
An Overview of 1,2,3-triazole-Containing Hybrids and Their Potential Anticholinesterase Activities
 , , and
, , and
Abstract
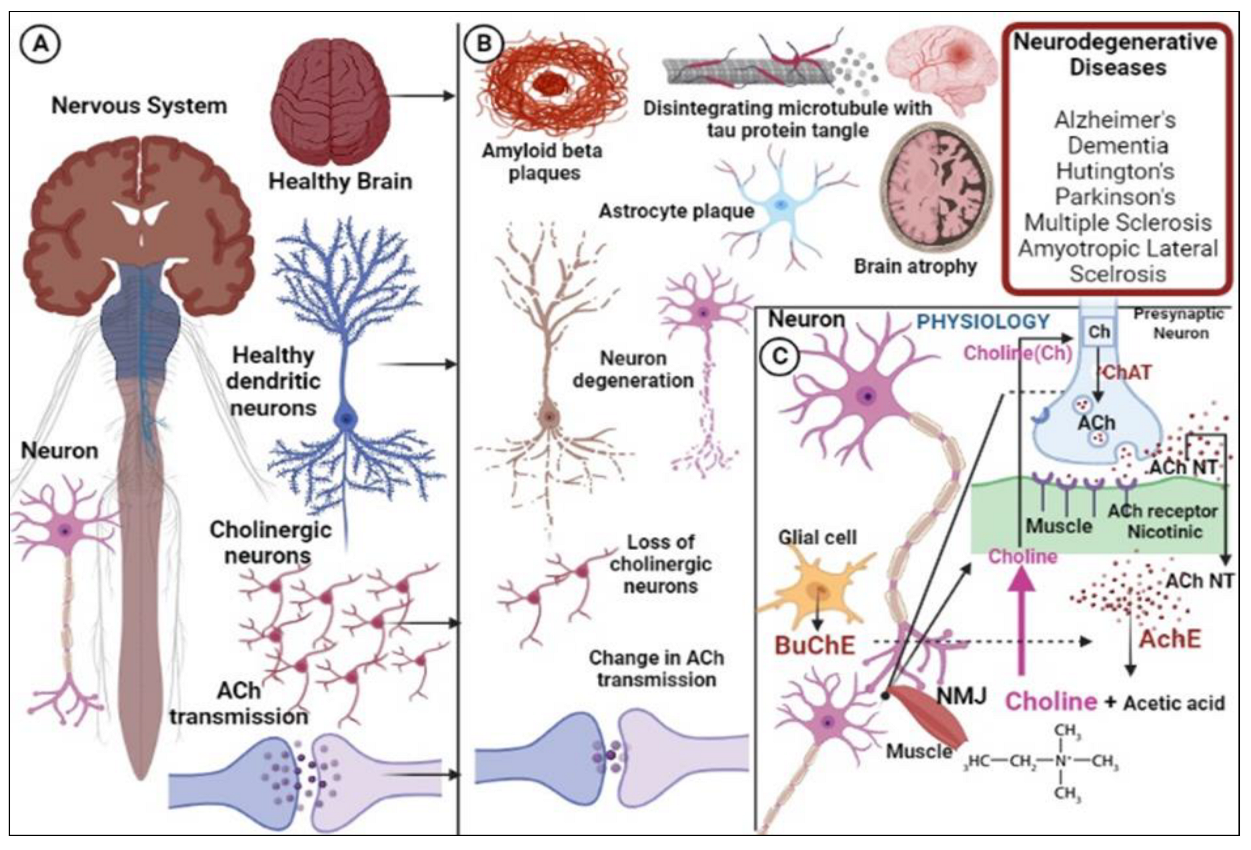
:1. Introduction
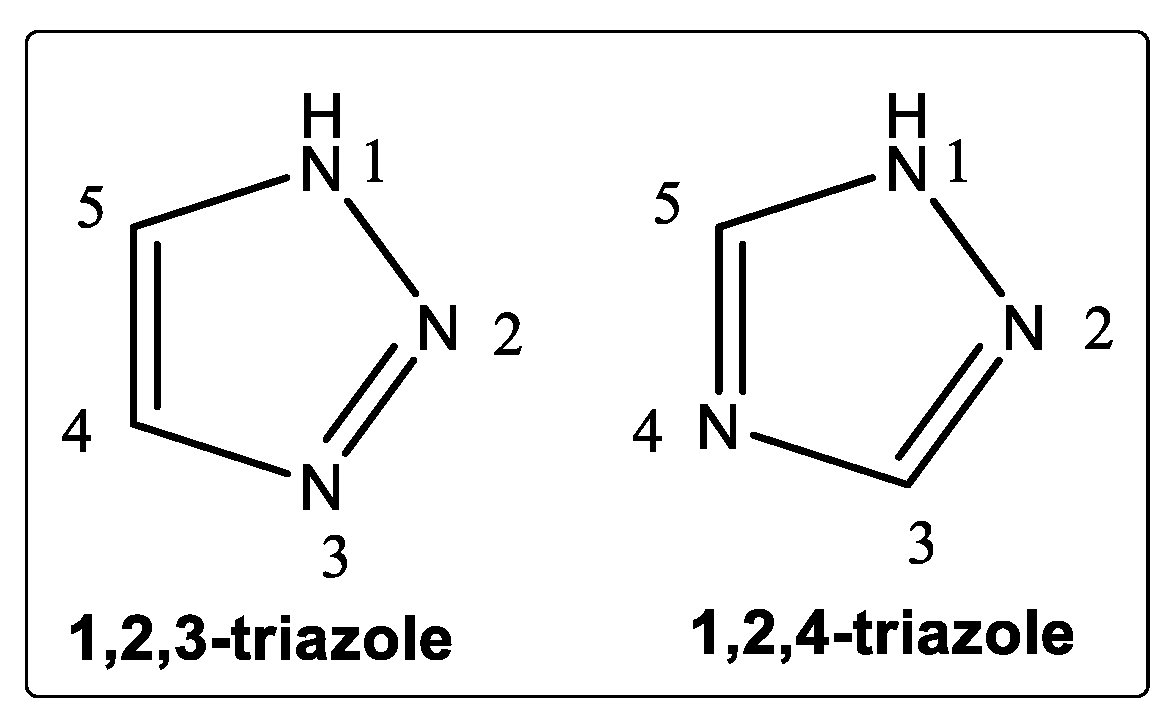
2. Chemistry and the Importance of the Triazole Ring System
2.1. Various Synthetic Routes for the Synthesis of 1,2,3-triazole Scaffold
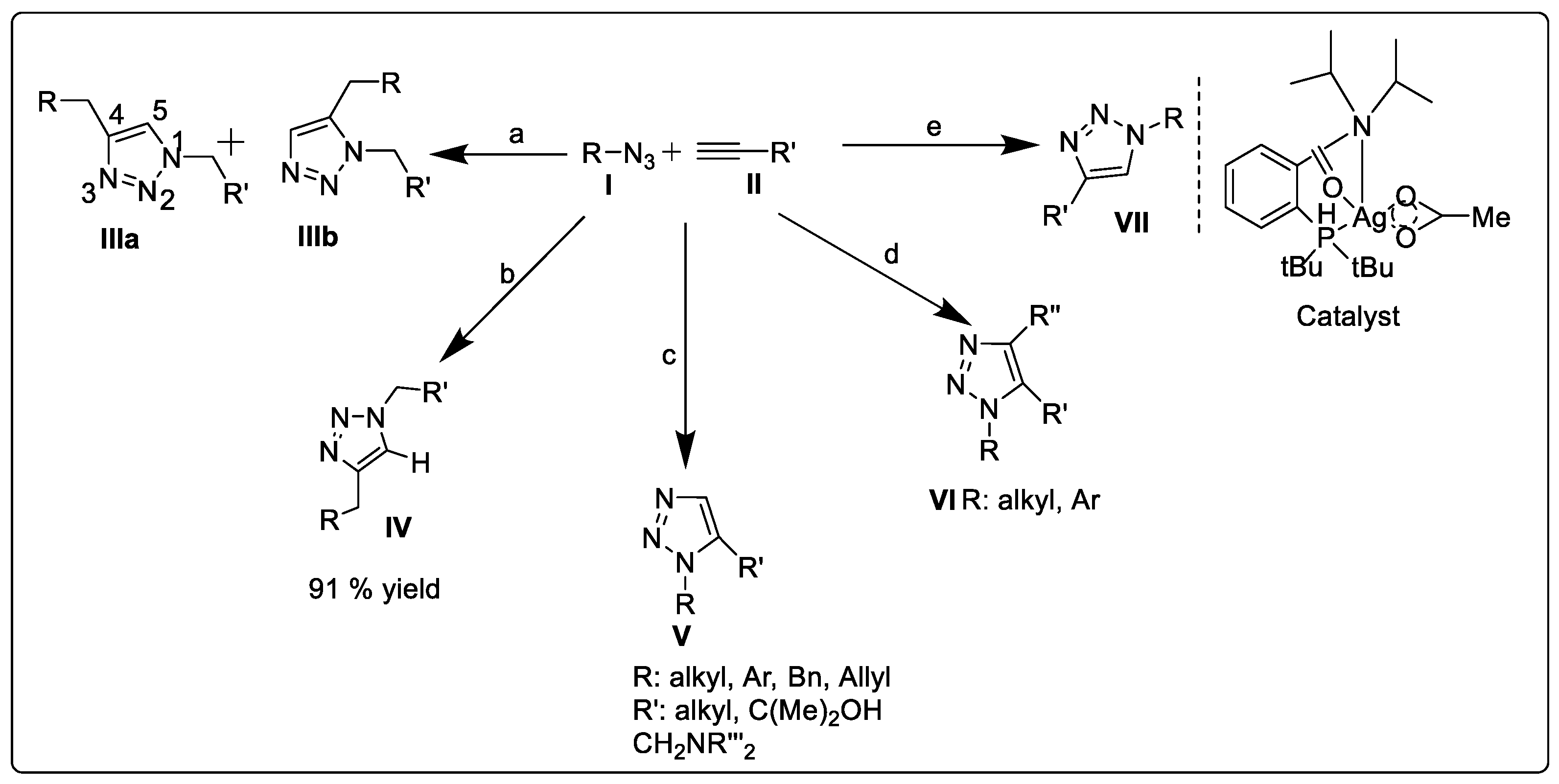
2.1.1. Huisgen 1,3-dipolar Cycloaddition
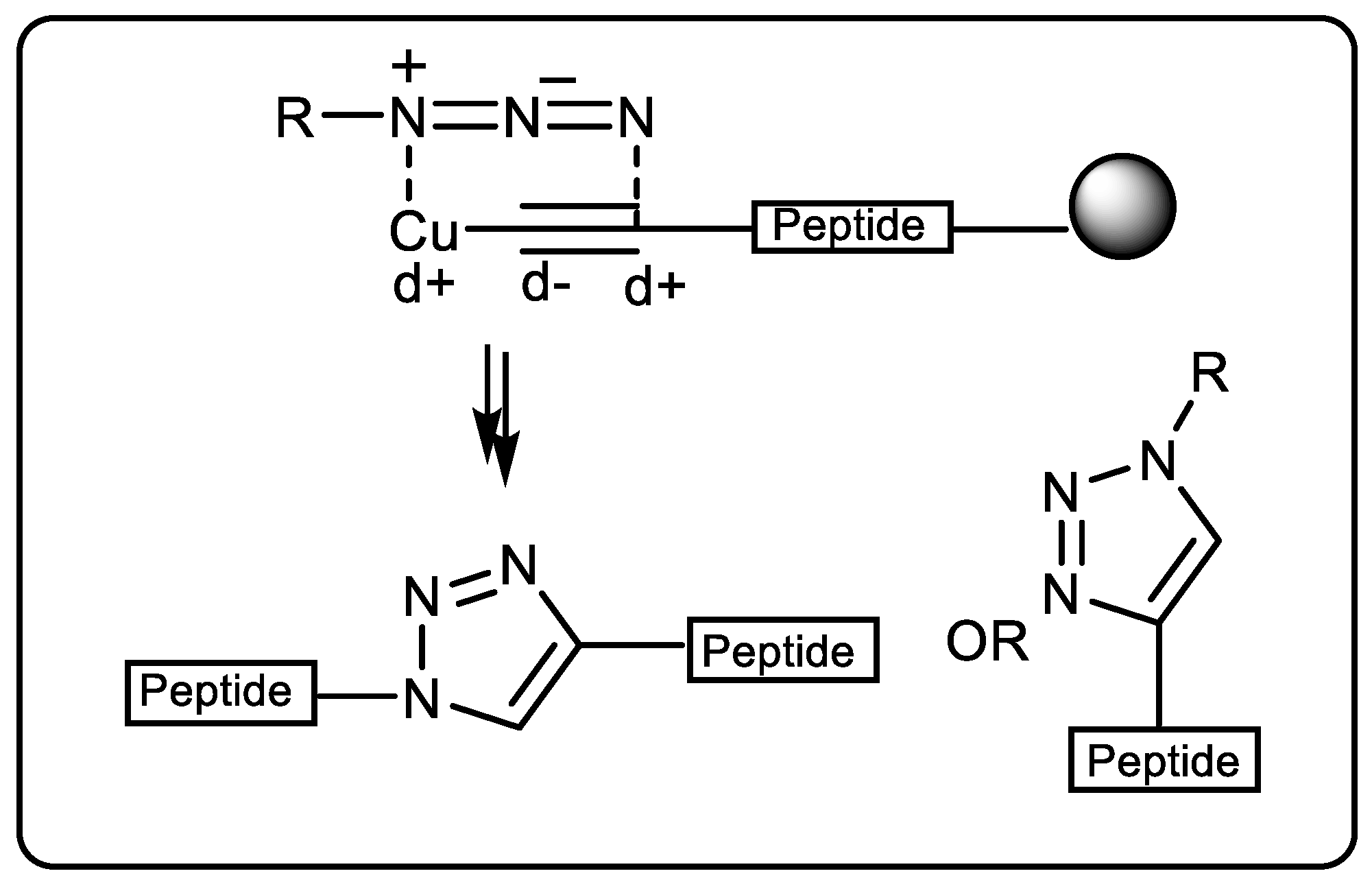
2.1.2. Copper-Catalyzed Azide–Alkyne Cycloaddition (Click Chemistry)
2.1.3. Ruthenium-Catalyzed Azide–Alkyne Cycloaddition (RuAAC)
2.1.4. Silver-Catalyzed Azide–Alkyne Cycloaddition (AgAAC)
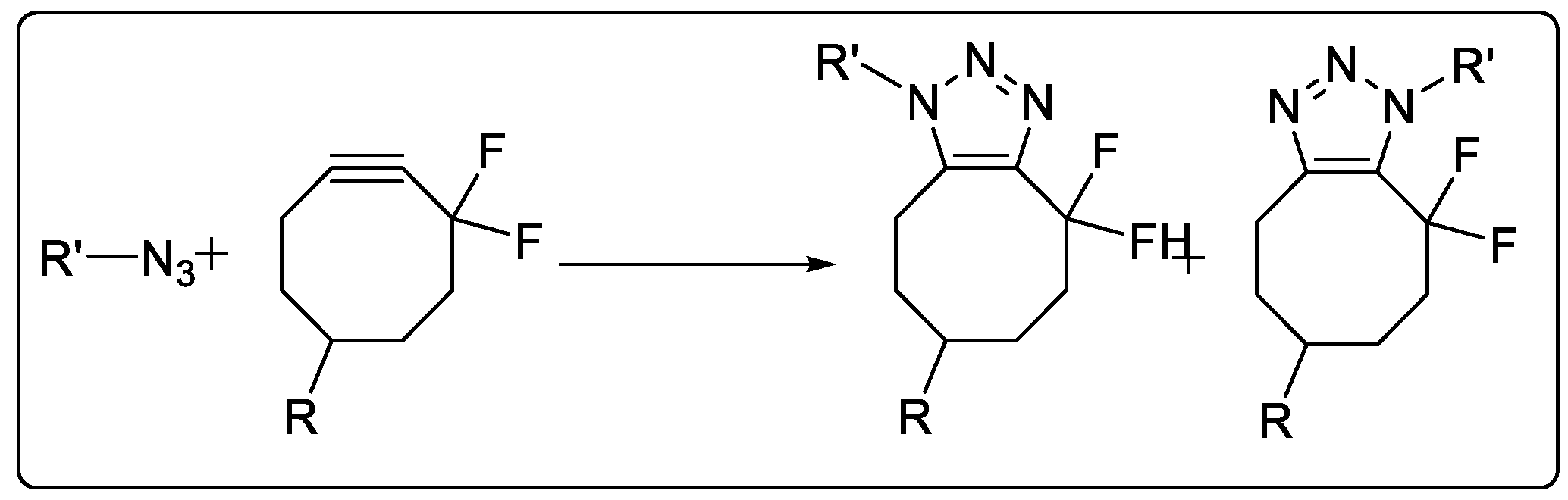
2.1.5. Strain-Promoted Azide–Alkyne Cycloaddition
2.1.6. Metal-Free Synthesis of 1,2,3-triazoles
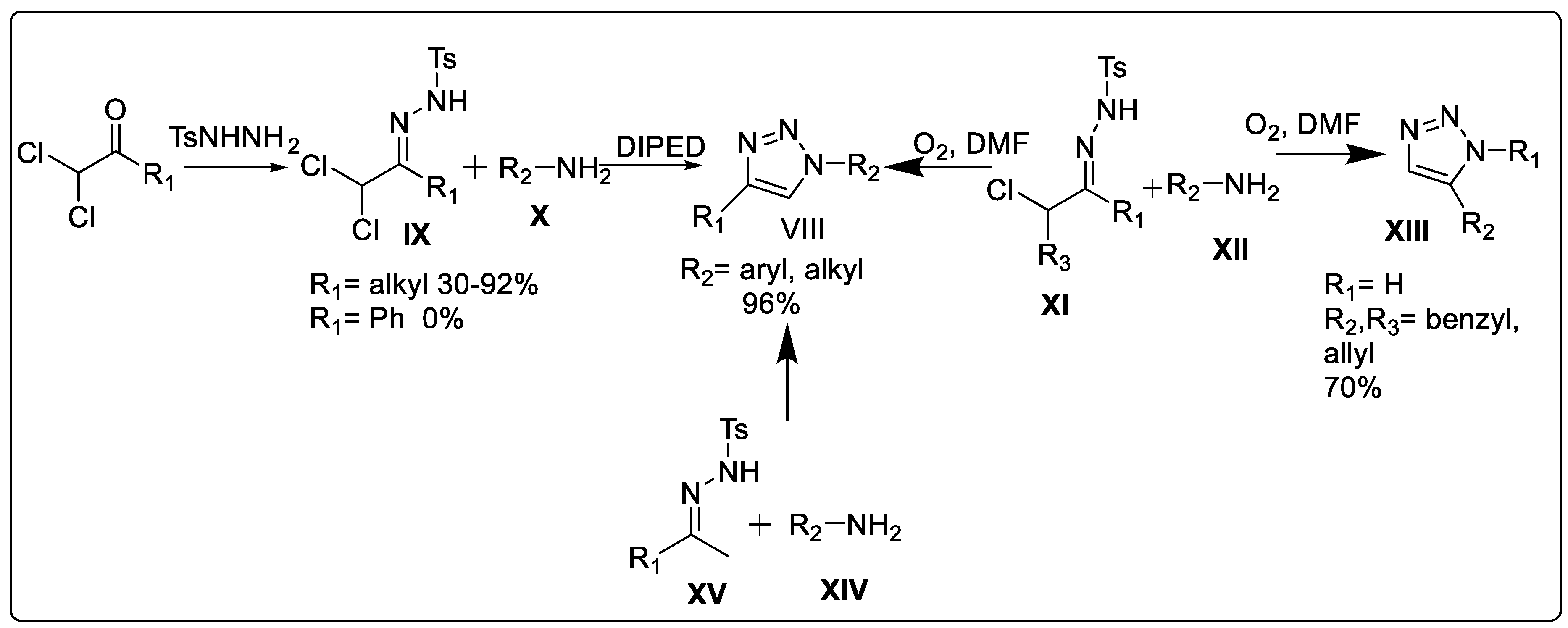
Synthesis of 1,2,3-triazole through α,α-dichlorotosylhydrazones
Synthesis of 1,2,3-triazole through α-chlorotosylhydrazones
Synthesis of 1,2,3-triazole through N-tosylhydrazones
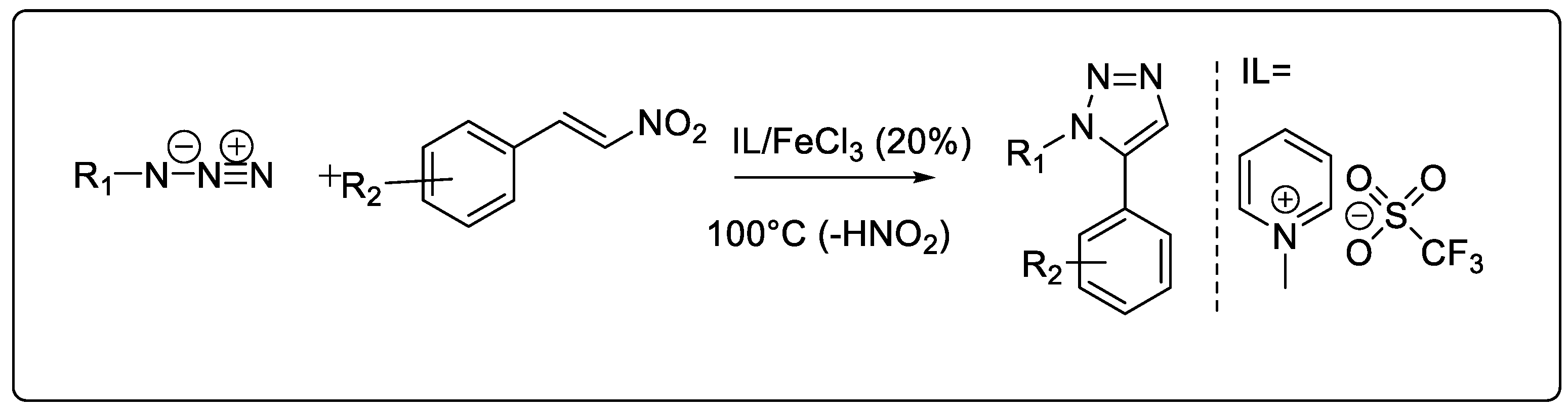
2.1.7. Ionic Liquid-Catalyzed Synthesis
2.2. Application of Triazoles in the Synthesis of Other Heterocyclic Compounds
3. Conjugates of 1,2,3-triazole as Potential AChE and BuChE Inhibitors
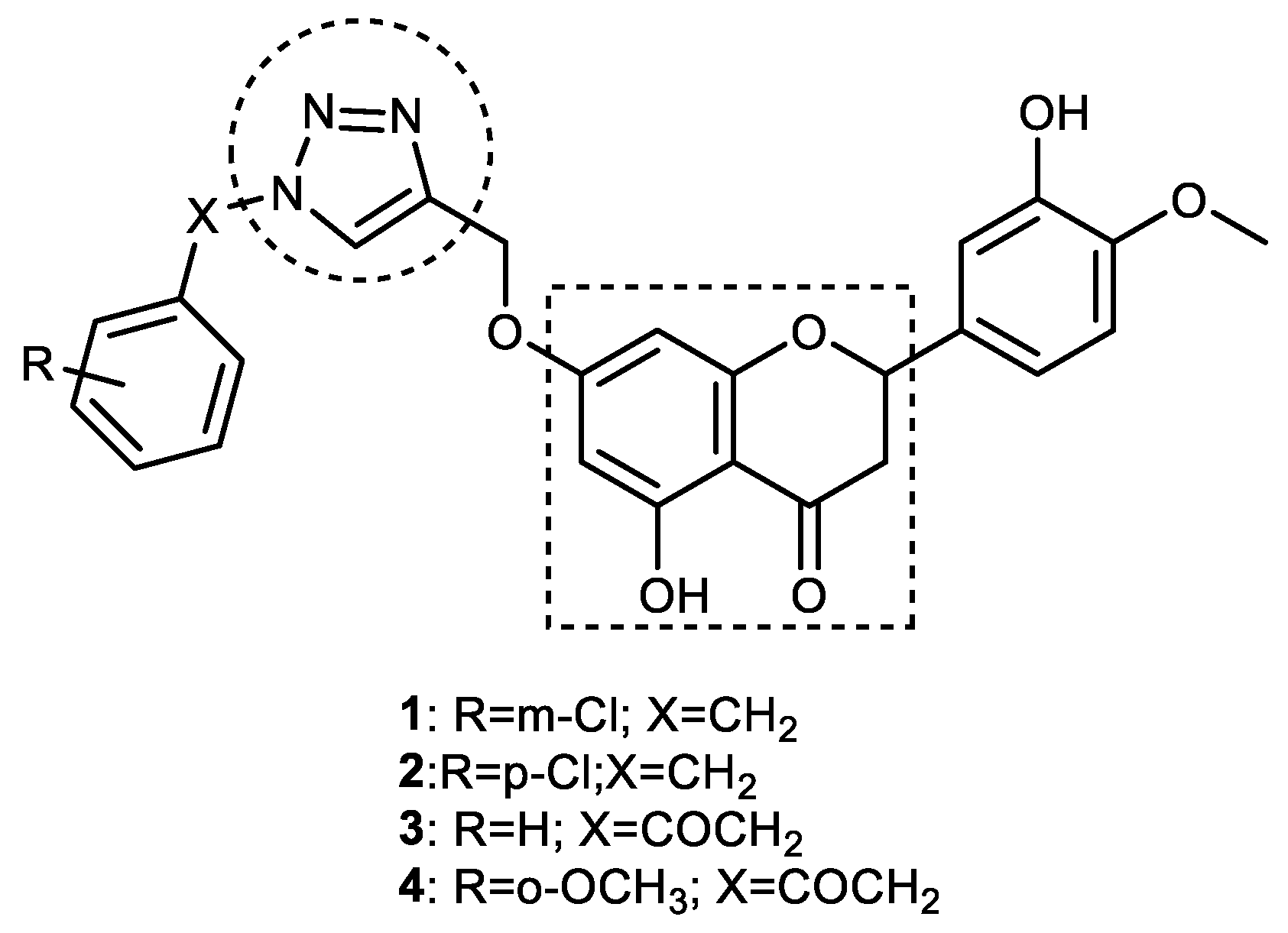
3.1. Hesperetin–1,2,3-triazole Hybrids
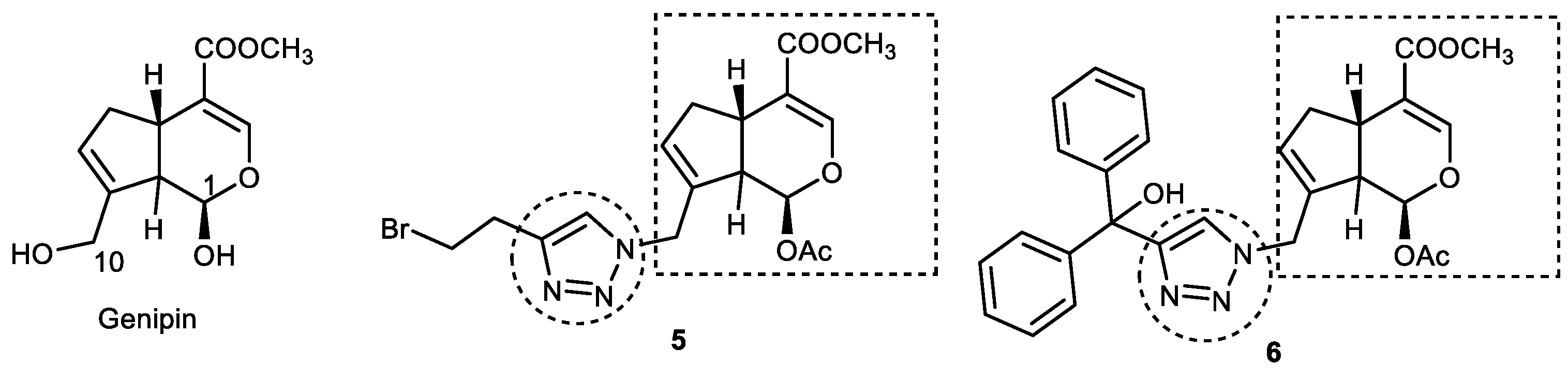
3.2. Genipin–1,2,3-triazole Hybrids
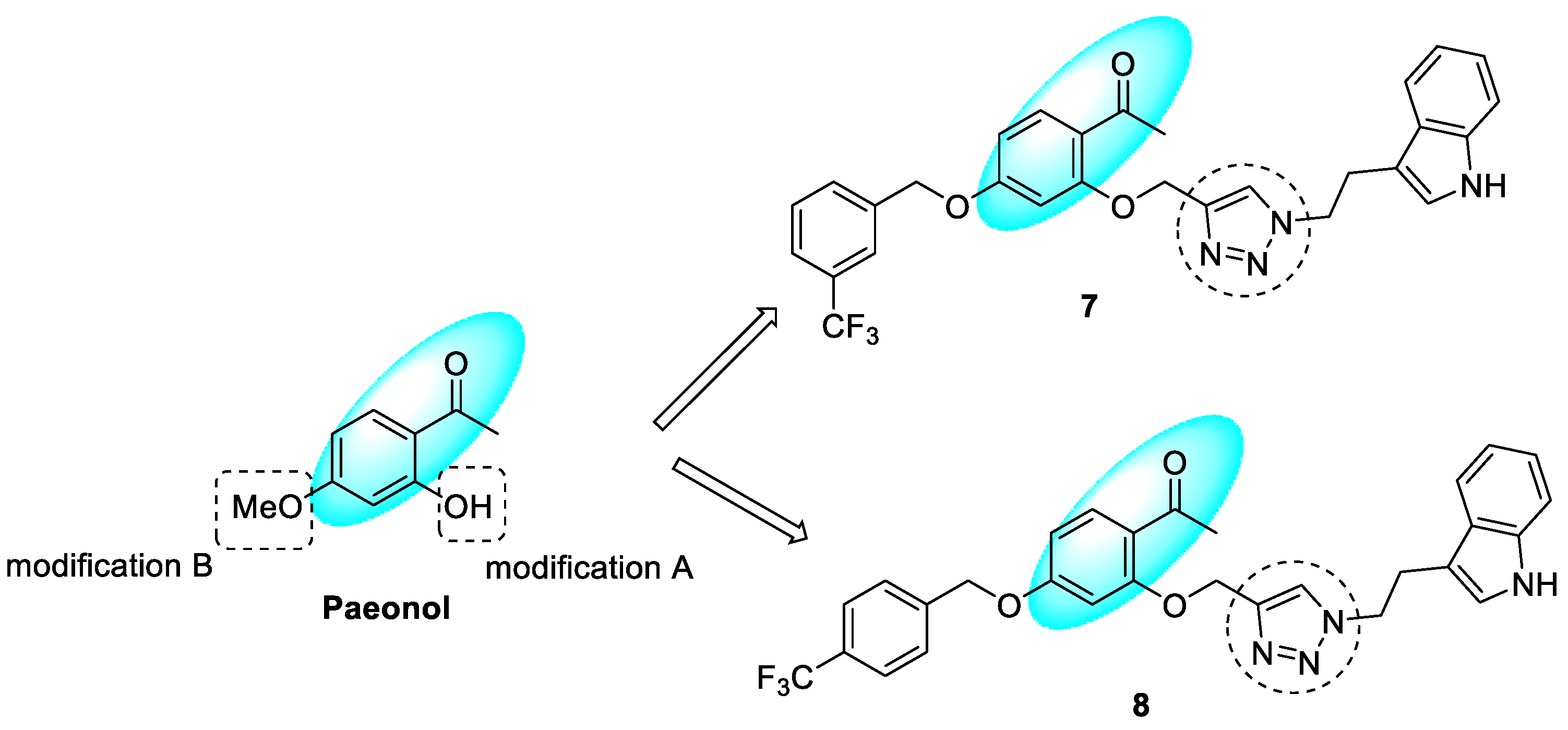
3.3. Paeonol–1,2,3-triazole Hybrids
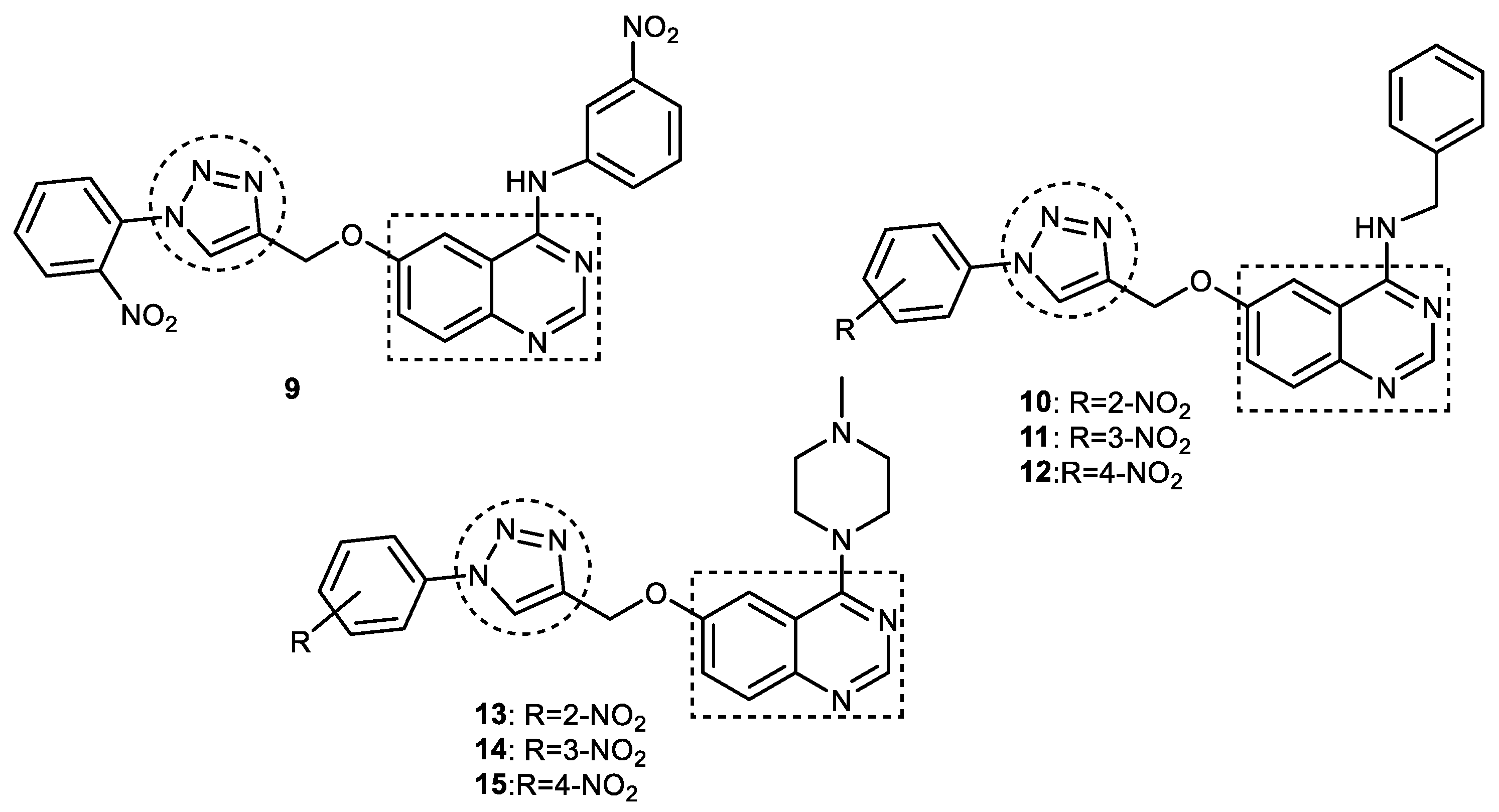
3.4. Quinazoline–1,2,3-triazole Hybrids
3.5. Quinoline-1,2,3-triazole Derivatives
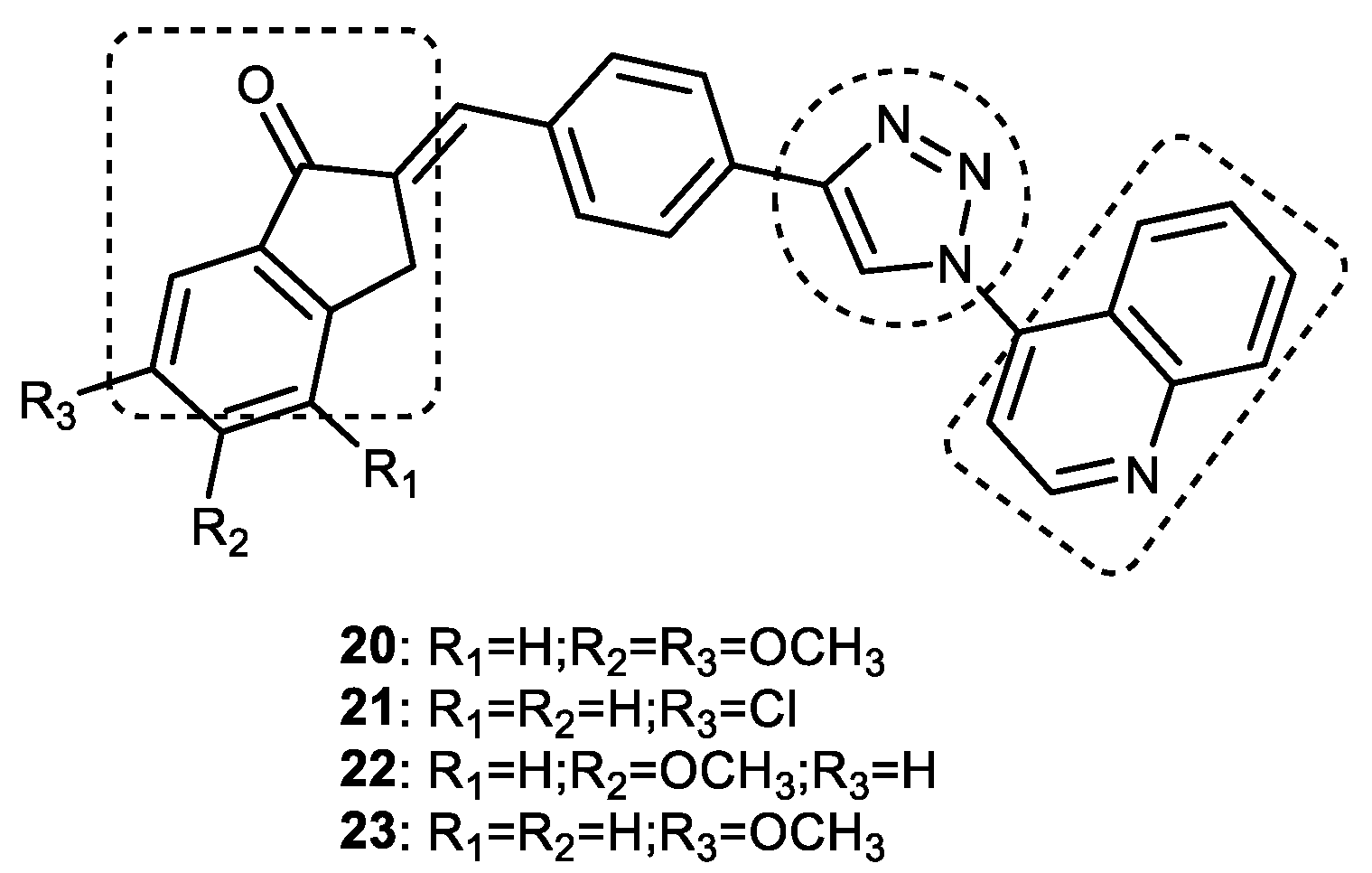
Quinoline–1,2,3-triazole–Indanone Conjugates
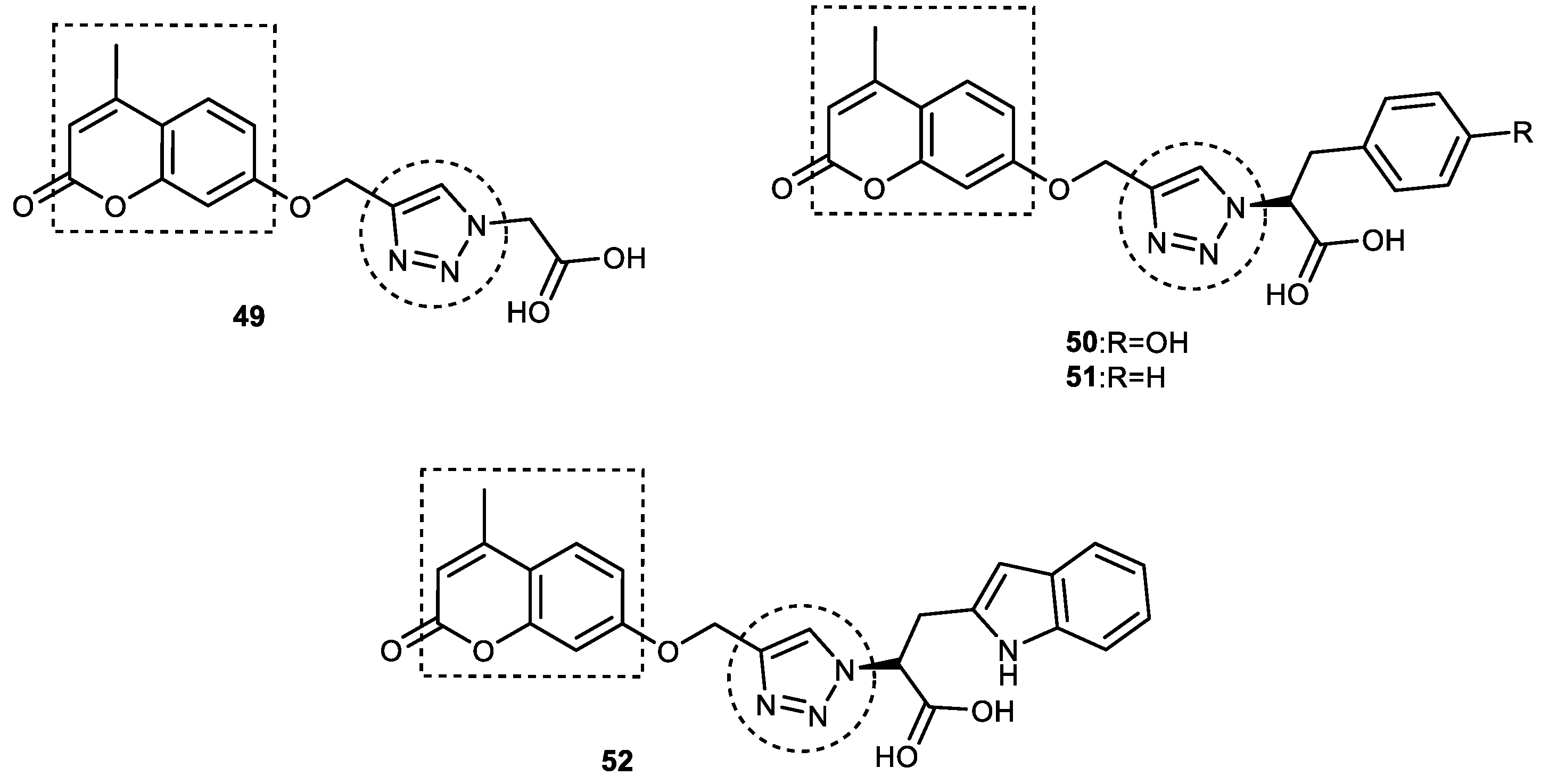
3.6. Coumarin–1,2,3-triazole Hybrids
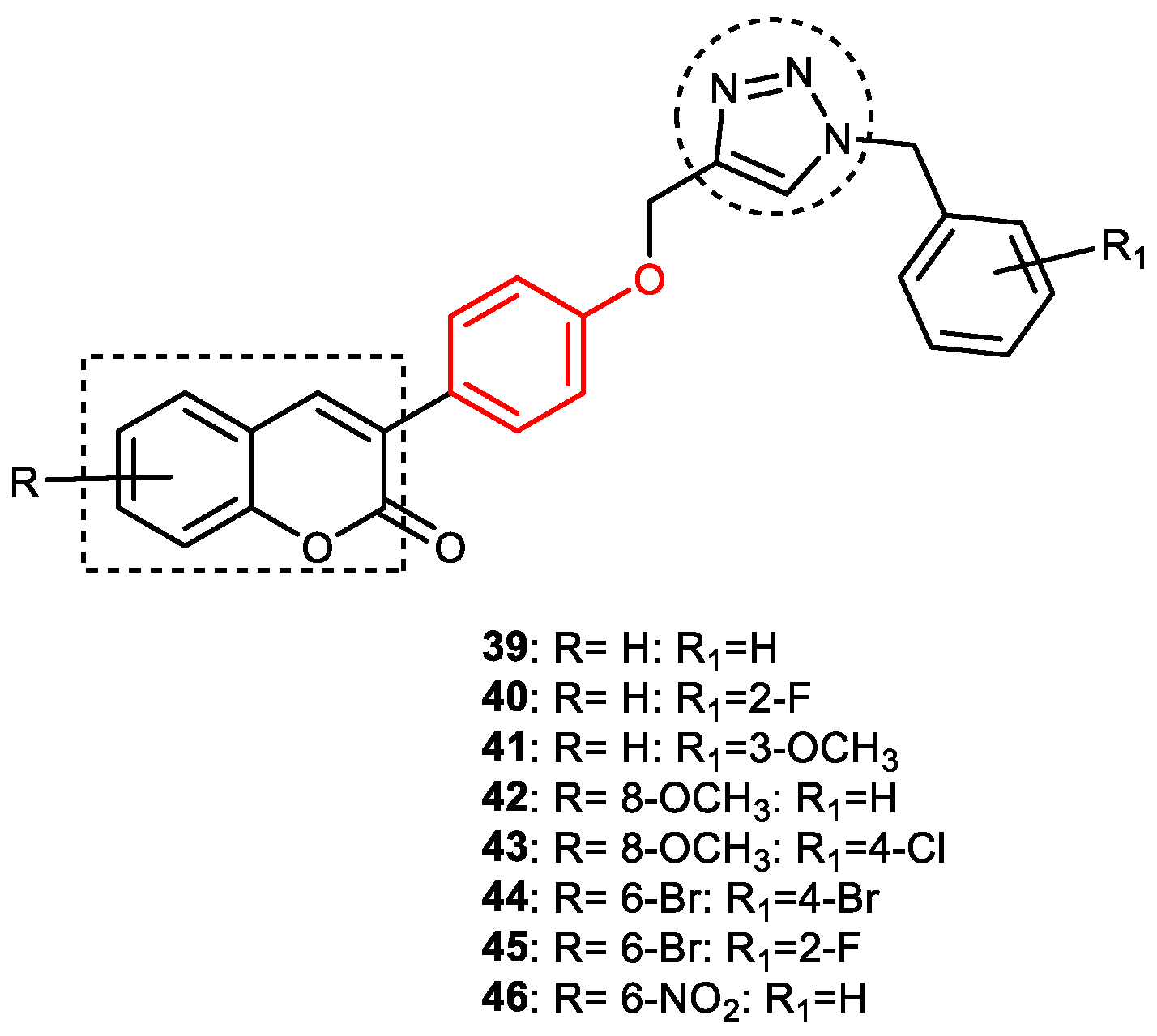
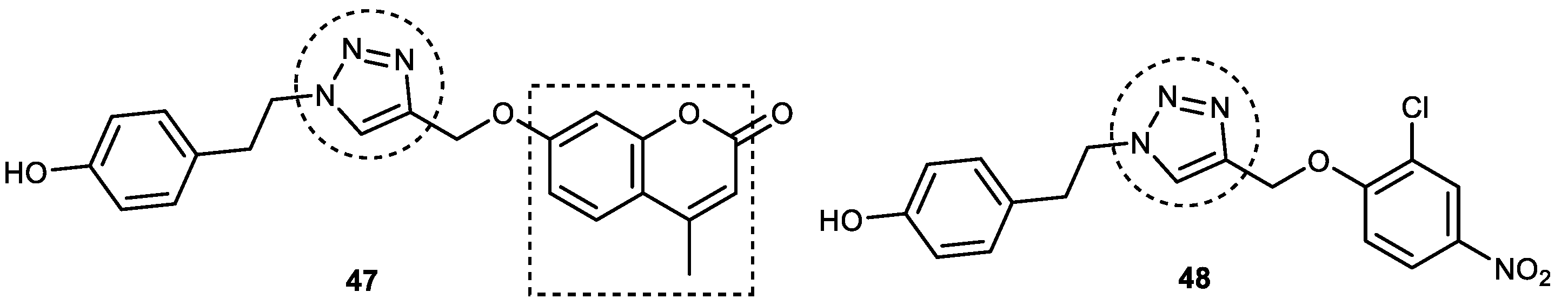
3.6.1. Coumarin–1,2,3-triazole–Tyrosol Trihybrids
3.6.2. Coumarin–1,2,3-triazole–Amino Acid Trihybrids
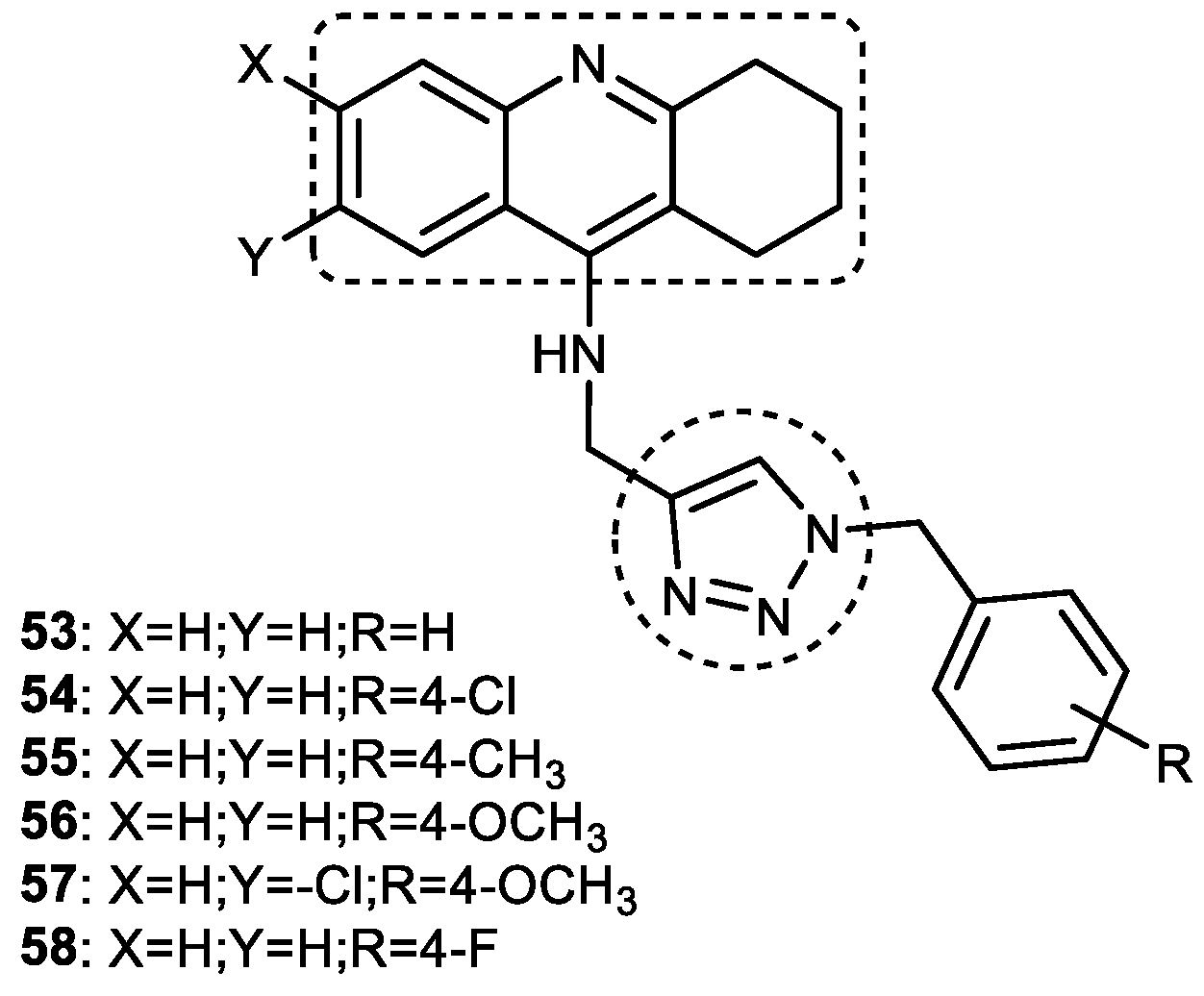
3.7. Tacrine–1,2,3-triazole Hybrids
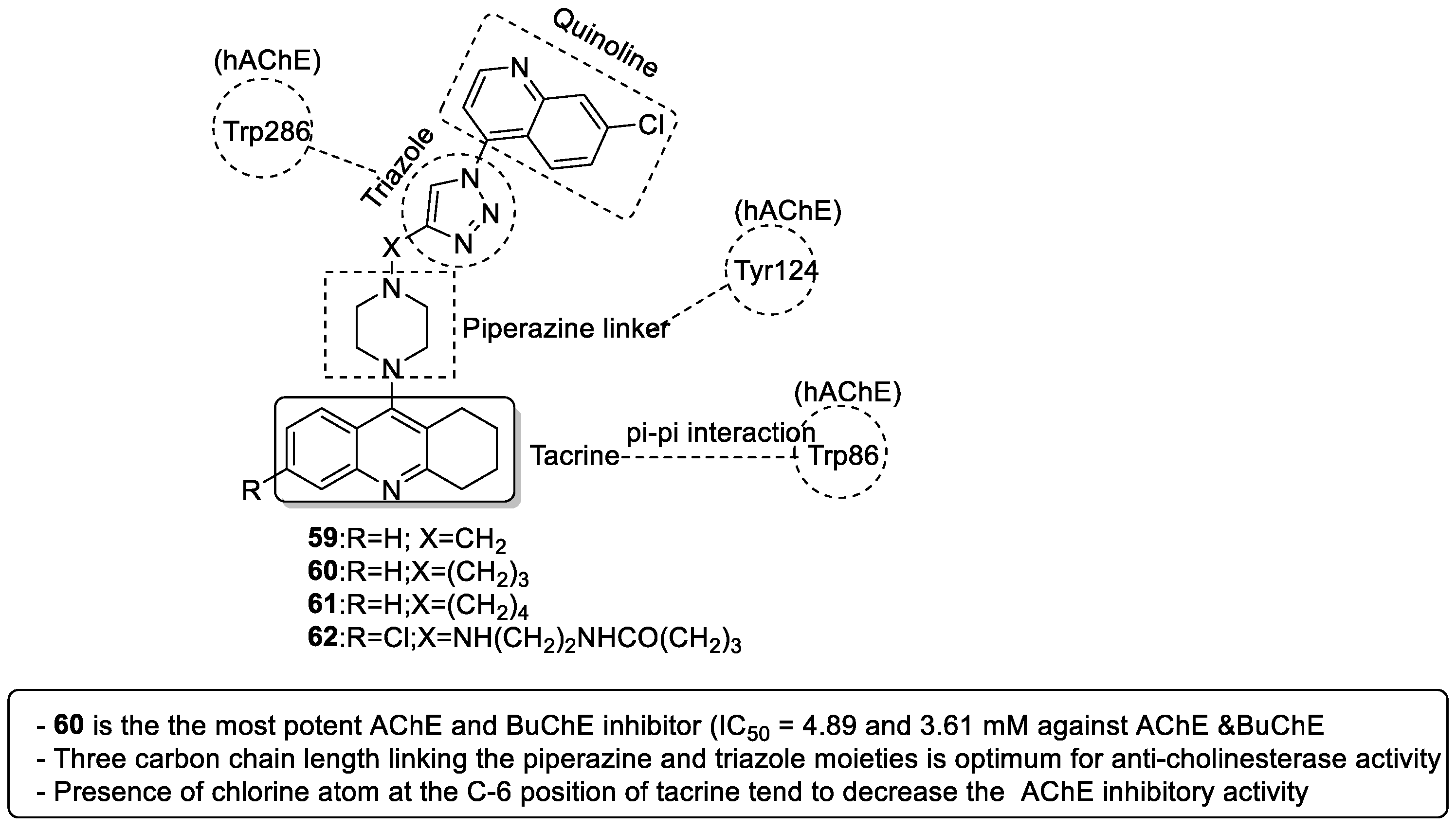
3.7.1. Tacrine–1,2,3-triazole–Quinoline Trihybrids
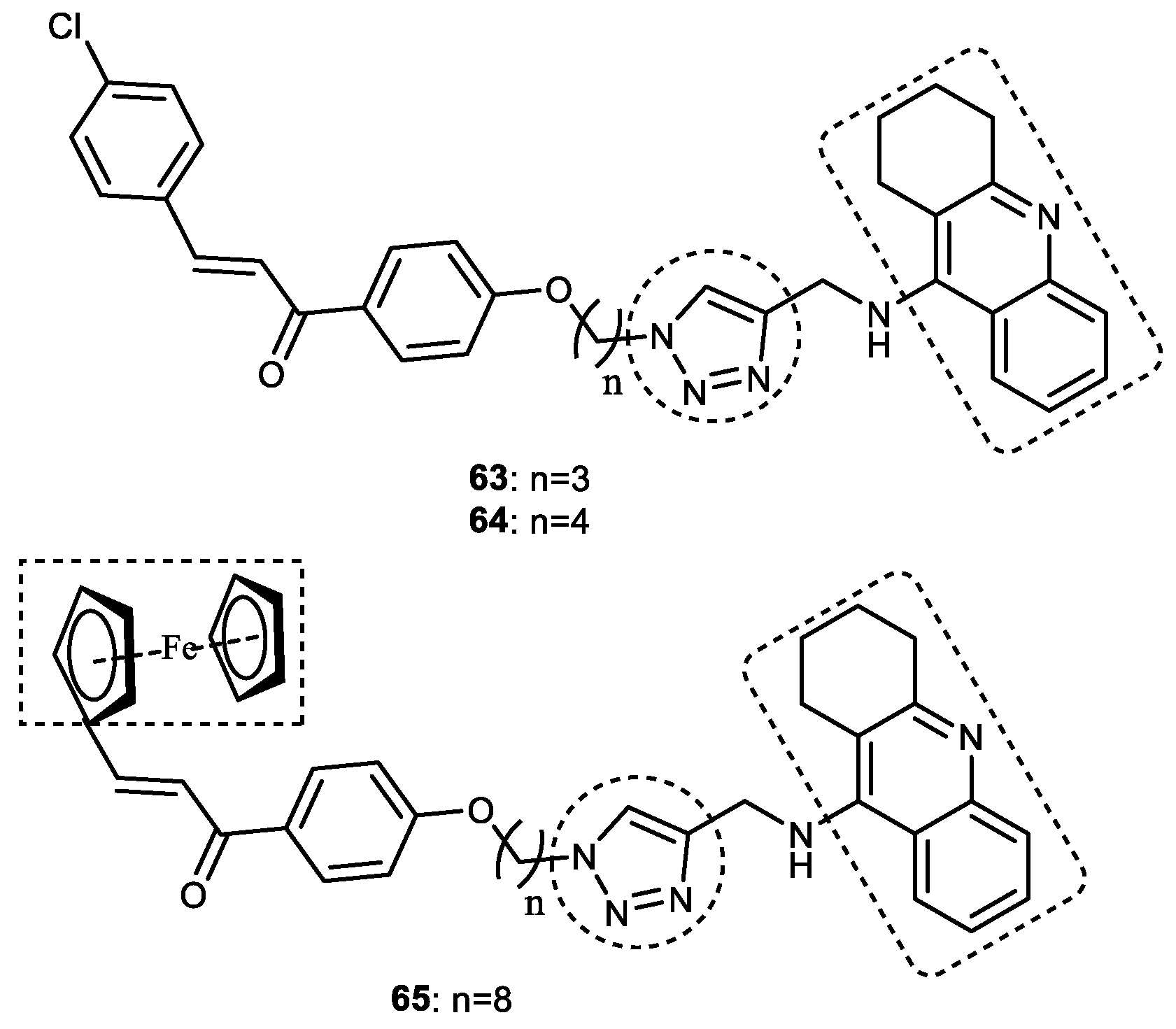
3.7.2. Tacrine–1,2,3-triazole–chalcone Hybrids
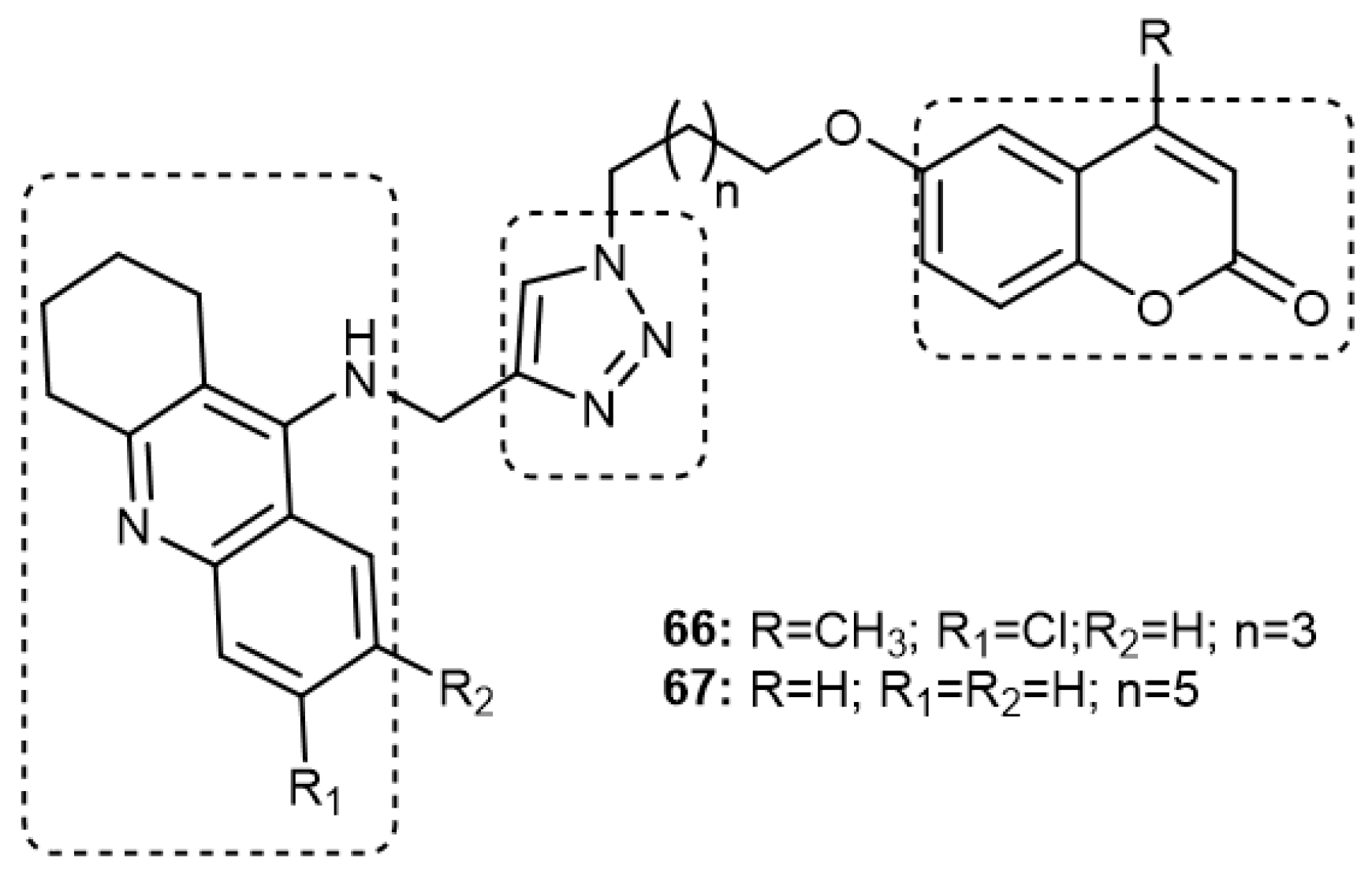
3.7.3. Tacrine–1,2,3-triazole–coumarin trihybrids
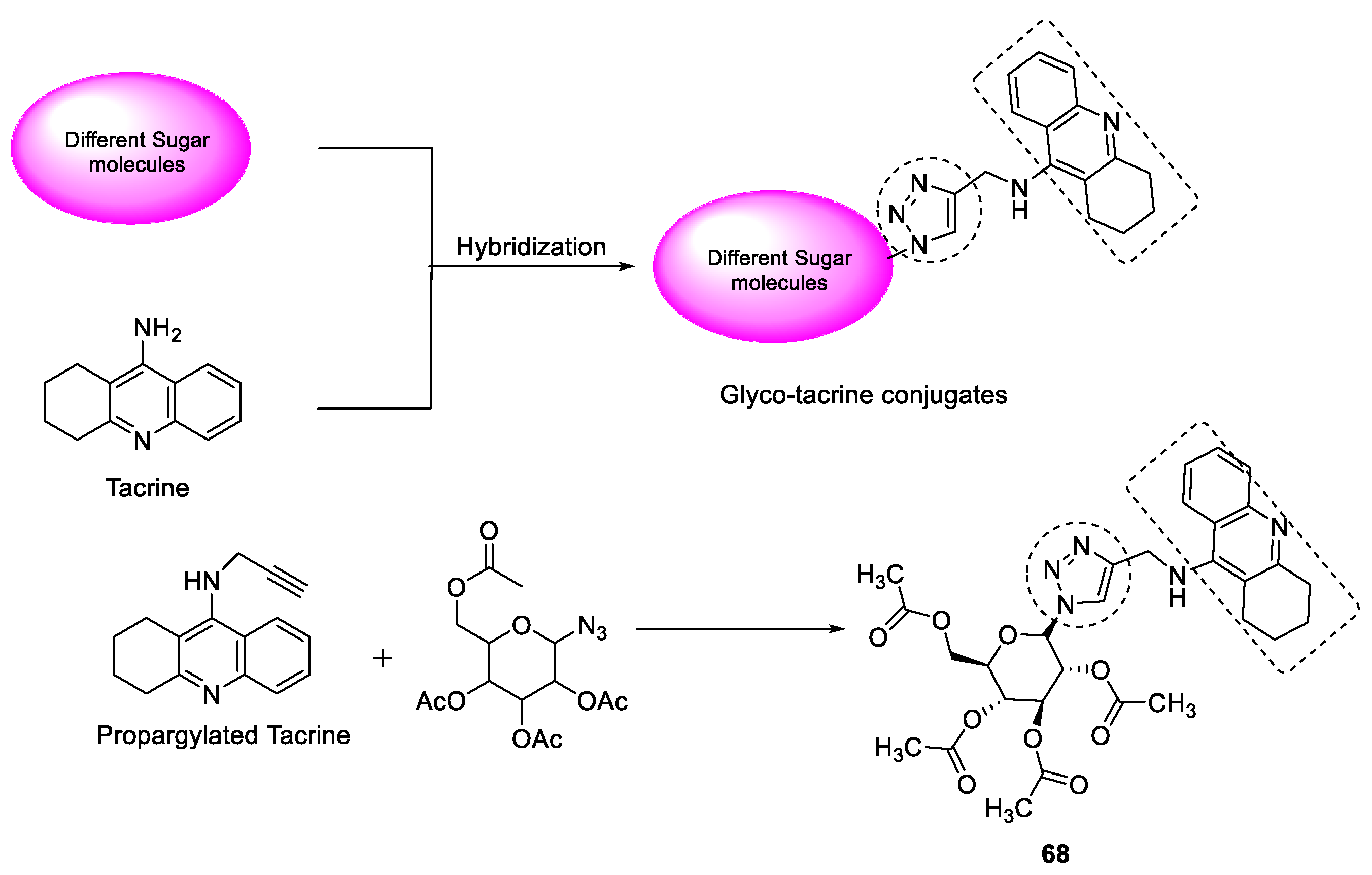
3.7.4. Tacrine–1,2,3-triazole Glycoconjugates
