochondrial Membrane Potential (MMP)
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Kubis-Kubiak, A.M.; Rorbach-Dolata, A.; Piwowar, A. Crucial players in Alzheimer’s disease and diabetes mellitus: Friends or foes? Mech. Ageing Dev. 2019, 181, 7–21. [Google Scholar] [CrossRef]
- Shieh, J.C.; Huang, P.T.; Lin, Y.F. Alzheimer’s Disease and Diabetes: Insulin Signaling as the Bridge Linking Two Pathologies. Mol. Neurobiol. 2020, 57, 1966–1977. [Google Scholar] [CrossRef]
- Wang, C.; Li, J.; Zhao, S.; Huang, L. Diabetic encephalopathy causes the imbalance of neural activities between hippocampal glutamatergic neurons and GABAergic neurons in mice. Brain Res. 2020, 1742, 146863. [Google Scholar] [CrossRef]
- Chou, P.S.; Wu, M.N.; Yang, C.C.; Shen, C.T.; Yang, Y.H. Effect of Advanced Glycation End Products on the Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 191–197. [Google Scholar] [CrossRef]
- Kuzan, A. Toxicity of advanced glycation end products (Review). Biomed Rep. 2021, 14, 46. [Google Scholar] [CrossRef]
- Ruiz, H.H.; Ramasamy, R.; Schmidt, A.M. Advanced Glycation End Products: Building on the Concept of the “Common Soil” in Metabolic Disease. Endocrinology 2020, 161, bqz006. [Google Scholar] [CrossRef]
- Lovestone, S.; Smith, U. Advanced glycation end products, dementia, and diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 4743–4744. [Google Scholar] [CrossRef] [Green Version]
- Kanfi, Y.; Shalman, R.; Peshti, V.; Pilosof, S.N.; Gozlan, Y.M.; Pearson, K.J.; Lerrer, B.; Moazed, D.; Marine, J.C.; de Cabo, R.; et al. Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett. 2008, 582, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xu, S.F.; Peng, Y.; Feng, N.; Wang, L.; Wang, X.L. Conversion and pharmacokinetics profiles of a novel pro-drug of 3-n-butylphthalide, potassium 2-(1-hydroxypentyl)-benzoate, in rats and dogs. Acta Pharmacol. Sin. 2018, 39, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Zhang, Y.; Peng, Y.; Zhao, Z.; Zhou, Y.; Wang, X.; Peng, Y. Protective effect of potassium 2-(l-hydroxypentyl)-benzoate on hippocampal neurons, synapses and dystrophic axons in APP/PS1 mice. Psychopharmacology 2019, 236, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Yin, H.J.; Li, J.; Wang, L.; Wang, W.P.; Wang, X.L. Studies of pathology and pharmacology of diabetic encephalopathy with KK-Ay mouse model. CNS Neurosci. Ther. 2020, 26, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Yin, H.; Sun, Y.; Shi, S.; Li, J.; Wang, X. The attenuation effect of potassium 2-(1-hydroxypentyl)-benzoate in a mouse model of diabetes-associated cognitive decline: The protein expression in the brain. CNS Neurosci. Ther. 2022, 28, 1108–1123. [Google Scholar] [CrossRef]
- Wang, H.; Chen, F.; Zhong, K.L.; Tang, S.S.; Hu, M.; Long, Y.; Miao, M.X.; Liao, J.M.; Sun, H.B.; Hong, H. PPARγ agonists regulate bidirectional transport of amyloid-β across the blood-brain barrier and hippocampus plasticity in db/db mice. Br. J. Pharmacol. 2016, 173, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J.; et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 338ra366. [Google Scholar] [CrossRef] [Green Version]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Di Loreto, S.; Zimmitti, V.; Sebastiani, P.; Cervelli, C.; Falone, S.; Amicarelli, F. Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. Int. J. Biochem. Cell Biol. 2008, 40, 245–257. [Google Scholar] [CrossRef]
