
Copper(II) Chelates of Schiff Bases Enriched with Aliphatic Fragments: Synthesis, Crystal Structure, In Silico Studies of ADMET Properties and a Potency against a Series of SARS-CoV-2 Proteins

Abstract
:1. Introduction
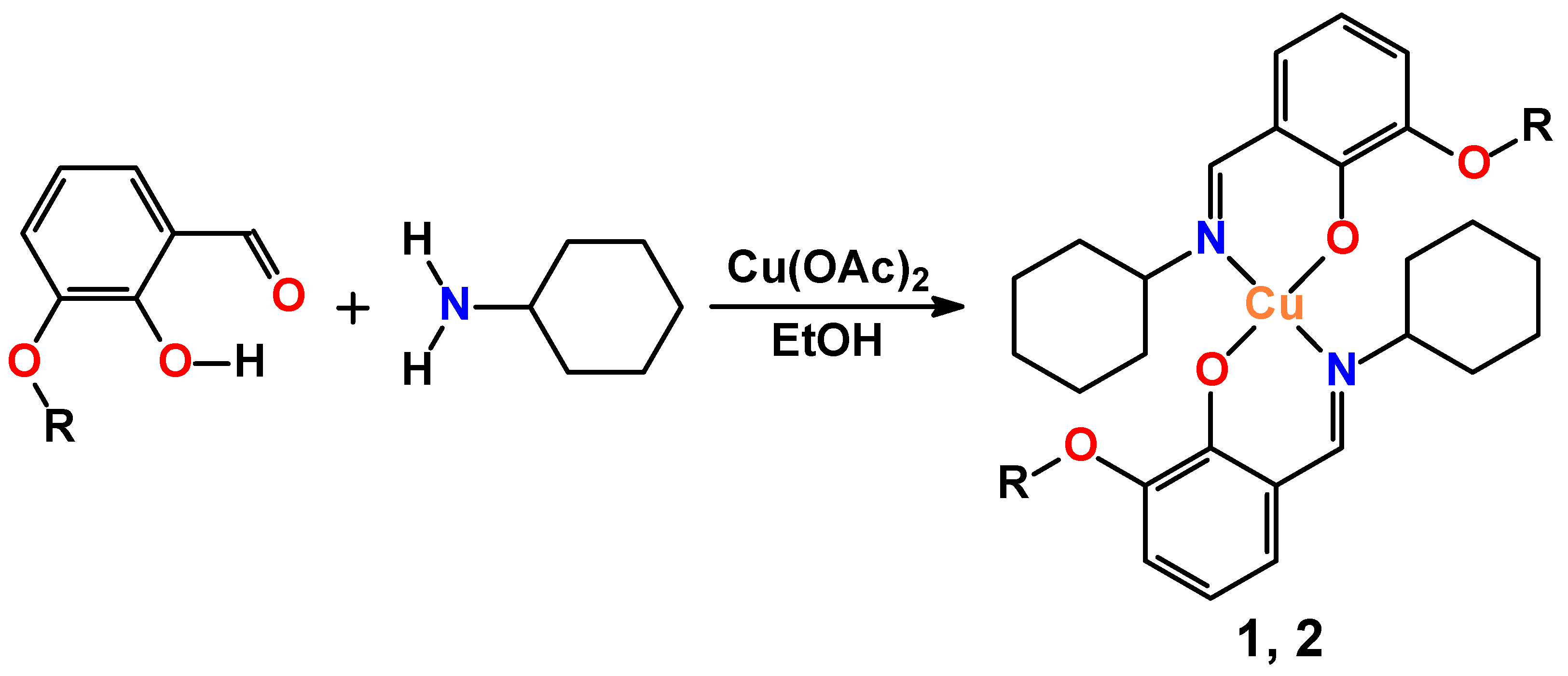
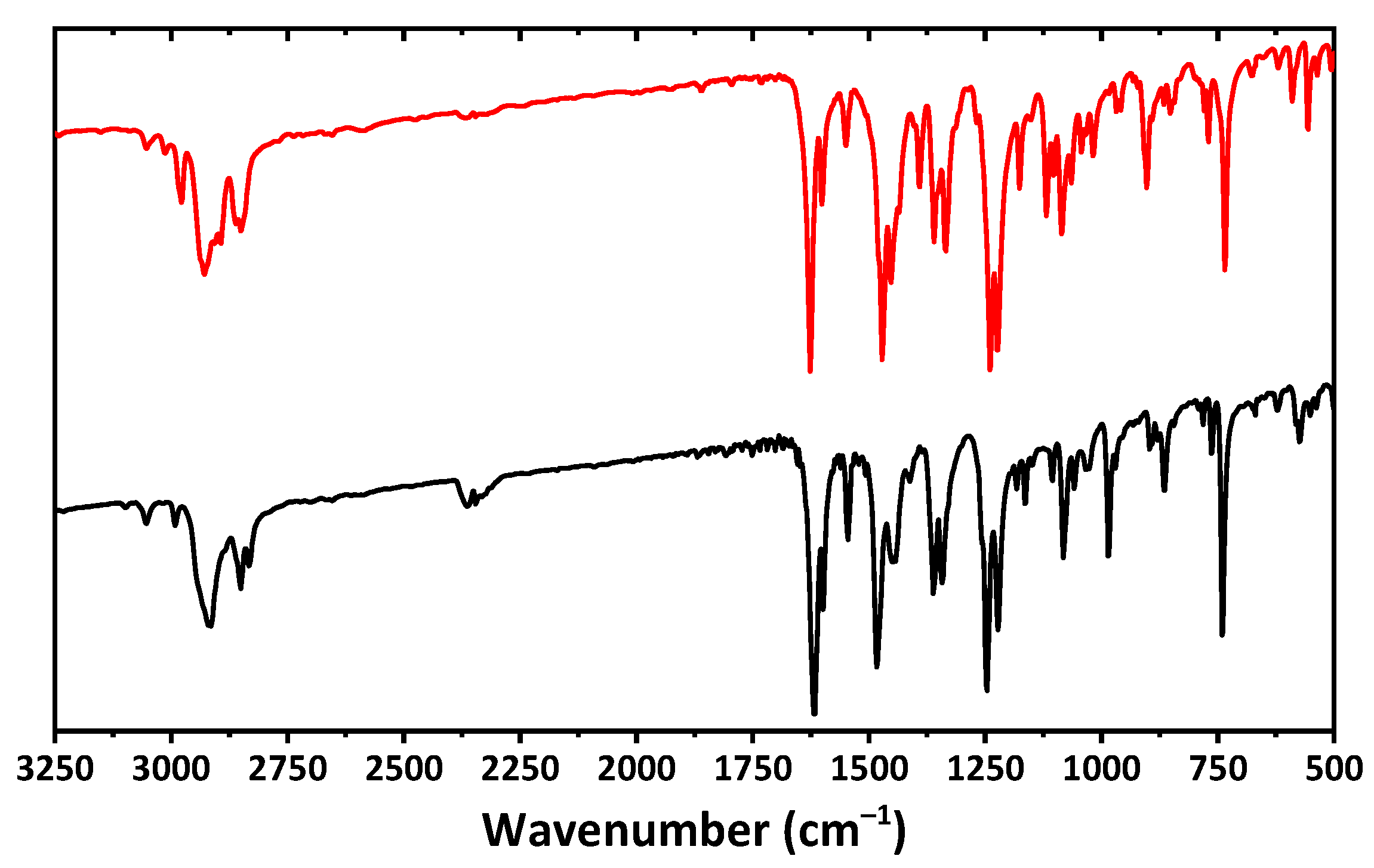
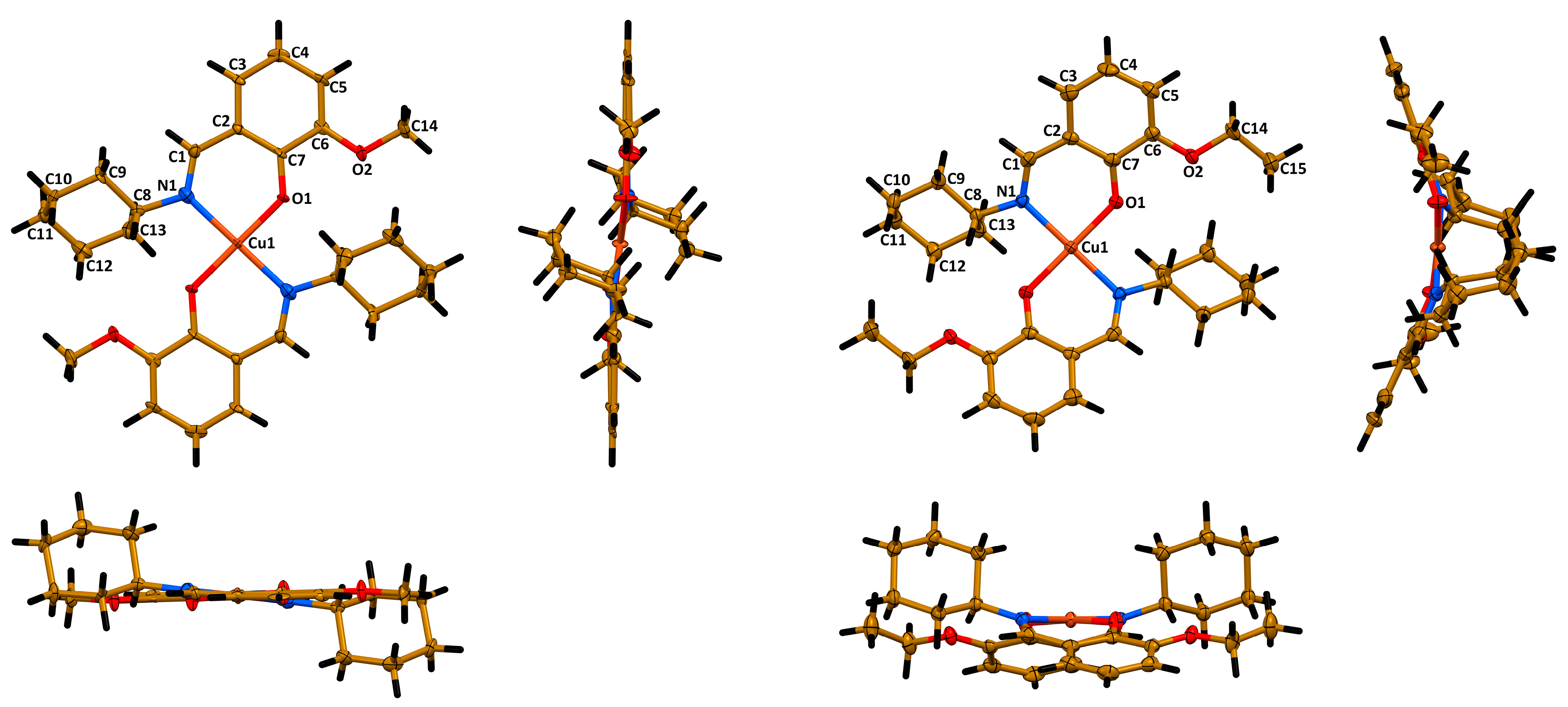
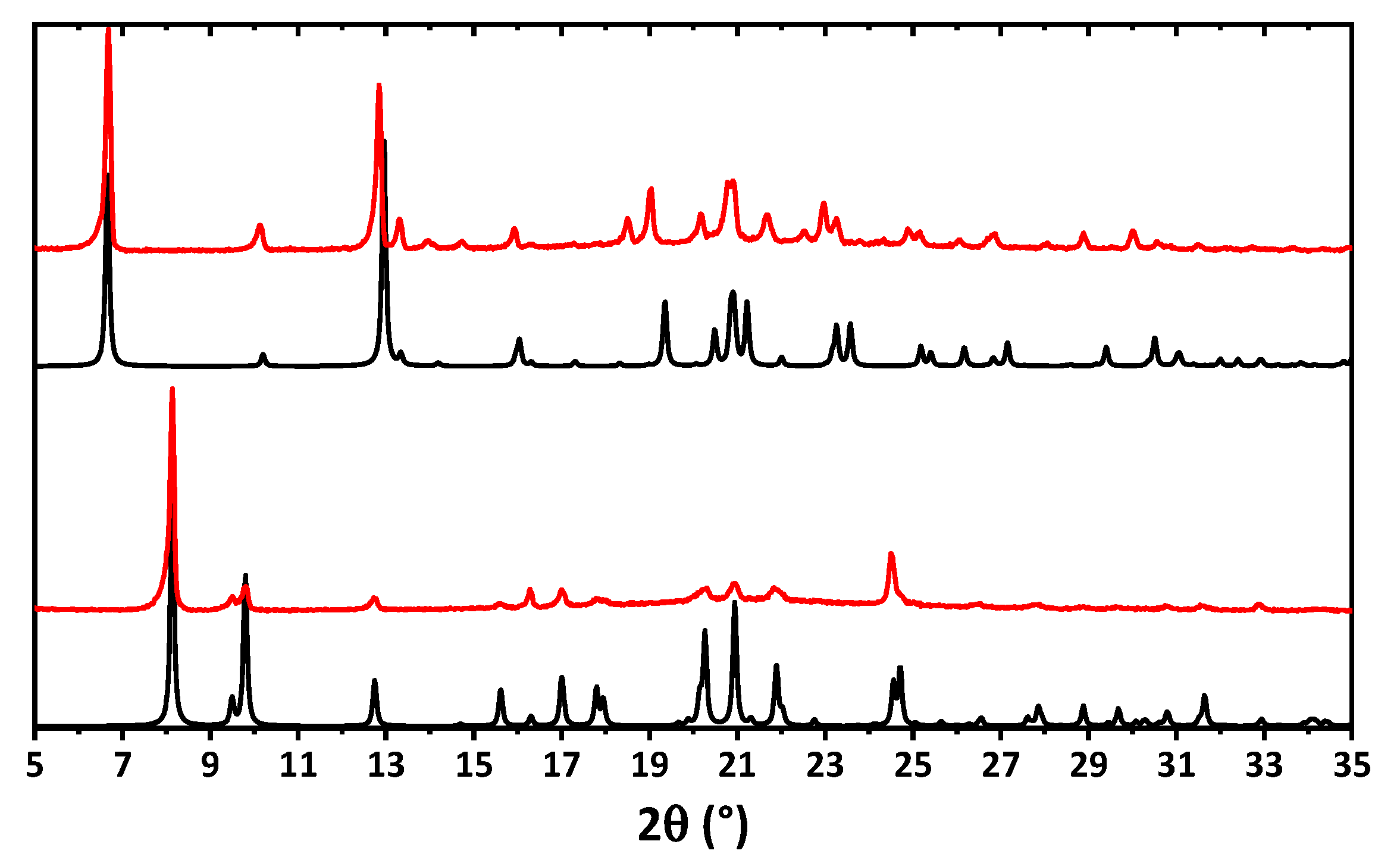
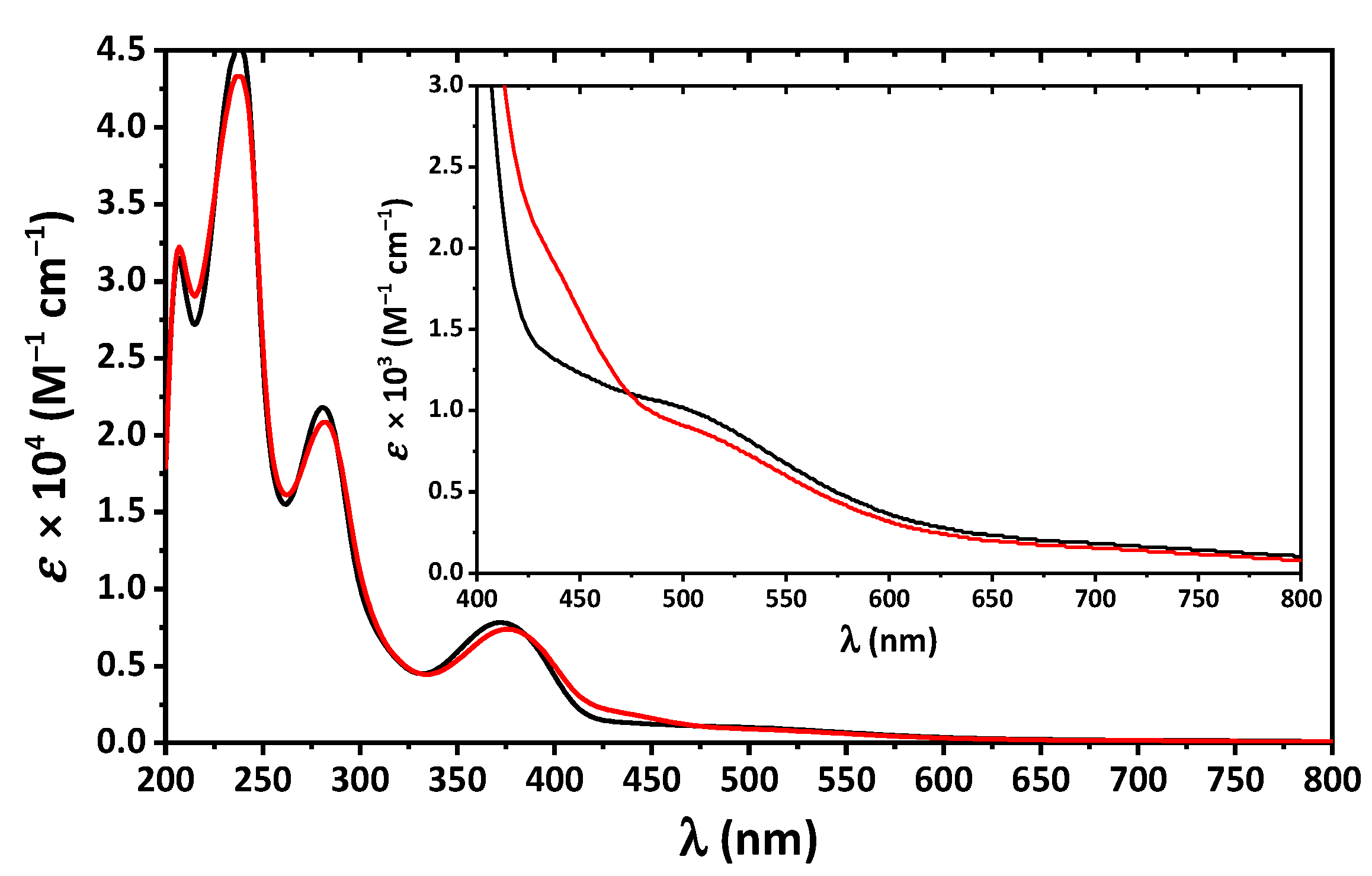
2. Results and Discussion
3. Materials and Methods
3.1. Physical Measurements
3.2. Synthesis
3.3. Single Crystal X-ray Diffraction
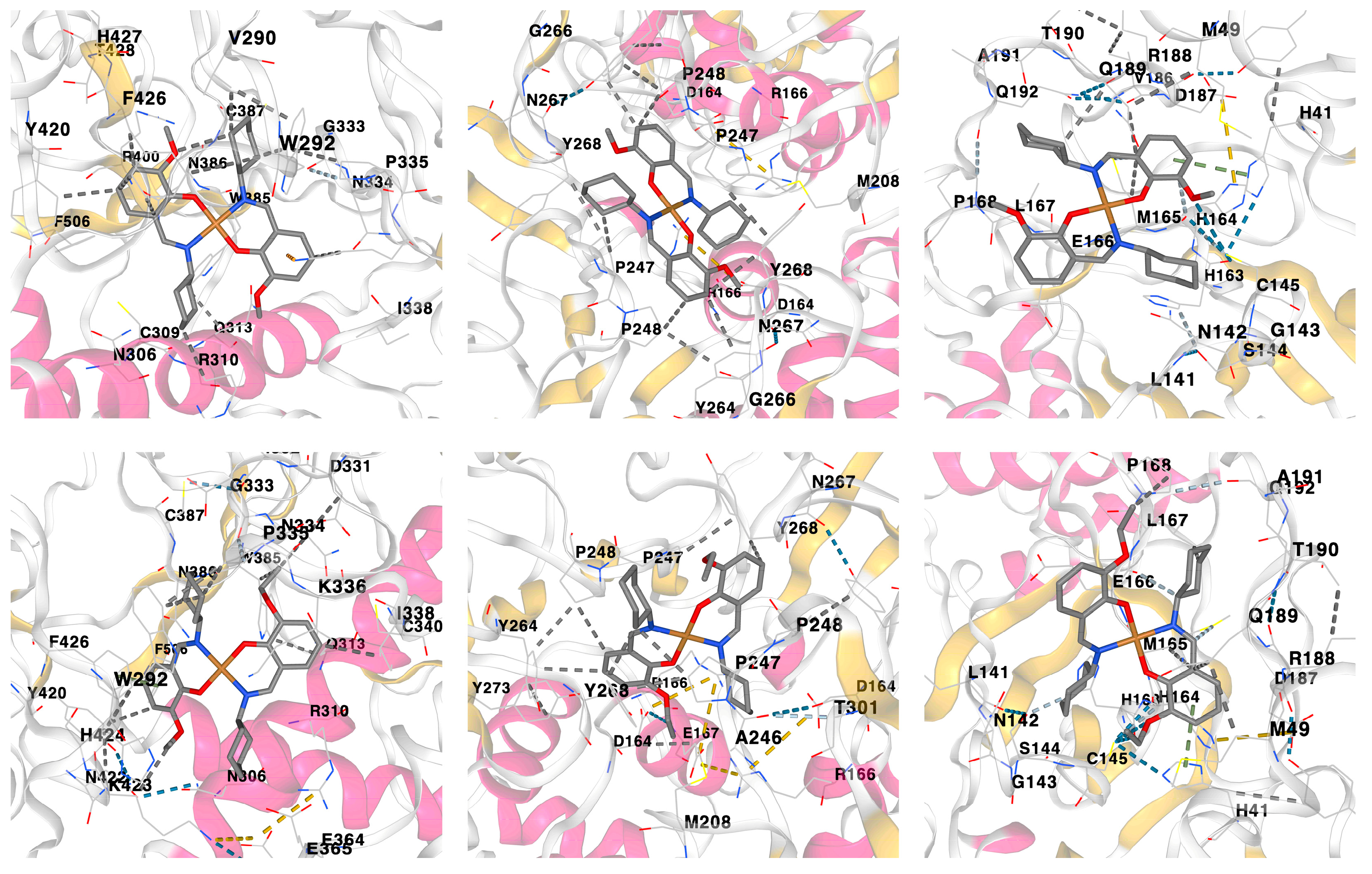
3.4. Molecular Docking
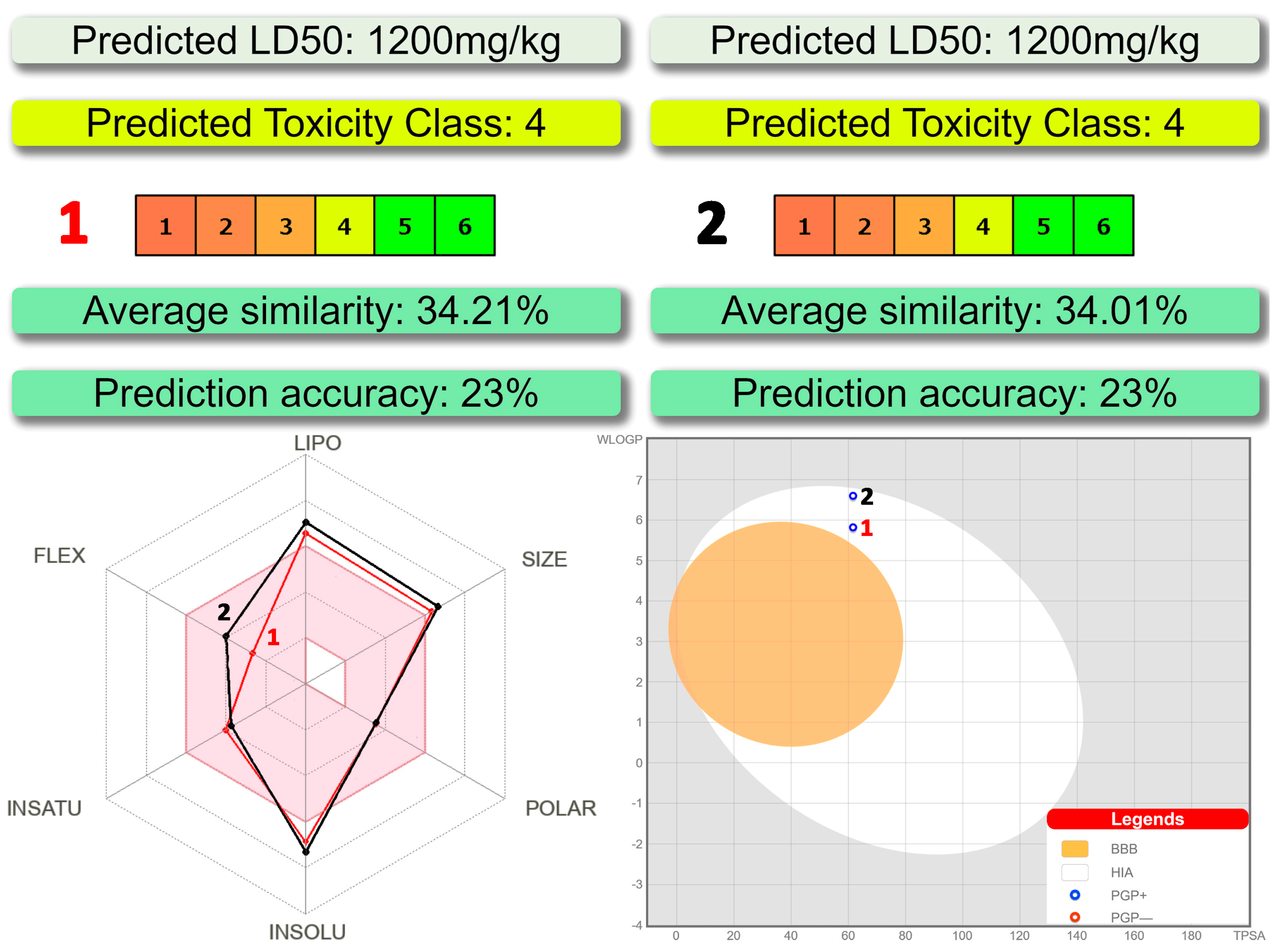
3.5. In Silico Drug-Likeness Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holm, R.H.; Kennepohl, P.; Solomon, E.I. Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev. 1996, 96, 2239–2314. [Google Scholar] [CrossRef] [PubMed]
- Klinman, J.P. Mechanisms Whereby Mononuclear Copper Proteins Functionalize Organic Substrates. Chem. Rev. 1996, 96, 2541–2561. [Google Scholar] [CrossRef]
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 7 February 2023).
- Andreou, A.; Trantza, S.; Filippou, D.; Sipsas, M.; Tsiodras, S. COVID-19: The potential role of copper and N-acetylcysteine (NAC) in a combination of candidate antiviral treatments against SARS-CoV-2. Vivo 2020, 34, 1567–1588. [Google Scholar] [CrossRef]