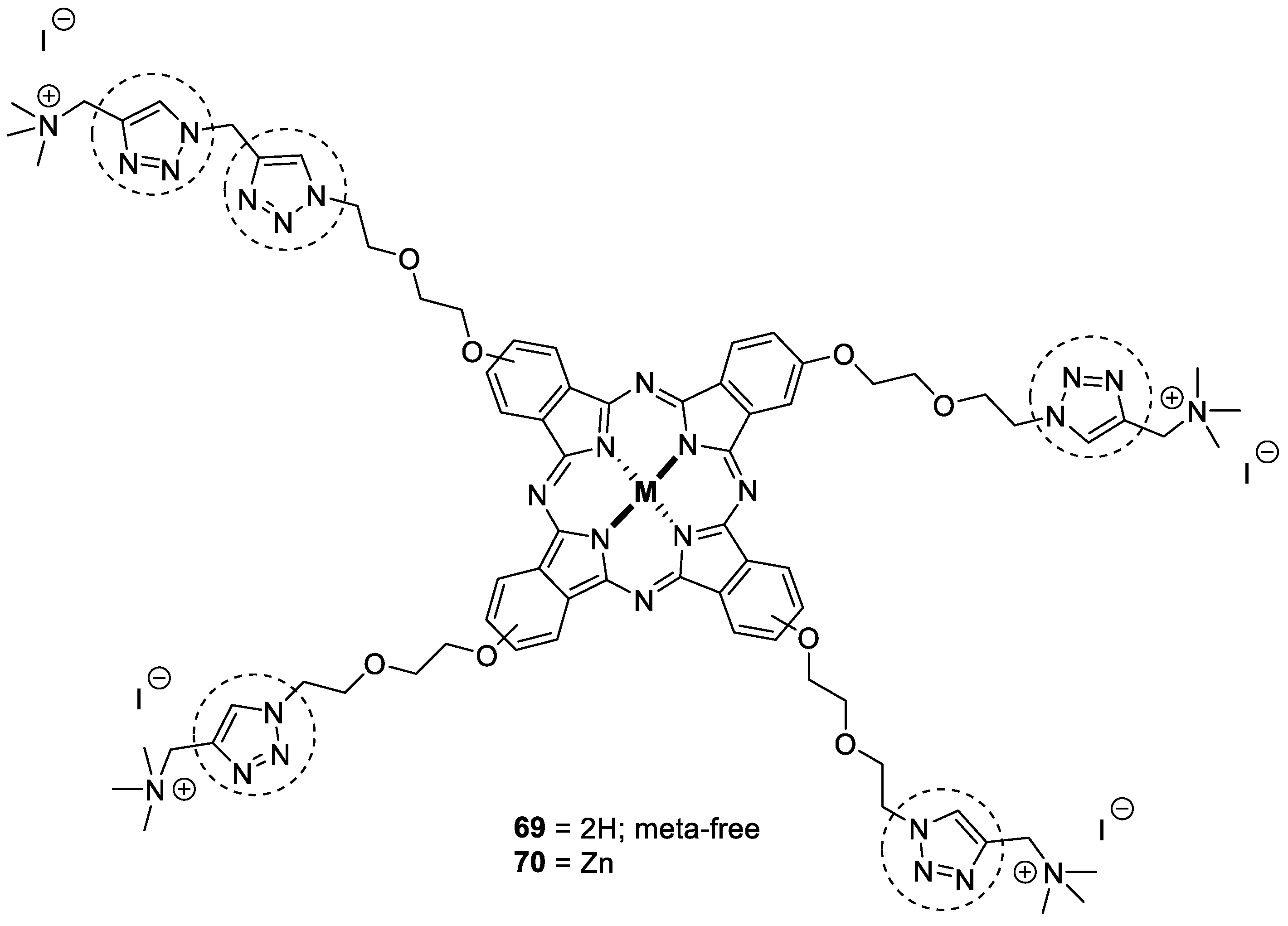
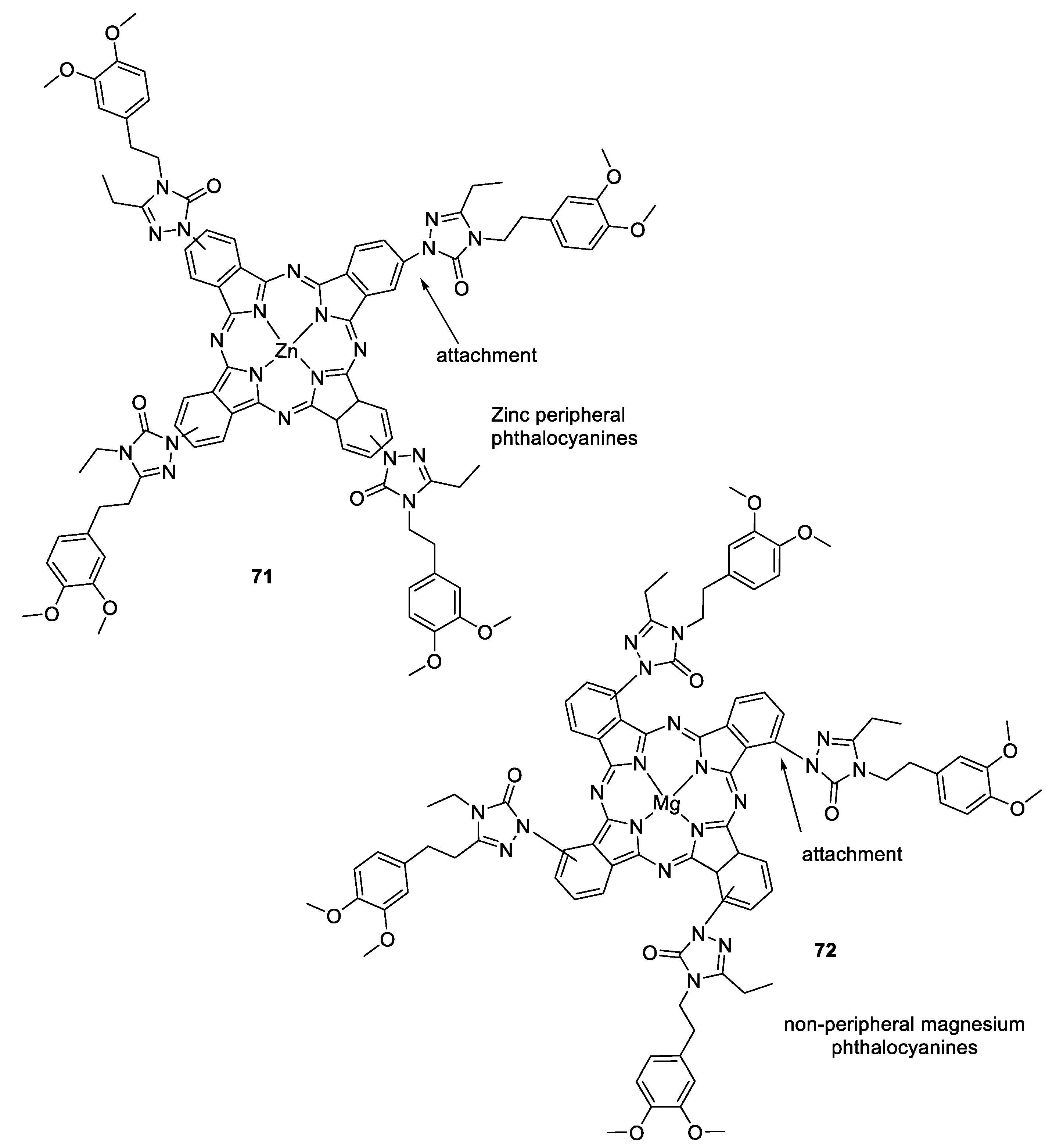
3.8. Metalophthalcyanines–1,2,3-triazole Hybrids
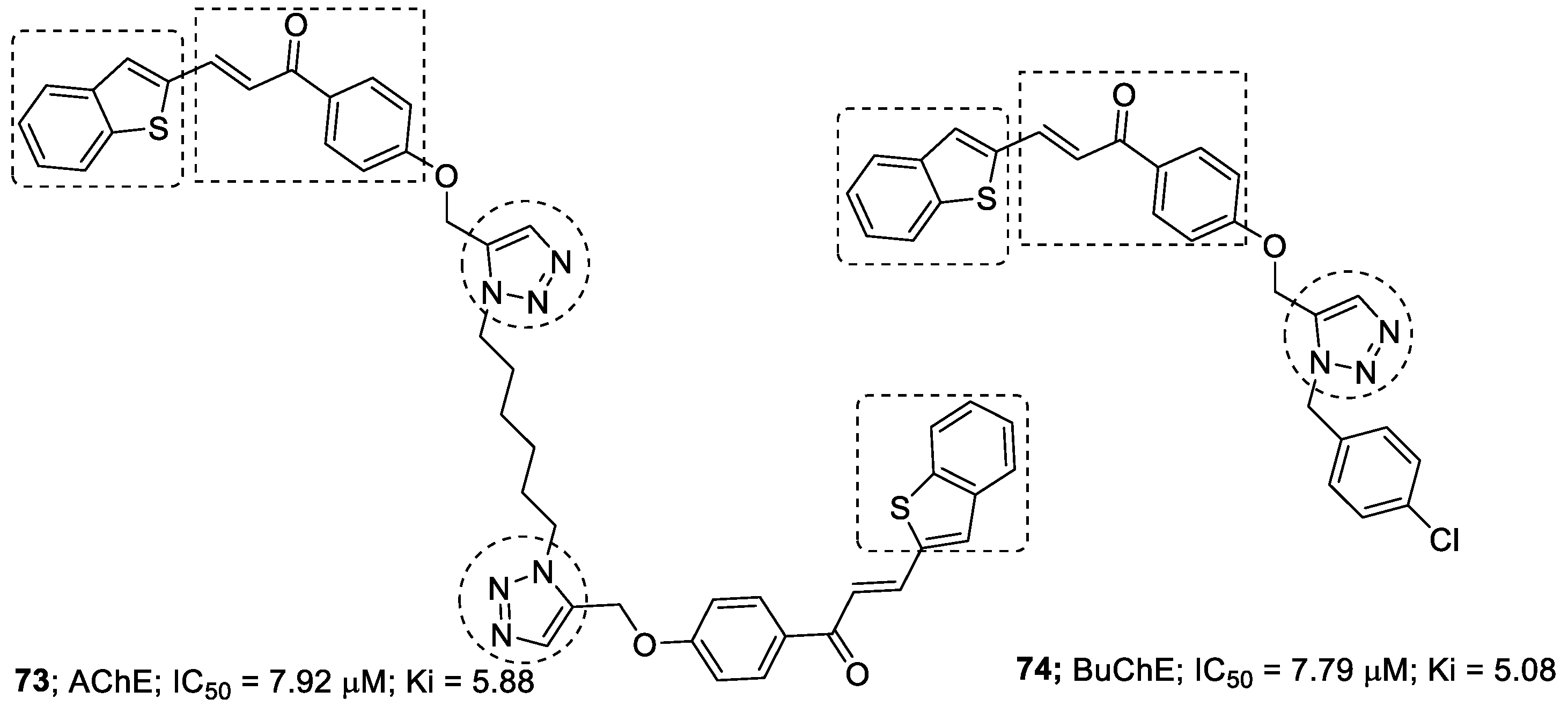
3.9. Chalcone–1,2,3-triazole Hybrids
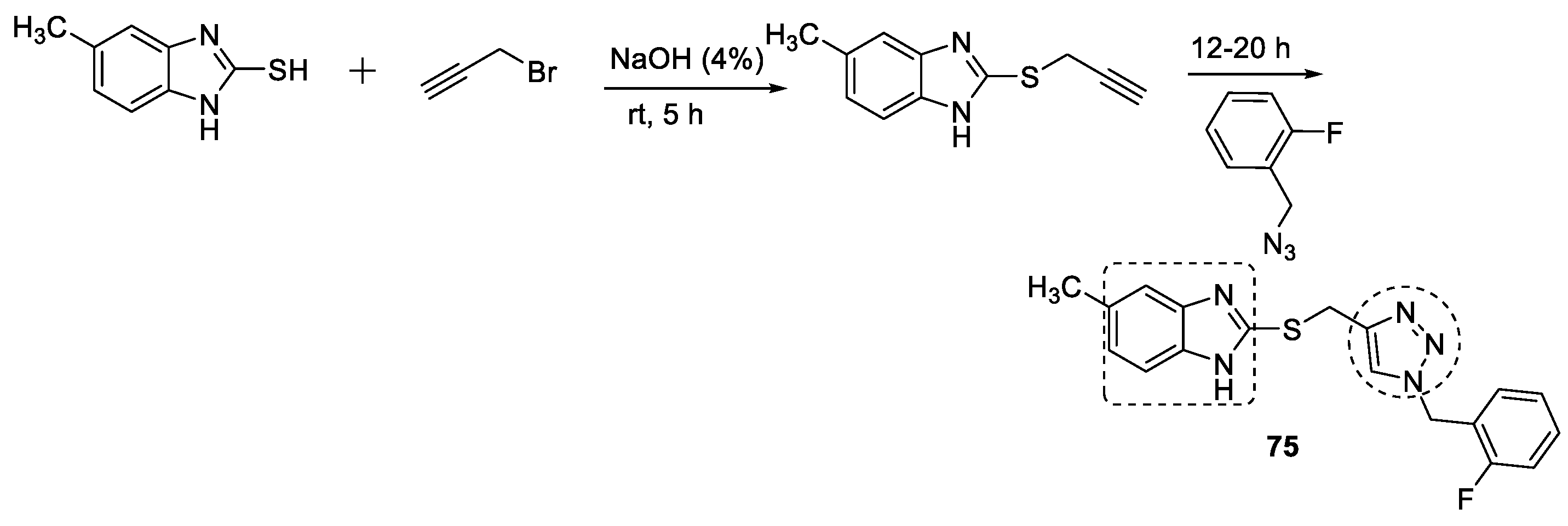
3.10. Benzimidazole–1,2,3-triazole Hybrids
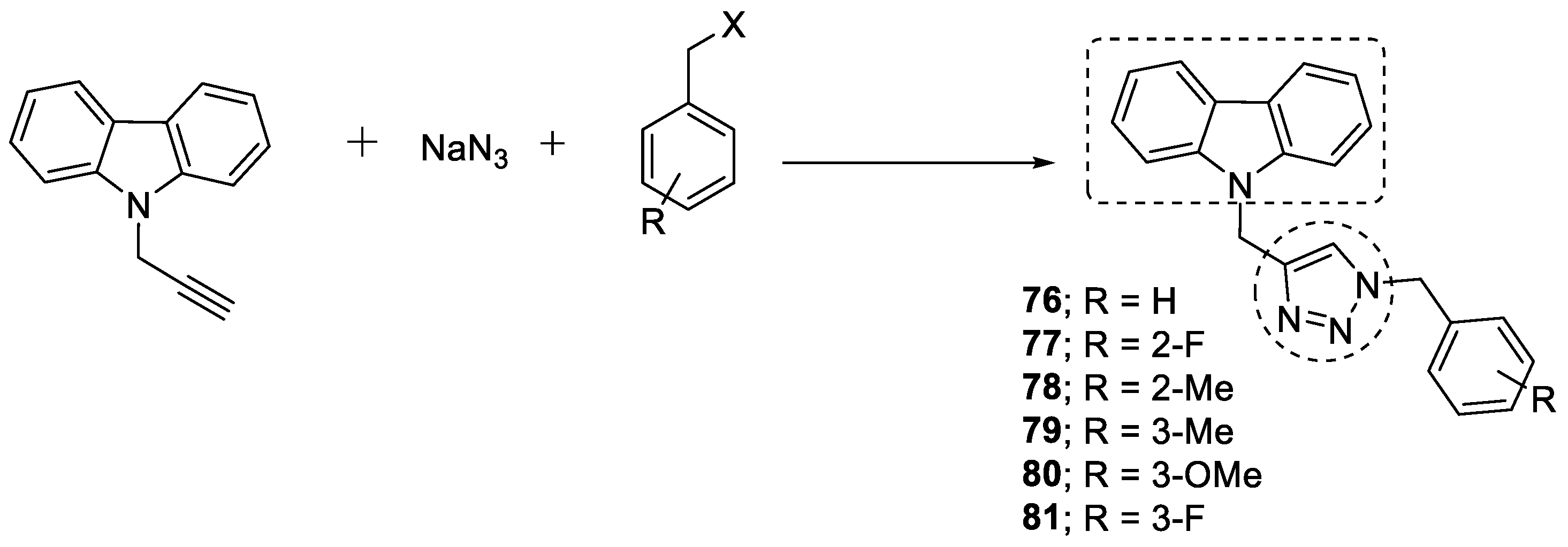
3.11. Carbazole–1,2,3-triazole Hybrids
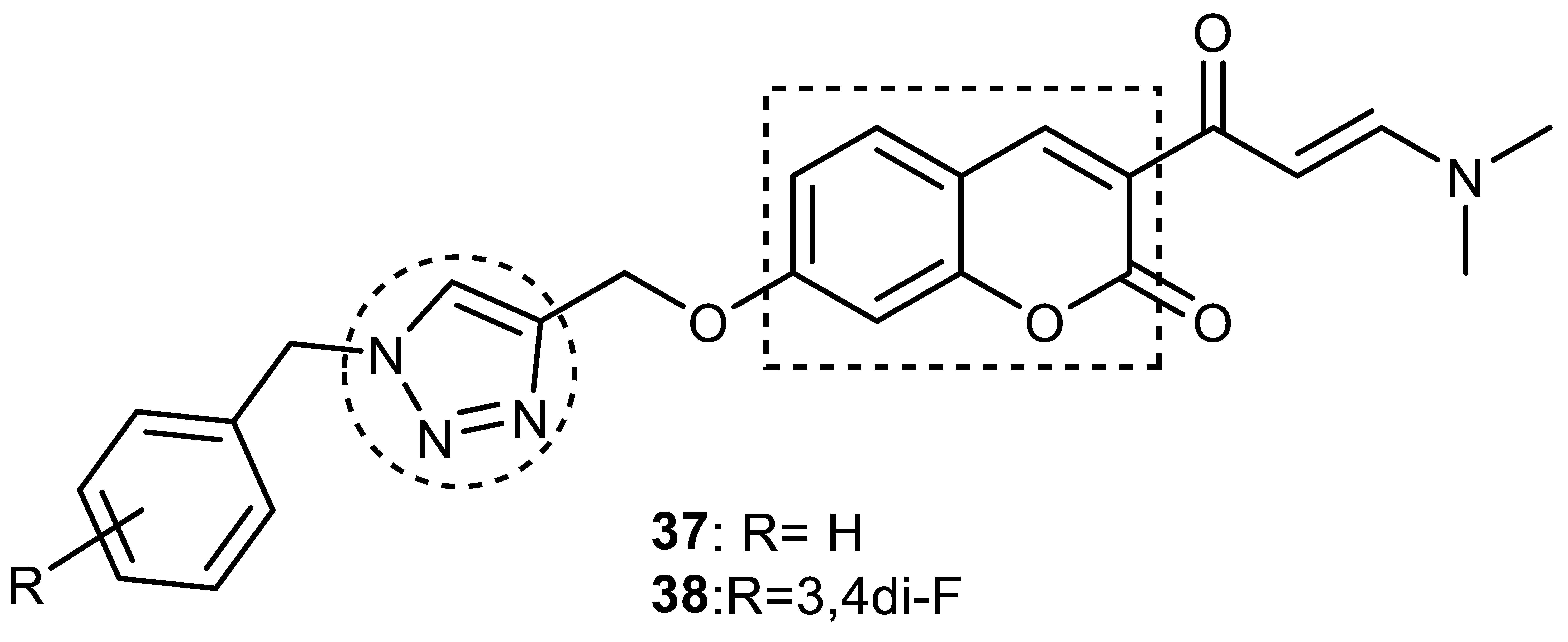
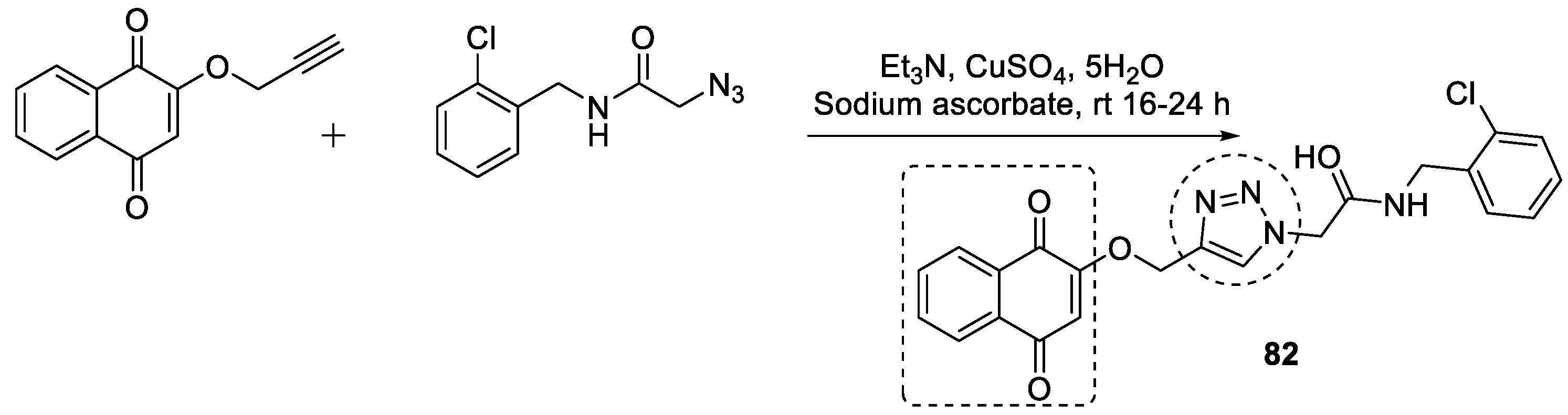
3.12. 1,4-Naphthoquinone–1,2,3-triazole Hybrids
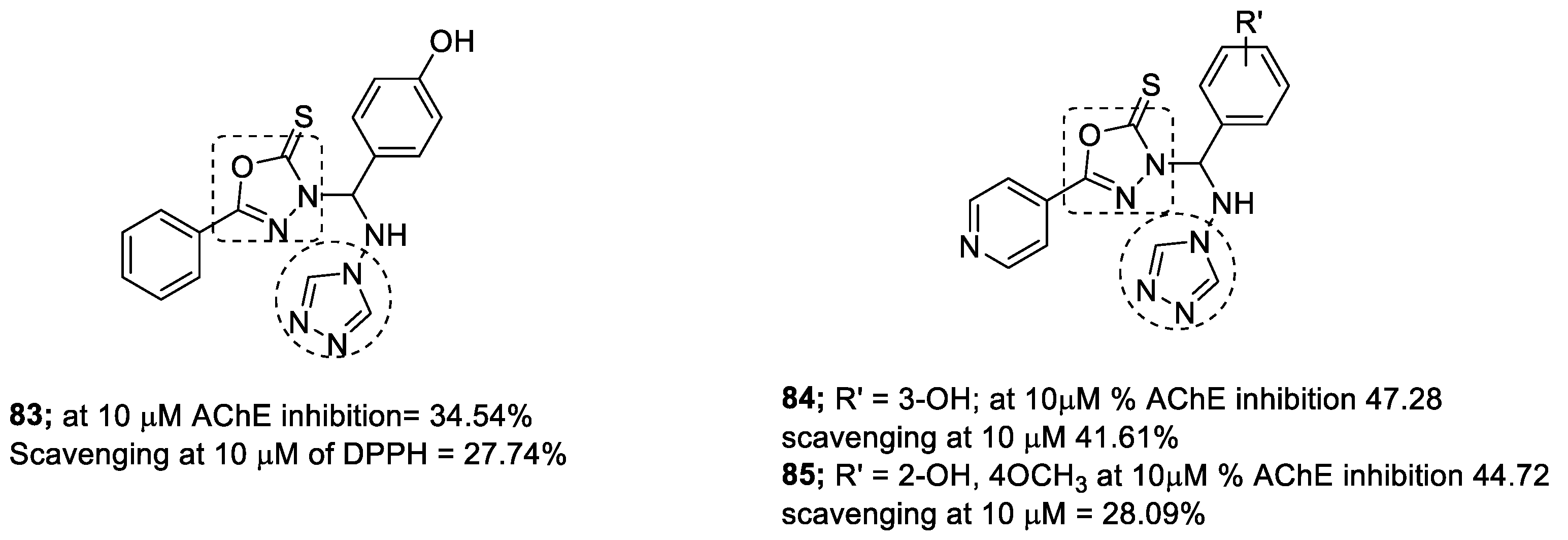
3.13. Oxadiazole–1,2,3-triazole Hybrids
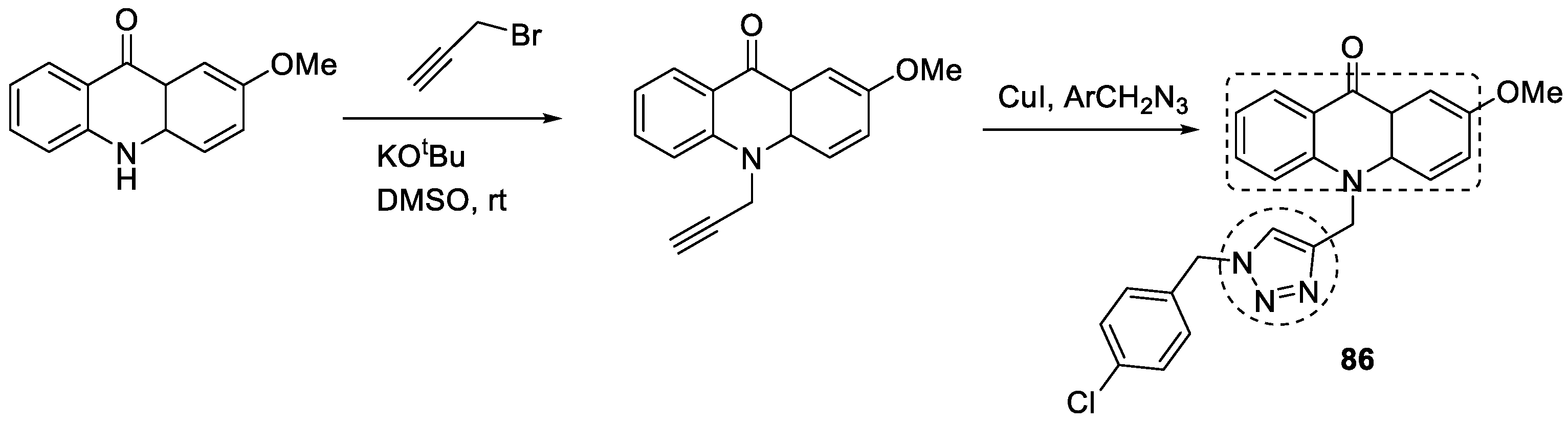
3.14. Acridone–1,2,3-triazole Hybrids
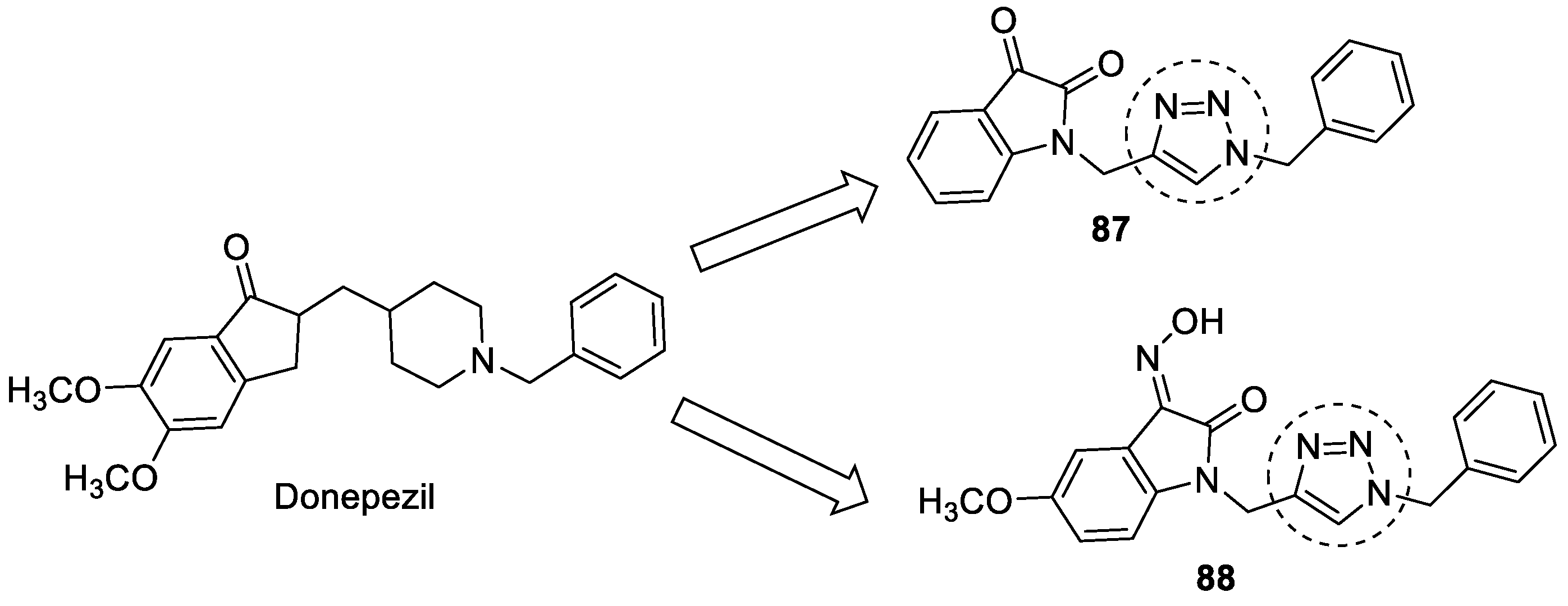
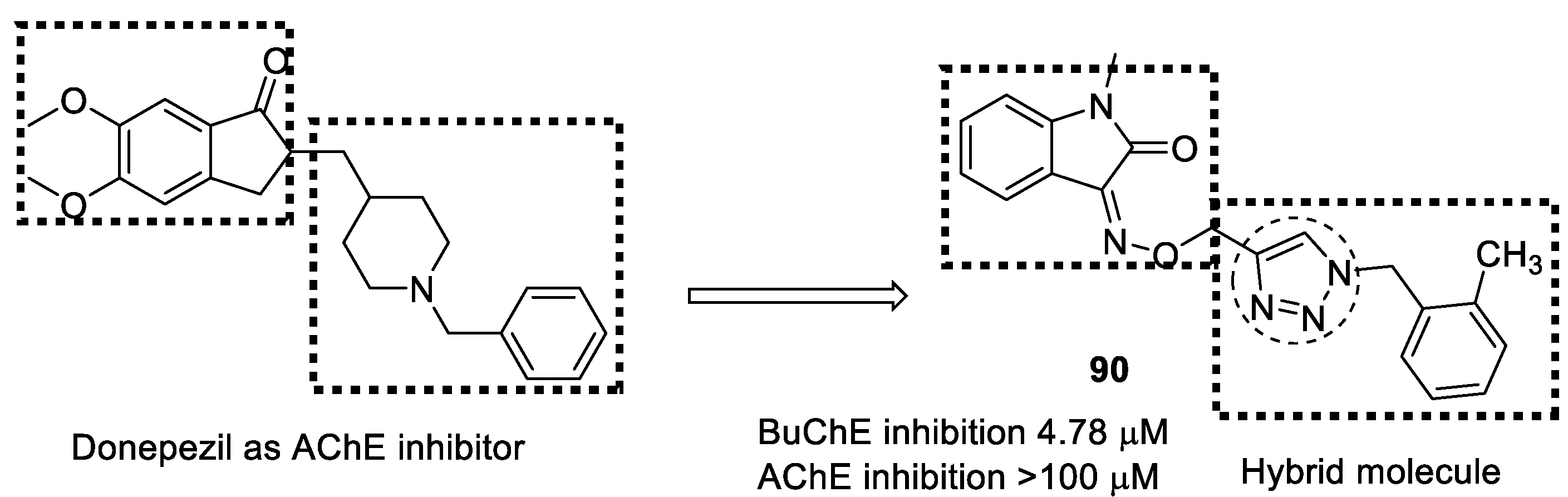
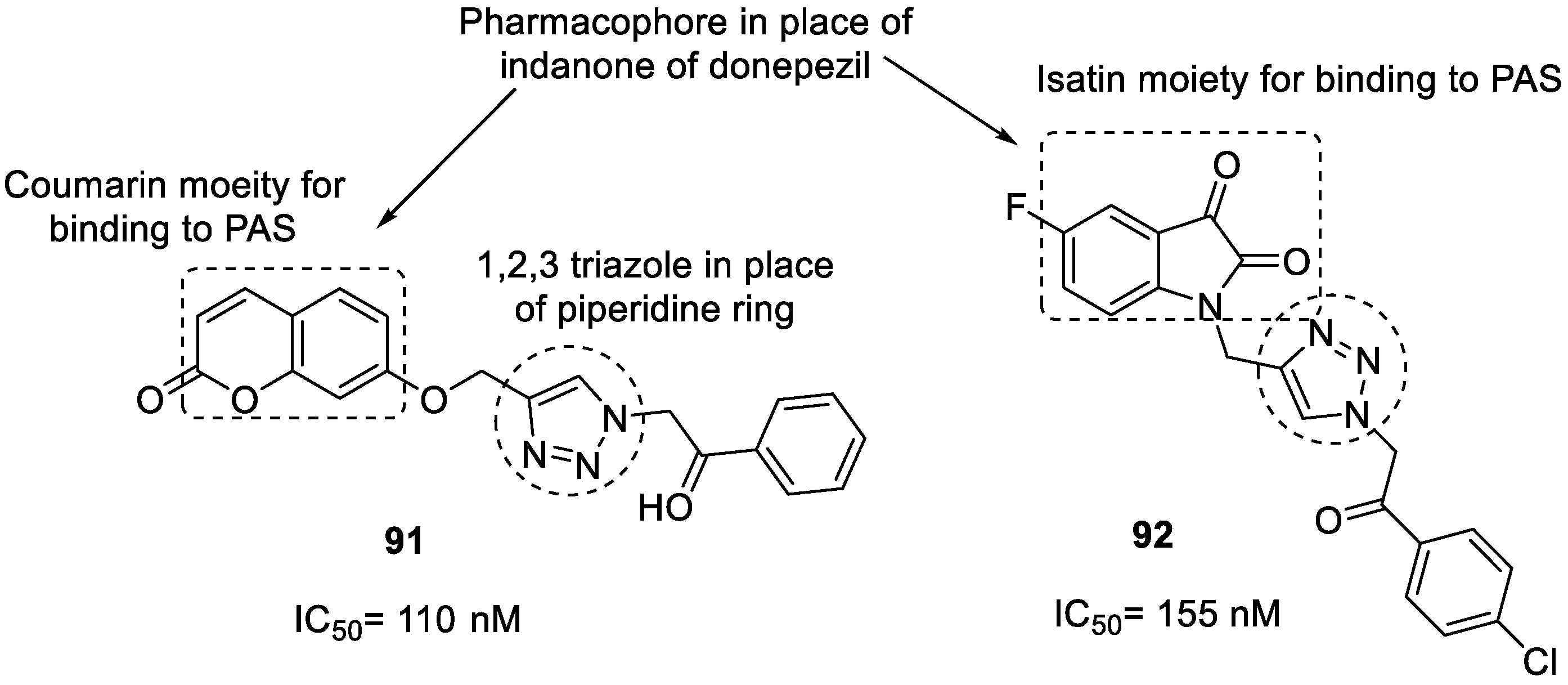
3.15. Donepezil–1,2,3-triazole Hybrids
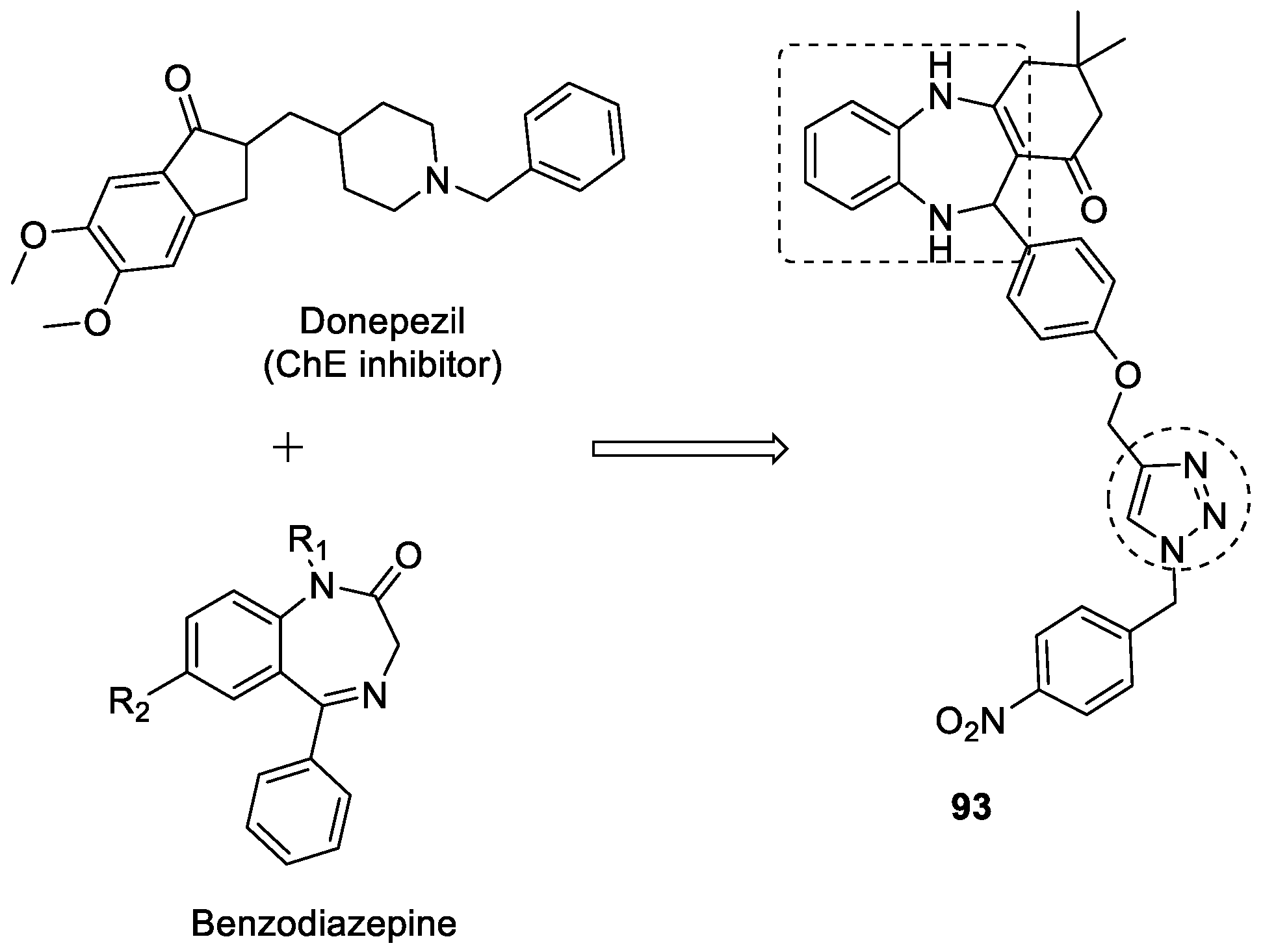
3.16. Benzodiazepine–1,2,3-triazole Hybrids
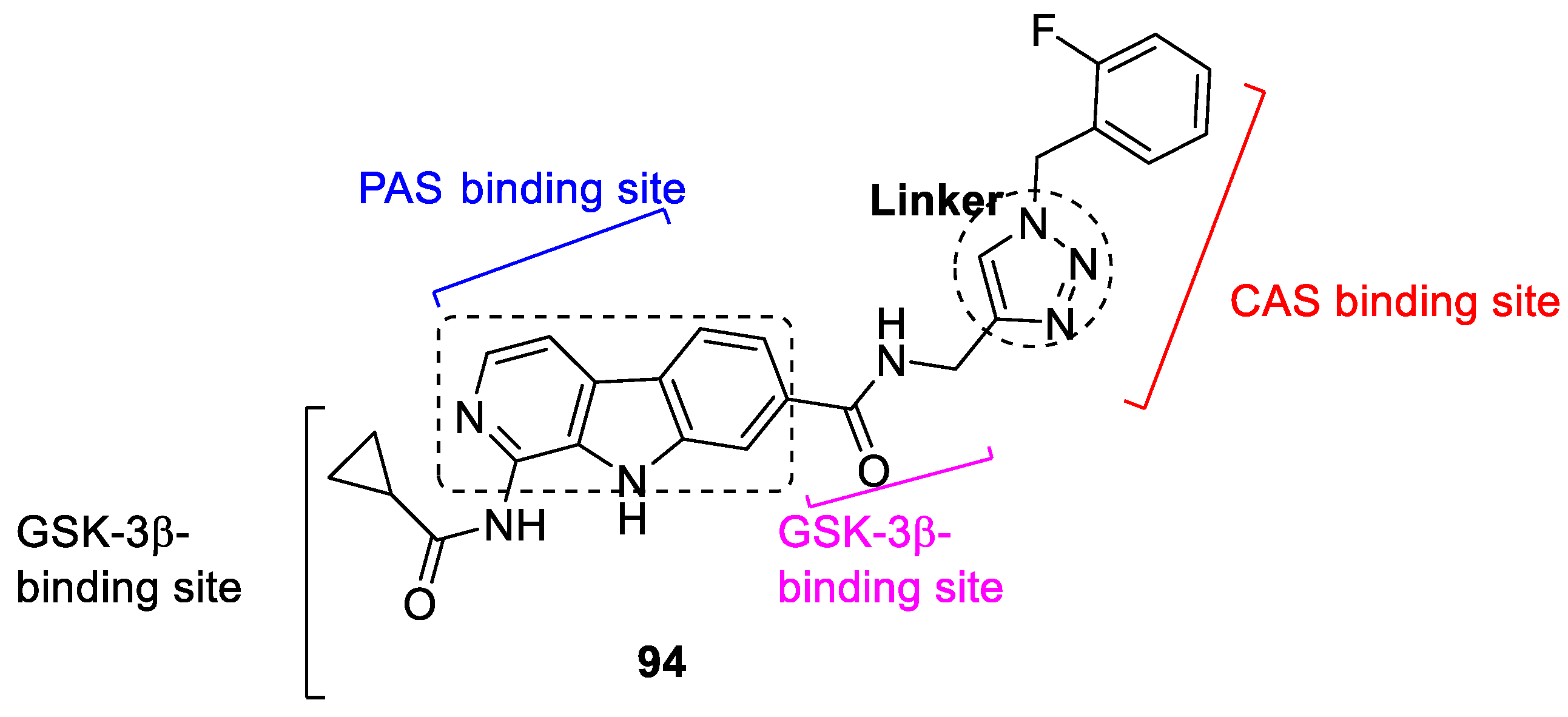
3.17. Carboline–1,2,3-triazole Hybrids
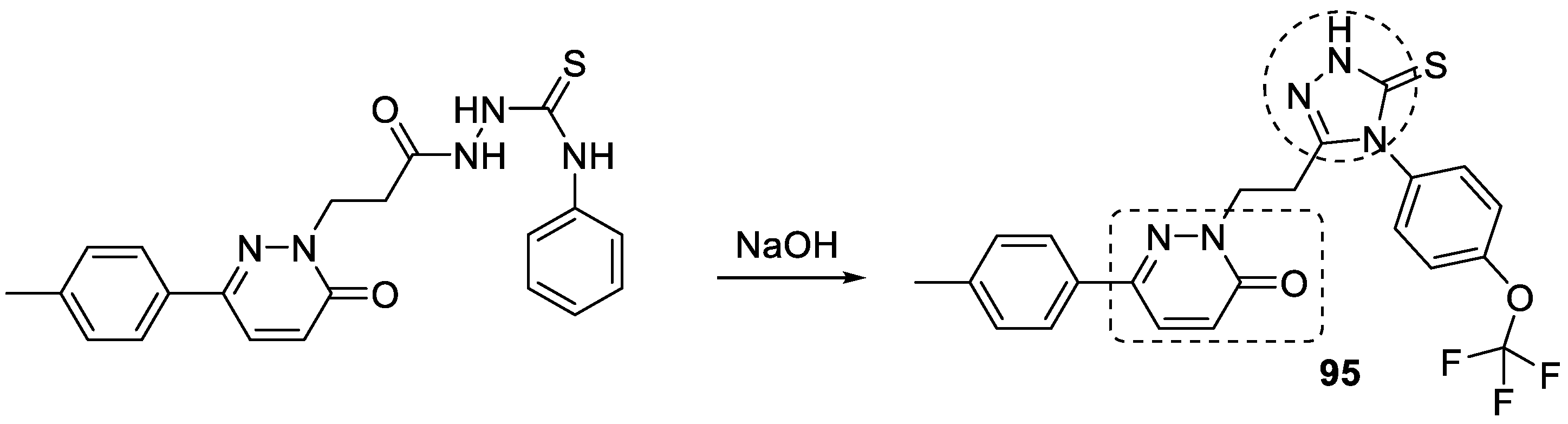
3.18. Pyridazinone–1,2,3-triazole Hybrids
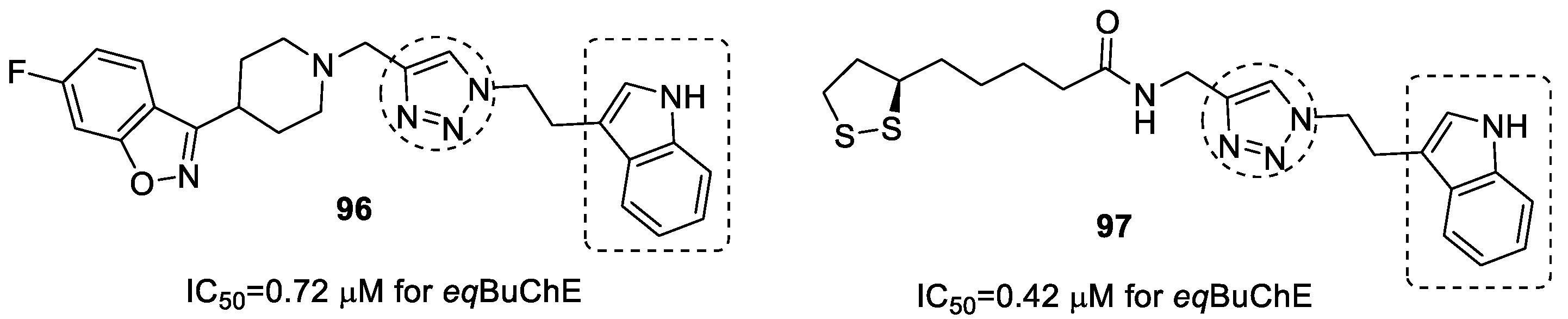
3.19. Tryptamine–1,2,3-triazole Hybrids
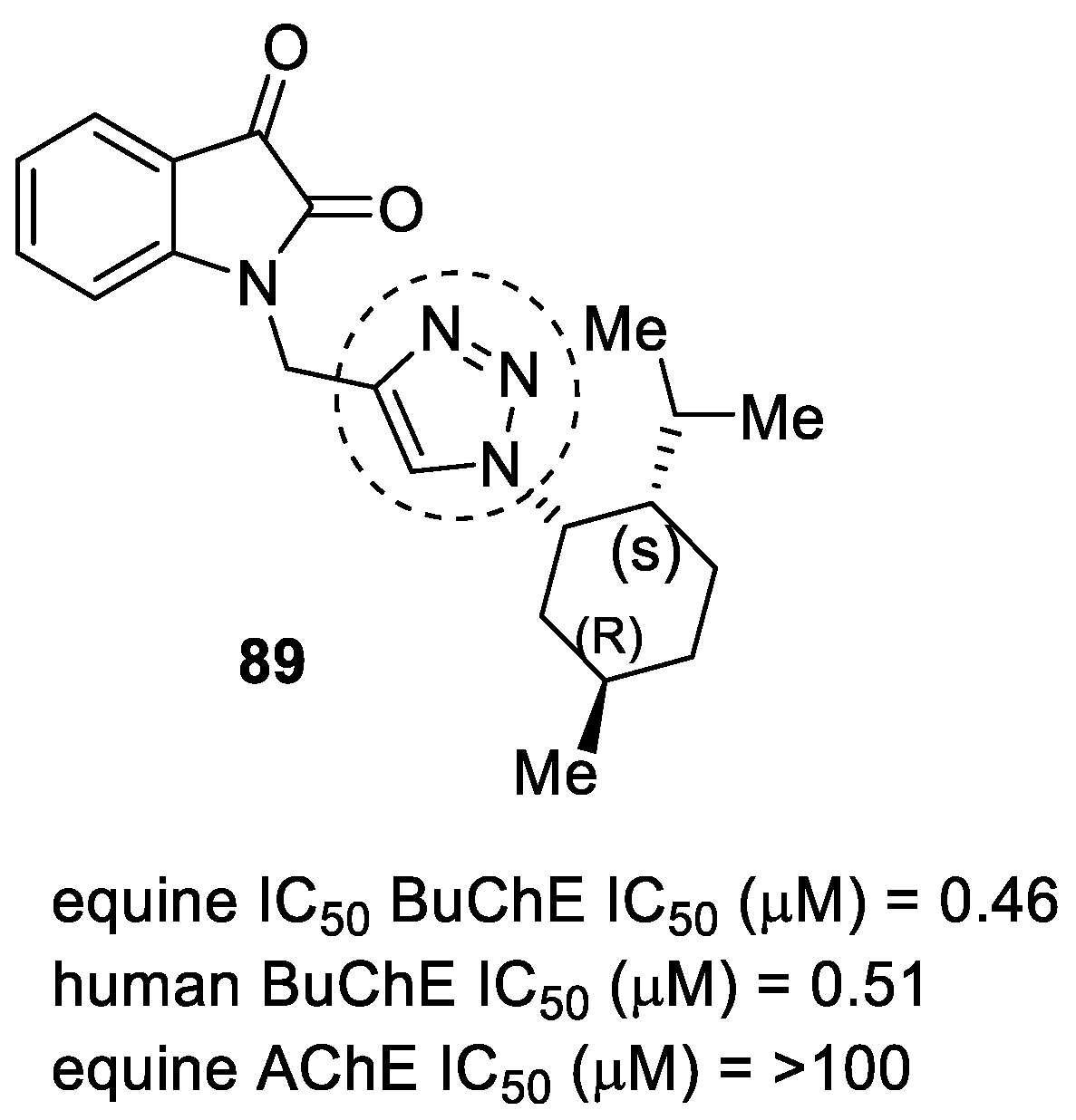
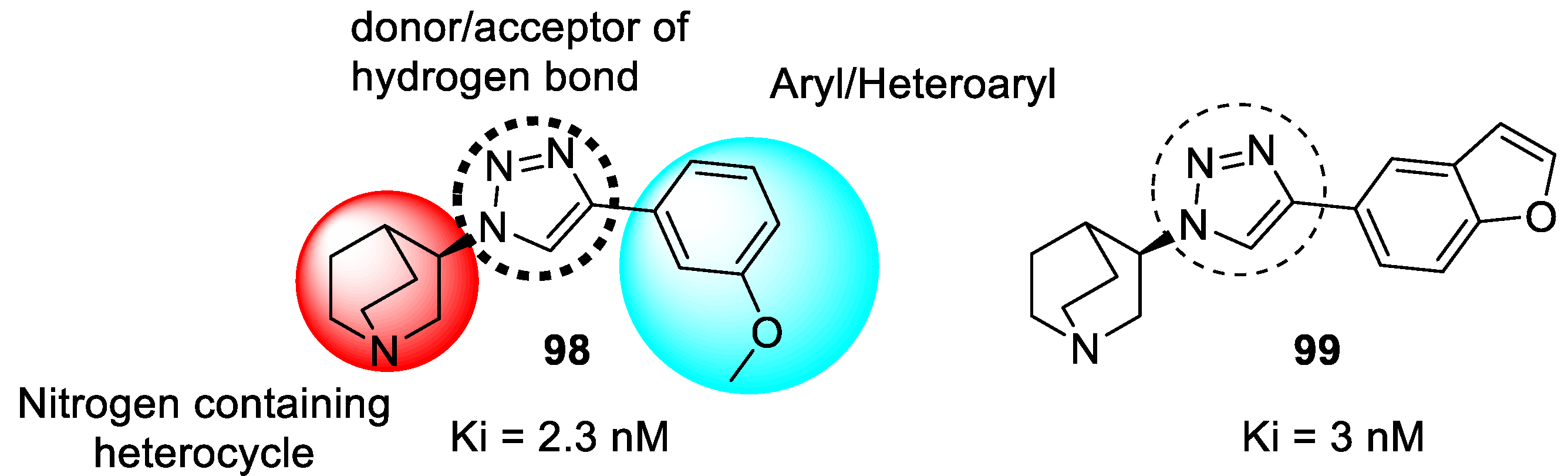
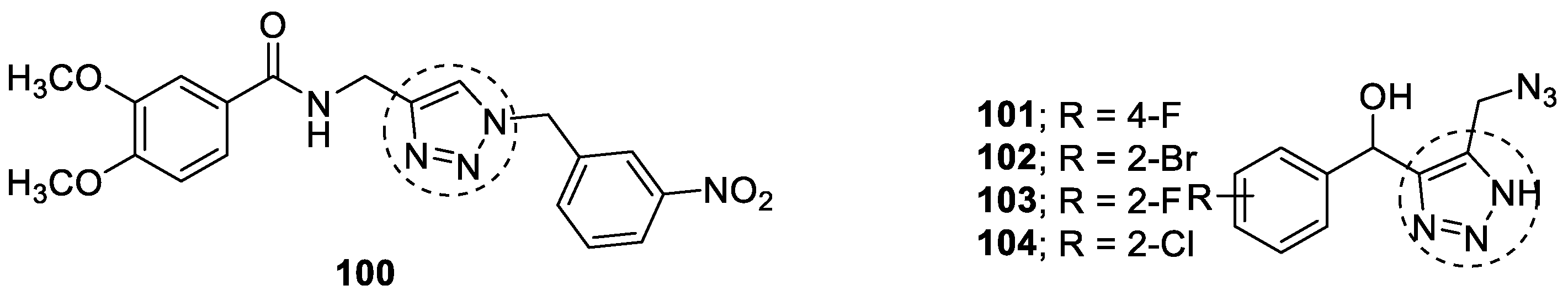
3.20. Miscellaneous Hybrids of 1,2,3-triazoles
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations:
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| eeAChE | eel AChE |
| hAChE | human AChE |
| AChEI | Acetylcholinesterase inhibitor |
| AD | Alzheimer’s disease |
| Aβ | Amyloid-β |
| BACE1 | β-site APP cleaving enzyme-1 |
| BBB | Blood-brain-barrier |
| BHT | Butylated hydroxytoluene |
| BuChE | Butyrylcholinesterase |
| eqBuChE | equine serum BuChE |
| CAS | Catalytic active (or anionic) site |
| ChAT | Choline acetyltransferase |
| ChE | Cholinesterase |
| CINs | Cholinergic interneurons |
| CuAAC | Cu(I))-catalyzed alkyne–azide 1,3-dipolar cycloaddition |
| DIPEA | N,N-Diisopropylethylamine or Hunig’s base |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| 5HT3 | 5—hydroxy tryptamine (Serotonin)- receptor-3 |
| LDH | Lactate dehydrogenase |
| LOX | Lipoxygenase |
| MAO | Monoamine oxidase |
| MG | Myasthenia gravis |
| MTDL | Multi-target-directed-ligand |
| NDs | Neurodegenerative disease |
| NFT | Neurofibrillary tangles |
| NMJ | Neuromuscular junction |
| NO | Nitric oxide |
| NT | Neurotransmitter |