- Hansen, F.; Pandolfo, P.; Galland, F.; Torres, F.V.; Dutra, M.F.; Batassini, C.; Guerra, M.C.; Leite, M.C.; Gonçalves, C.A. Methylglyoxal can mediate behavioral and neurochemical alterations in rat brain. Physiol. Behav. 2016, 164, 93–101. [Google Scholar] [CrossRef]
- Rodrigues, T.; Borges, P.; Mar, L.; Marques, D.; Albano, M.; Eickhoff, H.; Carrêlo, C.; Almeida, B.; Pires, S.; Abrantes, M.; et al. GLP-1 improves adipose tissue glyoxalase activity and capillarization improving insulin sensitivity in type 2 diabetes. Pharmacol. Res. 2020, 161, 105198. [Google Scholar] [CrossRef]
- Zhu, X.; Cheng, Y.Q.; Lu, Q.; Du, L.; Yin, X.X.; Liu, Y.W. Enhancement of glyoxalase 1, a polyfunctional defense enzyme, by quercetin in the brain in streptozotocin-induced diabetic rats. Naunyn Schmiedebergs Arch. Pharm. 2018, 391, 1237–1245. [Google Scholar] [CrossRef]
- de Matos, A.M.; de Macedo, M.P.; Rauter, A.P. Bridging Type 2 Diabetes and Alzheimer’s Disease: Assembling the Puzzle Pieces in the Quest for the Molecules With Therapeutic and Preventive Potential. Med. Res. Rev. 2018, 38, 261–324. [Google Scholar] [CrossRef]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. 2009, 14, 373–379. [Google Scholar]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Münch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J. Neurochem. 2005, 92, 255–263. [Google Scholar] [CrossRef]
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef]
- Dariya, B.; Nagaraju, G.P. Advanced glycation end products in diabetes, cancer and phytochemical therapy. Drug Discov. Today 2020, 25, 1614–1623. [Google Scholar] [CrossRef]
- Gandhi, G.K.; Ball, K.K.; Cruz, N.F.; Dienel, G.A. Hyperglycaemia and diabetes impair gap junctional communication among astrocytes. ASN Neuro 2010, 2, e00030. [Google Scholar] [CrossRef] [Green Version]
- Ball, K.K.; Harik, L.; Gandhi, G.K.; Cruz, N.F.; Dienel, G.A. Reduced gap junctional communication among astrocytes in experimental diabetes: Contributions of altered connexin protein levels and oxidative-nitrosative modifications. J. Neurosci. Res. 2011, 89, 2052–2067. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Aponte, D.E.; Méndez-González, M.P.; Rivera-Pagán, A.F.; Kucheryavykh, Y.V.; Kucheryavykh, L.Y.; Skatchkov, S.N.; Eaton, M.J. Hyperglycemia reduces functional expression of astrocytic Kir4.1 channels and glial glutamate uptake. Neuroscience 2015, 310, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Wang, W.; Yu, W.; Li, J.; Feng, N.; Wang, L.; Wang, X. Changes in Synaptic Plasticity and Glutamate Receptors in Type 2 Diabetic KK-Ay Mice. J. Alzheimers Dis. 2017, 57, 1207–1220. [Google Scholar] [CrossRef]
- Lubitz, I.; Ricny, J.; Atrakchi-Baranes, D.; Shemesh, C.; Kravitz, E.; Liraz-Zaltsman, S.; Maksin-Matveev, A.; Cooper, I.; Leibowitz, A.; Uribarri, J.; et al. High dietary advanced glycation end products are associated with poorer spatial learning and accelerated Aβ deposition in an Alzheimer mouse model. Aging Cell 2016, 15, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M.; Vlassara, H.; Kooney, A.; Ulrich, P.; Cerami, A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science 1986, 232, 1629–1632. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, P.; Sun, H.; Cai, R.; Xia, W.; Wang, S. Advanced glycation end product-induced astrocytic differentiation of cultured neurospheres through inhibition of Notch-Hes1 pathway-mediated neurogenesis. Int. J. Mol. Sci. 2013, 15, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Thornalley, P.J. Dicarbonyl intermediates in the maillard reaction. Ann. N.Y. Acad. Sci. 2005, 1043, 111–117. [Google Scholar] [CrossRef]