- Raha, S.; Mallick, R.; Basak, S.; Duttaroy, A.K. Is copper beneficial for COVID-19 patients? Med. Hypotheses 2020, 142, 10981. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.A.; Zuñiga, J.M. The use of copper to help prevent transmission of SARS-coronavirus and influenza viruses. A general review. Diagn. Microbiol. Infect. Dis. 2020, 98, 115176. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Keck, J.G.; Lien, E.J.; Lai, M.M.C. Design, synthesis, testing, and quantitative structure-activity relationship analysis of substituted salicylaldehyde Schiff bases of 1-amino-3-hydroxyguanidine tosylate as new antiviral agents against coronavirus. J. Med. Chem. 1990, 33, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Mir, J.M.; Majid, S.A.; Shalla, A.H. Enhancement of Schiff base biological efficacy by metal coordination and introduction of metallic compounds as anticovid candidates: A simple overview. Rev. Inorg. Chem. 2021, 41, 199–211. [Google Scholar] [CrossRef]
- Abd El-Lateef, H.M.; El-Dabea, T.; Khalaf, M.M.; Abu-Dief, A.M. Development of Metal Complexes for Treatment of Coronaviruses. Int. J. Mol. Sci. 2022, 23, 6418. [Google Scholar] [CrossRef]
- Soroceanu, A.; Bargan, A. Advanced and Biomedical Applications of Schiff-Base Ligands and Their Metal Complexes: A Review. Crystals 2022, 12, 1436. [Google Scholar] [CrossRef]
- Sinn, E.; Harris, C.M. Schiff base metal complexes as ligands. Coord. Chem. Rev. 1969, 4, 391–422. [Google Scholar] [CrossRef]
- Liu, X.; Manzur, C.; Novoa, N.; Celedón, S.; Carillo, D.; Hamon, J.-R. Multidentate unsymmetrically-substituted Schiff bases and their metal complexes: Synthesis, functional materials properties, and applications to catalysis. Coord. Chem. Rev. 2018, 357, 144–172. [Google Scholar] [CrossRef]