| PAS | Peripheral anionic site |
| PD | Parkinson’s disease |
| ROS | Reactive oxygen species |
| SAR | Structure activity relationship |
| TBPB | tert-butyl peroxybenzoate |
| THF | Tetrahydrofuran |
References
- Ahmed, N.Y.; Knowles, R.; Dehorter, N. New insights into cholinergic neuron diversity. Front. Mol. Neurosci. 2019, 12, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef] [PubMed]
- De Boer, D.; Nguyen, N.; Mao, J.; Moore, J.; Sorin, E. A Comprehensive Review of Cholinesterase Modeling and Simulation. Biomolecules 2021, 11, 580. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, D.; Rehman, M.T.; Bin Dukhyil, A.; Rizvi, S.M.D.; Al Ajmi, M.F.; Alshehri, B.M.; Banawas, S.; Khan, M.S.; Alturaiki, W.; Alsaweed, M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals 2021, 14, 937. [Google Scholar] [CrossRef]
- Bekdash, R.A. The cholinergic system, the adrenergic system and the neuropathology of alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 1273. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Paolone, G. From the gut to the brain and back: Therapeutic approaches for the treatment of network dysfunction in Parkinson’s disease. Front. Neurol. 2020, 11, 557928. [Google Scholar] [CrossRef]
- Amalric, M.; Pattij, T.; Sotiropoulos, I.; Silva, J.M.; Sousa, N.; Ztaou, S.; Chiamulera, C.; Wahlberg, L.U.; Emerich, D.F.; Paolone, G. Where dopaminergic and cholinergic systems interact: A gateway for tuning neurodegenerative disorders. Front. Behav. Neurosci. 2021, 15, 147. [Google Scholar] [CrossRef]
- Al-Hamed, F.S.; Maria, O.M.; Phan, J.; Al Subaie, A.; Gao, Q.; Mansour, A.; Abu Nada, L.; Boukhatem, I.; Elkashty, O.A.; Tran, S.D. Postoperative administration of the acetylcholinesterase inhibitor, donepezil, interferes with bone healing and implant osseointegration in a rat model. Biomolecules 2020, 10, 1318. [Google Scholar] [CrossRef]
- Zhou, S.; Huang, G. The biological activities of butyrylcholinesterase inhibitors. Biomed. Pharmacother. 2022, 146, 112556. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Korabecny, J.; Soukup, O. Cholinesterase Research. Biomolecules 2021, 11, 1121. [Google Scholar] [CrossRef] [PubMed]