- Miyata, T.; van Ypersele de Strihou, C.; Imasawa, T.; Yoshino, A.; Ueda, Y.; Ogura, H.; Kominami, K.; Onogi, H.; Inagi, R.; Nangaku, M.; et al. Glyoxalase I deficiency is associated with an unusual level of advanced glycation end products in a hemodialysis patient. Kidney Int. 2001, 60, 2351–2359. [Google Scholar] [CrossRef] [Green Version]
- Brouwers, O.; Niessen, P.M.; Ferreira, I.; Miyata, T.; Scheffer, P.G.; Teerlink, T.; Schrauwen, P.; Brownlee, M.; Stehouwer, C.D.; Schalkwijk, C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011, 286, 1374–1380. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, S.; Wang, C.Y.; Wang, Y.; Liu, H.X.; Cui, Y.; Zhang, L.D. Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. In Vitro Cell Dev. Biol. Anim. 2015, 51, 204–209. [Google Scholar] [CrossRef]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Jiao, Z.; Sun, X.; Zhao, Y.; Ren, J.; Xu, G. Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain Res. 2011, 1383, 300–306. [Google Scholar] [CrossRef]
- Kung, J.; Henry, R.R. Thiazolidinedione safety. Expert Opin. Drug Saf. 2012, 11, 565–579. [Google Scholar] [CrossRef]
- Sarathlal, C.S.; Kakoty, V.; Marathe, S.; Chitkara, D.; Taliyan, R. Exploring the Neuroprotective Potential of Rosiglitazone Embedded Nanocarrier System on Streptozotocin Induced Mice Model of Alzheimer’s Disease. Neurotox Res. 2021, 39, 240–255. [Google Scholar] [CrossRef]
- Jacob, R.J.; Fan, X.; Evans, M.L.; Dziura, J.; Sherwin, R.S. Brain glucose levels are elevated in chronically hyperglycemic diabetic rats: No evidence for protective adaptation by the blood brain barrier. Metabolism 2002, 51, 1522–1524. [Google Scholar] [CrossRef]
- Jensen, C.J.; Demol, F.; Bauwens, R.; Kooijman, R.; Massie, A.; Villers, A.; Ris, L.; De Keyser, J. Astrocytic β2 Adrenergic Receptor Gene Deletion Affects Memory in Aged Mice. PLoS ONE 2016, 11, e0164721. [Google Scholar] [CrossRef] [Green Version]
- Chaplen, F.W.; Fahl, W.E.; Cameron, D.C. Method for determination of free intracellular and extracellular methylglyoxal in animal cells grown in culture. Anal. Biochem. 1996, 238, 171–178. [Google Scholar] [CrossRef]
- Dhar, A.; Desai, K.; Liu, J.; Wu, L. Methylglyoxal, protein binding and biological samples: Are we getting the true measure? J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1093–1100. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Xu, S.; Wang, L.; Wang, X. PHPB Attenuated Cognitive Impairment in Type 2 Diabetic KK-Ay Mice by Modulating SIRT1/Insulin Signaling Pathway and Inhibiting Generation of AGEs. Pharmaceuticals 2023, 16, 305. https://doi.org/10.3390/ph16020305
Li J, Xu S, Wang L, Wang X. PHPB Attenuated Cognitive Impairment in Type 2 Diabetic KK-Ay Mice by Modulating SIRT1/Insulin Signaling Pathway and Inhibiting Generation of AGEs. Pharmaceuticals. 2023; 16(2):305. https://doi.org/10.3390/ph16020305
Chicago/Turabian StyleLi, Jiang, Shaofeng Xu, Ling Wang, and Xiaoliang Wang. 2023. "PHPB Attenuated Cognitive Impairment in Type 2 Diabetic KK-Ay Mice by Modulating SIRT1/Insulin Signaling Pathway and Inhibiting Generation of AGEs" Pharmaceuticals 16, no. 2: 305. https://doi.org/10.3390/ph16020305