- Pessoa, J.C.; Correia, I. Salan vs. salen metal complexes in catalysis and medicinal applications: Virtues and pitfalls. Coord. Chem. Rev. 2019, 388, 227–247. [Google Scholar] [CrossRef]
- Liu, X.; Hamon, J.-R. Recent developments in penta-, hexa- and heptadentate Schiff base ligands and their metal complexes. Coord. Chem. Rev. 2019, 389, 94–118. [Google Scholar] [CrossRef]
- Freire, C.; Nunes, M.; Pereira, C.; Fernandes, D.M.; Peixoto, A.F.; Rocha, M. Metallo(salen) complexes as versatile building blocks for the fabrication of molecular materials and devices with tuned properties. Coord. Chem. Rev. 2019, 394, 104–134. [Google Scholar] [CrossRef]
- Verma, C.; Quraishi, M.A. Recent progresses in Schiff bases as aqueous phase corrosion inhibitors: Design and applications. Coord. Chem. Rev. 2021, 446, 214105. [Google Scholar] [CrossRef]
- Pervaiz, M.; Sadiq, S.; Sadiq, A.; Younas, U.; Ashraf, A.; Saeed, Z.; Zuber, M.; Adnan, A. Azo-Schiff base derivatives of transition metal complexes as antimicrobial agents. Coord. Chem. Rev. 2021, 447, 214128. [Google Scholar] [CrossRef]
- Kaur, M.; Kumar, S.; Yusuf, M.; Lee, J.; Brown, R.J.C.; Kim, K.-H.; Malik, A.K. Post-synthetic modification of luminescent metal-organic frameworks using schiff base complexes for biological and chemical sensing. Coord. Chem. Rev. 2021, 449, 214214. [Google Scholar] [CrossRef]