- George, N.; Akhtar, M.J.; Al Balushi, K.A.; Khan, S.A. Rational drug design strategies for the development of promising multi-target directed Indole hybrids as Anti-Alzheimer agents. Bioorganic Chem. 2022, 105941. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Al Balushi, K.; Akhtar, M.J.; Khan, S.A. Coumarin linked heterocyclic hybrids: A promising approach to develop multi target drugs for Alzheimer’s disease. J. Mol. Struct. 2021, 1241, 130618. [Google Scholar] [CrossRef]
- Anil, D.A.; Aydin, B.O.; Demir, Y.; Turkmenoglu, B. Design, synthesis, biological evaluation and molecular docking studies of novel 1H-1,2,3-Triazole derivatives as potent inhibitors of carbonic anhydrase, acetylcholinesterase and aldose reductase. J. Mol. Struct. 2022, 1257, 132613. [Google Scholar] [CrossRef]
- Sahu, J.K.; Ganguly, S.; Kaushik, A. Triazoles: A valuable insight into recent developments and biological activities. Chin. J. Nat. Med. 2013, 11, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, A.; Silakari, O. Triazoles: Multidimensional 5-membered nucleus for designing multitargeting agents. In Key Heterocycle Cores for Designing Multitargeting Molecules; Elsevier: Amsterdam, The Netherlands, 2018; pp. 323–342. [Google Scholar]
- Potts, K. The Chemistry of 1,2,4-Triazoles. Chem. Rev. 1961, 61, 87–127. [Google Scholar] [CrossRef]
- Jiali, M.; Chenghe, Z.; Xue, B. Advances in Triazole Antimicrobial Agents. Chin. J. Antibiotics 2010, 32, 587–593. [Google Scholar]
- Lass-Flörl, C. Triazole antifungal agents in invasive fungal infections. Drugs 2011, 71, 2405–2419. [Google Scholar] [CrossRef]
- Strzelecka, M.; Świątek, P. 1,2,4-Triazoles as important antibacterial agents. Pharmaceuticals 2021, 14, 224. [Google Scholar] [CrossRef]
- Liu, P.; Zhu, S.; Li, P.; Xie, W.; Jin, Y.; Sun, Q.; Wu, Q.; Sun, P.; Zhang, Y.; Yang, X. Synthesis and SAR studies of biaryloxy-substituted triazoles as antifungal agents. Bioorganic Med. Chem. Lett. 2008, 18, 3261–3265. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Z.; Gao, C.; Ren, Q.-C.; Chang, L.; Lv, Z.-S.; Feng, L.-S. Triazole derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 138, 501–513. [Google Scholar] [CrossRef]
- Al-Humaidi, J.Y.; Shaaban, M.M.; Rezki, N.; Aouad, M.R.; Zakaria, M.; Jaremko, M.; Hagar, M.; Elwakil, B.H. 1,2,3-Triazole-Benzofused Molecular Conjugates as Potential Antiviral Agents against SARS-CoV-2 Virus Variants. Life 2022, 12, 1341. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, W.; Yang, G.; Wang, R.; Ma, L. Design and synthesis of novel diosgenin-triazole hybrids targeting inflammation as potential neuroprotective agents. Bioorganic Med. Chem. Lett. 2021, 43, 128092. [Google Scholar] [CrossRef]
- Kumbhare, R.M.; Kosurkar, U.B.; Ramaiah, M.J.; Dadmal, T.L.; Pushpavalli, S.; Pal-Bhadra, M. Synthesis and biological evaluation of novel triazoles and isoxazoles linked 2-phenyl benzothiazole as potential anticancer agents. Bioorganic Med. Chem. Lett. 2012, 22, 5424–5427. [Google Scholar] [CrossRef]
- Aljohani, F.S.; Rezki, N.; Aouad, M.R.; Elwakil, B.H.; Hagar, M.; Sheta, E.; Hussein Mogahed, N.M.F.; Bardaweel, S.K.; Hagras, N.A.-E. Synthesis, Characterization and Nanoformulation of Novel Sulfonamide-1,2,3-triazole Molecular Conjugates as Potent Antiparasitic Agents. Int. J. Mol. Sci. 2022, 23, 4241. [Google Scholar] [CrossRef]
- Fallah, Z.; Tajbakhsh, M.; Alikhani, M.; Larijani, B.; Faramarzi, M.A.; Hamedifar, H.; Mohammadi-Khanaposhtani, M.; Mahdavi, M. A review on synthesis, mechanism of action, and structure-activity relationships of 1,2,3-triazole-based α-glucosidase inhibitors as promising anti-diabetic agents. J. Mol. Struct. 2022, 1255, 132469. [Google Scholar] [CrossRef]
- Huisgen, R.; Szeimies, G.; Möbius, L. 1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen. Chem. Ber. 1967, 100, 2494–2507. [Google Scholar] [CrossRef]
- Li, Y.-J.; Li, X.; Zhang, S.-X.; Zhao, Y.-L.; Liu, Q. Copper (II)-catalyzed oxidative [3+2] cycloaddition reactions of secondary amines with α-diazo compounds: A facile and efficient synthesis of 1,2,3-triazoles. Chem. Commun. 2015, 51, 11564–11567. [Google Scholar] [CrossRef]
- Dommerholt, J.; Rutjes, F.P.; Delft, F.L.v. Strain-promoted 1,3-dipolar cycloaddition of cycloalkynes and organic azides. Cycloaddit. Bioorthogonal Chem. 2016, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Giel, M.C.; Smedley, C.J.; Mackie, E.R.; Guo, T.; Dong, J.; Soares da Costa, T.P.; Moses, J.E. Metal-Free Synthesis of Functional 1-Substituted-1,2,3-Triazoles from Ethenesulfonyl Fluoride and Organic Azides. Angew. Chem. 2020, 132, 1197–1202. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide− alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase:[1–3]-triazoles by regiospecific copper (I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.; Straub, B.F. Advancements in the mechanistic understanding of the copper-catalyzed azide–alkyne cycloaddition. Beilstein J. Org. Chem. 2013, 9, 2715–2750. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Singh, G.; George, N.; Singh, G.; Gupta, S.; Singh, H.; Kaur, G.; Singh, J. Copper-Based Metal–Organic Frameworks (MOFs) as an Emerging Catalytic Framework for Click Chemistry. Catalysts 2023, 13, 130. [Google Scholar] [CrossRef]