- Boswell, J.S.; Reedy, B.J.; Kulathila, R.; Merkler, D.; Blackburn, N.J. Structural Investigations on the Coordination Environment of the Active-Site Copper Centers of Recombinant Bifunctional Peptidylglycine α-Amidating Enzyme. Biochemistry 1996, 35, 12241–12250. [Google Scholar] [CrossRef]
- Shiryaev, A.A.; Burkhanova, T.M.; Mahmoudi, G.; Babashkina, M.G.; Safin, D.A. Photophysical properties of ethyl N-(5-bromosalicylidene)glycinate and ethyl N-(5-nitrosalicylidene)glycinate in CH2Cl2. J. Lumin. 2020, 226, 117454. [Google Scholar] [CrossRef]
- Shapenova, D.S.; Shiryaev, A.A.; Bolte, M.; Kukułka, M.; Szczepanik, D.W.; Hooper, J.; Babashkina, M.G.; Mahmoudi, G.; Mitoraj, M.P.; Safin, D.A. Resonance Assisted Hydrogen Bonding Phenomenon Unveiled from Both Experiment and Theory–an Example of New Family of Ethyl N-salicylideneglycinate Dyes. Chem. Eur. J. 2020, 26, 12987–12995. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Bolte, M.; Ptaszek, A.L.; Kukułka, M.; Mitoraj, M.P. Novel sterically demanding Schiff base dyes: An insight from experimental and theoretical calculations. J. Lumin. 2021, 238, 118264. [Google Scholar] [CrossRef]
- Babashkina, M.G.; Panova, E.V.; Alkhimova, L.E.; Safn, D.A. Salen: Insight into the crystal structure, Hirshfeld surface analysis, optical properties, DFT, and molecular docking studies. Polycycl. Aromat. Comp. 2022. [Google Scholar] [CrossRef]
- Sharov, A.V.; Burkhanova, T.M.; Taskın Tok, T.; Babashkina, M.G.; Safn, D.A. Computational analysis of molnupiravir. Int. J. Mol. Sci. 2022, 23, 1508. [Google Scholar] [CrossRef]