- Saini, P.; Singh, G.; Kaur, G.; Singh, J.; Singh, H. Robust and Versatile Cu (I) metal frameworks as potential catalysts for azide-alkyne cycloaddition reactions. Mol. Catal. 2021, 504, 111432. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar] [CrossRef]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V.V. Ruthenium-catalyzed azide− alkyne cycloaddition: Scope and mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar] [CrossRef]
- McNulty, J.; Keskar, K.; Vemula, R. The First Well-Defined Silver (I)-Complex-Catalyzed Cycloaddition of Azides onto Terminal Alkynes at Room Temperature. Chem. A Eur. J. 2011, 17, 14727–14730. [Google Scholar] [CrossRef] [PubMed]
- Sultana, J.; Sarma, D. Ag-catalyzed azide-alkyne cycloaddition: Copper free approaches for synthesis of 1,4-disubstituted 1,2,3-triazoles. Catal. Rev. 2020, 62, 96–117. [Google Scholar] [CrossRef]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3+2] azide− alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Baskin, J.M.; Prescher, J.A.; Lo, A.; Bertozzi, C.R. A comparative study of bioorthogonal reactions with azides. ACS Chem. Biol. 2006, 1, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Hida, N.; Kondo, K. Reactions of α-polyhalo ketone tosylhydrazones with sulfide ion and primary amines. Cyclization to 1,2,3-thiadiazoles and 1,2,3-triazoles. Bull. Chem. Soc. Jpn. 1986, 59, 179–183. [Google Scholar] [CrossRef] [Green Version]
- van Berkel, S.S.; Brauch, S.; Gabriel, L.; Henze, M.; Stark, S.; Vasilev, D.; Wessjohann, L.A.; Abbas, M.; Westermann, B. Traceless Tosylhydrazone-Based Triazole Formation: A Metal-Free Alternative to Strain-Promoted Azide—Alkyne Cycloaddition. Angew. Chem. Int. Ed. 2012, 51, 5343–5346. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.-W.; Cai, Z.-J.; Wang, S.-Y.; Ji, S.-J. Aerobic oxidative cycloaddition of α-chlorotosylhydrazones with arylamines: General chemoselective construction of 1,4-disubstituted and 1,5-disubstituted 1,2,3-triazoles under metal-free and azide-free conditions. Org. Lett. 2015, 17, 2898–2901. [Google Scholar] [CrossRef]
- Cai, Z.-J.; Lu, X.-M.; Zi, Y.; Yang, C.; Shen, L.-J.; Li, J.; Wang, S.-Y.; Ji, S.-J. I2/TBPB mediated oxidative reaction of N-tosylhydrazones with anilines: Practical construction of 1,4-disubstituted 1,2,3-triazoles under metal-free and azide-free conditions. Org. Lett. 2014, 16, 5108–5111. [Google Scholar] [CrossRef]
- Holbrey, J.D.; Seddon, K.R. The phase behaviour of 1-alkyl-3-methylimidazolium tetrafluoroborates; ionic liquids and ionic liquid crystals. J. Chem. Soc. Dalton Trans. 1999, 2133–2140. [Google Scholar] [CrossRef]
- Liu, X.-G.; Zhao, X.-L.; Zhang, Y.; Gao, J.-R. An Efficient Three-Component Reaction of Sodium Azide, Haloalkane and Alkyne for the Synthesis of 1,2,3-triazoles Catalyzed by the Bifunctional Ionic Liquid Catalyst Choline Chloride-CuCl in Water. Lett. Org. Chem. 2016, 13, 224–230. [Google Scholar] [CrossRef]
- Xu, Z. 1,2,3-Triazole-containing hybrids with potential antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem. 2020, 206, 112686. [Google Scholar] [CrossRef] [PubMed]
- Tozkoparan, B.; Küpeli, E.; Yeşilada, E.; Ertan, M. Preparation of 5-aryl-3-alkylthio-l,2,4-triazoles and corresponding sulfones with antiinflammatory–analgesic activity. Bioorganic Med. Chem. 2007, 15, 1808–1814. [Google Scholar] [CrossRef] [PubMed]
- Kinfe, H.H.; Belay, Y.H.; Joseph, J.S.; Mukwevho, E. Evaluation of the Influence of thiosemicarbazone–triazole hybrids on genes implicated in lipid oxidation and accumulation as potential anti-obesity agents. Bioorganic Med. Chem. Lett. 2013, 23, 5275–5278. [Google Scholar] [CrossRef]
- Chirke, S.S.; Krishna, J.S.; Rathod, B.B.; Bonam, S.R.; Khedkar, V.M.; Rao, B.V.; Sampath Kumar, H.M.; Shetty, P.R. Synthesis of Triazole Derivatives of 9-Ethyl-9H-carbazole and Dibenzo[b,d]furan and Evaluation of Their Antimycobacterial and Immunomodulatory Activity. ChemistrySelect 2017, 2, 7309–7318. [Google Scholar] [CrossRef]
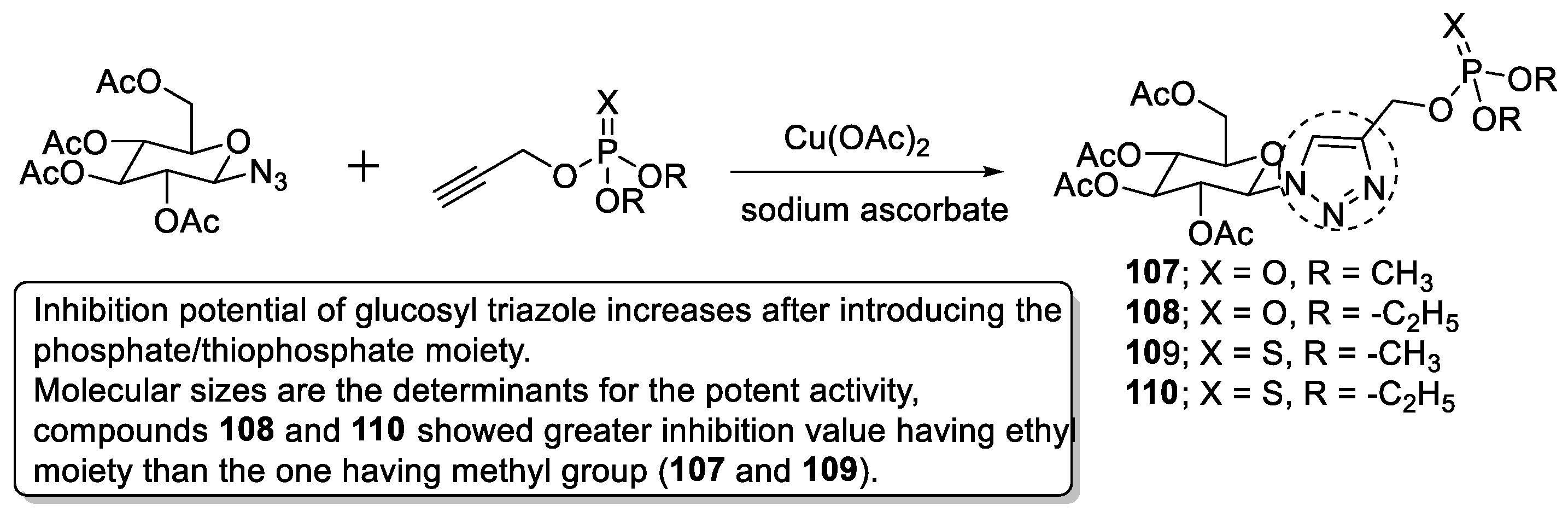
- Anjaneyulu, B.; Rao, G.D.; Bajaj, T. Click chemistry: In vitro evaluation of glycosyl hybrid phosphorylated/thiophosphorylated 1,2,3-triazole derivatives as irreversible acetyl cholinesterase (AChE) inhibitors. Results Chem. 2021, 3, 100093. [Google Scholar] [CrossRef]
- Li, B.; Huang, A.-L.; Zhang, Y.-L.; Li, Z.; Ding, H.-W.; Huang, C.; Meng, X.-M.; Li, J. Design, synthesis and evaluation of hesperetin derivatives as potential multifunctional anti-Alzheimer agents. Molecules 2017, 22, 1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Xing, S.; Chen, Y.; Liao, Q.; Xiong, B.; He, S.; Lu, W.; Liu, Y.; Yang, H.; Li, Q. Discovery and biological evaluation of a novel highly potent selective butyrylcholinsterase inhibitor. J. Med. Chem. 2020, 63, 10030–10044. [Google Scholar] [CrossRef]