- Burkhanova, T.M.; Krysantieva, A.I.; Babashkina, M.G.; Konyaeva, I.A.; Monina, L.N.; Goncharenko, A.N.; Safin, D.A. In silico analyses of betulin: DFT studies, corrosion inhibition properties, ADMET prediction and molecular docking with a series of SARS-CoV-2 and monkeypox proteins. Struct. Chem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Cline, S.J.; Wasson, J.R.; Hatfield, W.E.; Hodgson, D.J. Structure and Spectroscopic Properties of Bis(N-cyclohexyl-3-methoxysalicylideneiminato)copper(II). J. Chem. Soc., Dalton Trans. 1978, 9, 1051–1057. [Google Scholar] [CrossRef]
- Lin, H.-W. Crystal structure of trans-bis(N-cyclohexyl-3-methoxysalicylideneiminato)cobalt(II), Co(C14H18NO2)2. Z. Kristallogr. NCS 2006, 221, 485–486. [Google Scholar] [CrossRef]
- Wang, C. Bis[2-(cyclohexyliminomethyl)-6-methoxyphenolato]nickel(II). Acta Cryst. 2006, E62, m1754–m1755. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Robeyns, K.; Mitoraj, M.P.; Kubisiak, P.; Garcia, Y. Influence of the Homopolar Dihydrogen Bonding C–H⋅⋅⋅H–C on Coordination Geometry: Experimental and Theoretical Studies. Chem. Eur. J. 2015, 21, 16679–16687. [Google Scholar] [CrossRef]
- ProTox-II-Prediction Of Toxicity Of Chemicals. Available online: https://tox-new.charite.de/protox_II/index.php?site=home (accessed on 7 February 2023).
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, w257–w263. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Diana, A.; Zoete, V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Tok, T.T.; Tatar, G. Structures and functions of coronavirus proteins: Molecular modeling of viral nucleoprotein. Int. J. Virol. Infect. Dis. 2017, 2, 001–007. [Google Scholar]
- Tok, T.T.; Gowder, S.J.T. An updated review on Covid-19 with special reference to structural elucidation and functional properties. Biomed. J. Sci. Tech. Res. 2020, 31, 24345–24351. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Anwar, S.; Amani, S.; Khan, M.S.; Husain, F.M.; Rehman, M.T.; Islam, A.; Hassan, M.I. Potential drug targets of SARSCoV-2: From genomics to therapeutics. Int. J. Biol. Macromol. 2021, 177, 1–9. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Tounge, B.A.; Bembenek, S.D. Ligand Binding Efficiency: Trends, Physical Basis, and Implications. J. Med. Chem. 2008, 51, 2432–2438. [Google Scholar] [CrossRef]
- Schultes, S.; de Graaf, C.; Haaksma, E.E.; de Esch, I.J.P.; Leurs, R.; Krämer, O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discov. Today Technol. 2010, 7, e157–e162. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmaco. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligands efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Abdul-Hammed, M.; Adedotun, I.O.; Falade, V.A.; Adepoju, A.J.; Olasupo, S.B.; Akinboade, M.W. Target-based drug discovery, ADMET profiling and bioactivity studies of antibiotics as potential inhibitors of SARS-CoV-2 main protease (Mpro). VirusDis. 2021, 32, 642–656. [Google Scholar] [CrossRef]
- LogP-Octanol-Water Partition Coefficient. Available online: https://www.molinspiration.com/services/logp.html (accessed on 7 February 2023).
- Sheldrick, G.M. SADABS; Bruker AXS Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Cao Lab. CB-Dock2-Cavity Detection Guided Blind Docking. Available online: https://cadd.labshare.cn/cb-dock2/php/index.php (accessed on 7 February 2023).
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.-X.; Cao, Y. CB-Dock2: Improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Rose, Y.; Duarte, J.M.; Lowe, R.; Segura, J.; Bi, C.; Bhikadiya, C.; Chen, L.; Rose, A.S.; Bittrich, S.; Burley, S.K.; et al. RCSB protein data bank: Architectural advances towards integrated searching and efficient access to macromolecular structure data from the PDB archive. J. Mol. Biol. 2021, 433, 166704. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 1 | 2 | ||
|---|---|---|---|---|---|
| Bond length | |||||
| Cu1–N1 | 2.036(5) | 2.0008(17) | C1–N1 | 1.292(7) | 1.287(3) |
| Cu1–O1 | 1.875(4) | 1.8970(13) | C1–O1 | 1.311(7) | 1.304(2) |
| Bond angle | |||||
| N1–Cu1–O1 | 91.55(19) | 91.19(6) | N1–Cu1–N1′ | 180.00 | 179.00(6) |
| N1–Cu1–O1′ | 88.45(19) | 88.85(6) | O1–Cu1–O1′ | 180.00 | 175.27(6) |
| Dihedral angle | |||||
| N1–Cu1–O1–C1 | 8.3(5) | 25.20(16) | O1–Cu1–N1′–C1′ | −170.4(5) | −153.51(15) |
| O1–Cu1–N1–C1 | −9.6(5) | −21.76(15) | C6H3∙∙∙C6H3 | 0.00 | 44.35 |
| N1–Cu1–O1′–C′1 | 171.7(5) | 155.80(16) | |||
| Ligand Efficiency Score | Initial Ligand * | 1 | 2 |
|---|---|---|---|
| Main protease (Mpro) (PDB code 6LU7) | |||
| Binding energy (BE, kcal/mol) | −7.4(1) | −8.6(0) | −7.5(1) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 3.76 | 0.50 | 3.18 |
| miLogP | 2.32 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.151 | 0.246 | 0.203 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.233 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.649 | 0.821 | 0.707 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | 15.362 | 21.855 | 30.241 |
| Papain-like protease (PLpro) (PDB code 6WUU) | |||
| Binding energy (BE, kcal/mol) | −8.6(1) | −8.7(0) | −7.9(0) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 0.50 | 0.42 | 1.62 |
| miLogP | −1.61 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.239 | 0.249 | 0.214 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.293 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.816 | 0.831 | 0.745 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | −6.740 | 21.603 | 28.710 |
| Nonstructural protein 3 (Nsp3_range 207–379-AMP) (PDB code 6W6Y) | |||
| Binding energy (BE, kcal/mol) | −7.2(0) | −7.4(1) | −7.5(1) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 5.28 | 3.76 | 3.18 |
| miLogP | −1.52 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.313 | 0.211 | 0.203 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.418 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.749 | 0.706 | 0.707 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | −4.856 | 25.399 | 30.241 |
| Nonstructural protein 3 (Nsp3_range 207–379-MES) (PDB code 6W6Y) | |||
| Binding energy (BE, kcal/mol) | −5.8(0) | −7.7(0) | −7.4(0) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 56.05 | 2.27 | 3.76 |
| miLogP | −4.08 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.483 | 0.220 | 0.200 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.669 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.723 | 0.735 | 0.698 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | −8.441 | 24.409 | 30.650 |
| RdRp-RNA (PDB code 7BV2) | |||
| Binding energy (BE, kcal/mol) | −6.6(0) | −7.2(0) | −6.6(0) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 14.53 | 5.28 | 14.53 |
| miLogP | −1.55 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.264 | 0.206 | 0.178 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.391 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.676 | 0.687 | 0.622 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | 5.871 | 26.104 | 34.365 |
| Nonstructural protein 14 (N7-MTase) (PDB code 5C8S) | |||
| Binding energy (BE, kcal/mol) | −10.7(0) | −10.4(0) | −9.6(0) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 0.01 | 0.02 | 0.09 |
| miLogP | −4.67 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.214 | 0.297 | 0.259 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.230 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.932 | 0.993 | 0.905 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | −21.822 | 18.072 | 23.626 |
| Nonstructural protein 15 (endoribonuclease) (PDB code 6WLC) | |||
| Binding energy (BE, kcal/mol) | −7.5(1) | −7.8(0) | −7.6(0) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 3.18 | 1.92 | 2.69 |
| miLogP | −2.76 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.357 | 0.223 | 0.205 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.449 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.795 | 0.745 | 0.716 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | −7.728 | 24.096 | 29.843 |
| Nonstructural protein 16 (GTA site) (PDB code 6WVN) | |||
| Binding energy (BE, kcal/mol) | −8.7(1) | −7.7(0) | −6.9(0) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 0.42 | 2.27 | 8.75 |
| miLogP | −5.69 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.171 | 0.220 | 0.186 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.226 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.754 | 0.735 | 0.650 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | −33.355 | 24.409 | 32.871 |
| Nonstructural protein 16 (MGP site) (PDB code 6WVN) | |||
| Binding energy (BE, kcal/mol) | −6.7(0) | −6.3(0) | −6.3(1) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 12.27 | 24.10 | 24.10 |
| miLogP | −4.22 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.203 | 0.180 | 0.170 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.313 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.648 | 0.601 | 0.594 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | −20.785 | 29.833 | 36.002 |
| Nonstructural protein 16 (SAM site) (PDB code 6WVN) | |||
| Binding energy (BE, kcal/mol) | −7.3(1) | −7.2(1) | −7.3(1) |
| Inhibition constant (Ki = e(−BE/RT), μM) ** | 4.46 | 5.28 | 4.46 |
| miLogP | −5.01 | 5.37 | 6.13 |
| Ligand efficiency (LE = −BE/(Heavy atoms), kcal/(mol HA) | 0.270 | 0.206 | 0.197 |
| LE_Scale (0.0715 + 7.5328/(HA) + 25.7079/(HA2) − 361.4722/(HA3)) | 0.367 | 0.299 | 0.287 |
| Fit quality (FQ = LE/LE_Scale) | 0.736 | 0.687 | 0.688 |
| Ligand-efficiency-dependent lipophilicity (LELP = miLogP/LE) | −18.530 | 26.104 | 31.070 |
| Interaction | Distance (Å) | Bonding | Bonding Type |
|---|---|---|---|
| Nonstructural protein 14 (N7-MTase)–1 | |||
| D:CYS309–A:1 | 4.55383 | Hydrophobic | Alkyl |
| D:ARG310–A:1:C11 | 5.03726 | Hydrophobic | Alkyl |
| D:TRP292–A:1:C13 | 5.40534 | Hydrophobic | π∙∙∙Alkyl |
| D:TYR420–A:1:C4′ | 5.06410 | Hydrophobic | π∙∙∙Alkyl |
| D:PHE426–A:1 | 3.96141 | Hydrophobic | π∙∙∙Alkyl |
| Papain-like protease (PLpro)–1 | |||
| A:PRO248–A:1:C4 | 3.78832 | Hydrophobic | Alkyl |
| C:PRO248–A:1:C5′ | 4.73701 | Hydrophobic | Alkyl |
| C:TYR264–A:1:C5′ | 4.72694 | Hydrophobic | π∙∙∙Alkyl |
| Main protease (Mpro)–1 | |||
| A:1:C4′–A:HIS41 | 3.74205 | Hydrophobic | π∙∙∙Sigma |
| A:CYS145–A:1 | 4.99879 | Hydrophobic | Alkyl |
| A:1:C2–A:MET165 | 4.58696 | Hydrophobic | Alkyl |
| Nonstructural protein 14 (N7-MTase)–2 | |||
| A:2:C15–D:ASN334 | 3.58506 | Hydrogen Bond | Carbon Hydrogen Bond |
| A:2:C15–D:TRP385:O | 3.55155 | Hydrogen Bond | Carbon Hydrogen Bond |
| D:PRO335–A:2:C12 | 5.22535 | Hydrophobic | Alkyl |
| A:2:C15′–D:LYS423 | 4.53131 | Hydrophobic | Alkyl |
| D:TYR420–A:2:C5 | 5.24129 | Hydrophobic | π∙∙∙Alkyl |
| D:PHE426–A:2:C3 | 5.37133 | Hydrophobic | π∙∙∙Alkyl |
| A:2:C15–D:ASN334 | 4.03087 | Hydrophobic | π∙∙∙Alkyl |
| A:2:C15–D:TRP385:O | 5.14456 | Hydrophobic | π∙∙∙Alkyl |
| Papain-like protease (PLpro)–2 | |||
| A:PRO247–A:2:C13 | 5.16136 | Hydrophobic | Alkyl |
| A:PRO247–A:2 | 5.14188 | Hydrophobic | Alkyl |
| A:PRO248–A:2 | 4.46827 | Hydrophobic | Alkyl |
| C:PRO248–A:2 | 4.76755 | Hydrophobic | Alkyl |
| A:2:C14–A:MET208 | 4.24574 | Hydrophobic | Alkyl |
| C:TYR264–A:2:C4 | 4.56104 | Hydrophobic | π∙∙∙Alkyl |
| Main protease (Mpro)–2 | |||
| A:CYS145–A:2:O2 | 5.13188 | Hydrophobic | Alkyl |
| A:MET165–A:2:C1′ | 5.06805 | Hydrophobic | Alkyl |
| A:PRO168–A:2:C15 | 5.23131 | Hydrophobic | Alkyl |
| A:2:C15′–A:PRO168 | 4.16653 | Hydrophobic | Alkyl |
| A:2–A:MET49 | 5.08585 | Hydrophobic | Alkyl |
| A:2–A:CYS145 | 4.80241 | Hydrophobic | Alkyl |
| A:HIS41–A:2:C2 | 4.63051 | Hydrophobic | π∙∙∙Alkyl |
| A:HIS41–A:2:C15 | 3.89620 | Hydrophobic | π∙∙∙Alkyl |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panova, E.V.; Voronina, J.K.; Safin, D.A. Copper(II) Chelates of Schiff Bases Enriched with Aliphatic Fragments: Synthesis, Crystal Structure, In Silico Studies of ADMET Properties and a Potency against a Series of SARS-CoV-2 Proteins. Pharmaceuticals 2023, 16, 286. https://doi.org/10.3390/ph16020286
Panova EV, Voronina JK, Safin DA. Copper(II) Chelates of Schiff Bases Enriched with Aliphatic Fragments: Synthesis, Crystal Structure, In Silico Studies of ADMET Properties and a Potency against a Series of SARS-CoV-2 Proteins. Pharmaceuticals. 2023; 16(2):286. https://doi.org/10.3390/ph16020286
Chicago/Turabian StylePanova, Elizaveta V., Julia K. Voronina, and Damir A. Safin. 2023. "Copper(II) Chelates of Schiff Bases Enriched with Aliphatic Fragments: Synthesis, Crystal Structure, In Silico Studies of ADMET Properties and a Potency against a Series of SARS-CoV-2 Proteins" Pharmaceuticals 16, no. 2: 286. https://doi.org/10.3390/ph16020286