- Wang, M.; Fang, L.; Liu, T.; Chen, X.; Zheng, Y.; Zhang, Y.; Chen, S.; Li, Z. Discovery of 7-O-1,2,3-triazole hesperetin derivatives as multi-target-directed ligands against Alzheimer’s disease. Chem. Biol. Interact. 2021, 342, 109489. [Google Scholar] [CrossRef]
- Yamazaki, M.; Sakura, N.; Chiba, K.; Mohri, T. Prevention of the neurotoxicity of the amyloid β protein by genipin. Biol. Pharm. Bull. 2001, 24, 1454–1455. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Wang, Y.; Li, J.; Zhang, Y.; Ma, X.; Zhu, P.; Zhang, Y. Design, synthesis, and evaluation of genipin derivatives for the treatment of Alzheimer’s Disease. Chem. Biol. Drug Des. 2019, 93, 110–122. [Google Scholar] [CrossRef]
- Silalai, P.; Jaipea, S.; Tocharus, J.; Athipornchai, A.; Suksamrarn, A.; Saeeng, R. New 1,2,3-Triazole-genipin Analogues and Their Anti-Alzheimer’s Activity. ACS Omega 2022, 7, 24302–24316. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Kang, Y.; Hwang, J.H.; Park, J.-H.; Shin, W.-H.; Mun, S.-K.; Lee, J.U.; Yee, S.-T.; Kim, H. Synthesis of 4-substituted benzyl-2-triazole-linked-tryptamine-paeonol derivatives and evaluation of their selective inhibitions against butyrylcholinesterase and monoamine oxidase-B. Int. J. Biol. Macromol. 2022, 217, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, S.A.; Arshia Ishtiaq, M.; Khan, K.M. Schiff bases in medicinal chemistry: A patent review (2010–2015). Expert Opin. Ther. Pat 2018, 28, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Hong, J. Recent advancements of 4-aminoquinazoline derivatives as kinase inhibitors and their applications in medicinal chemistry. Eur. J. Med. Chem. 2019, 170, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, B.; Hou, J.-Q.; Huang, S.-L.; Ou, T.-M.; Tan, J.-H.; An, L.-K.; Li, D.; Gu, L.-Q.; Huang, Z.-S. 2-(2-indolyl-)-4(3H)-quinazolines derivates as new inhibitors of AChE: Design, synthesis, biological evaluation and molecular modelling. J. Enzym. Inhib. Med. Chem. 2013, 28, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, J.; Polo, S.; Insuasty, B.; Gutiérrez, M.; Cáceres, D.; Alzate-Morales, J.H.; De-la-Torre, P.; Quiroga, J. Design, facile synthesis, and evaluation of novel spiro-and pyrazolo[1,5-c]quinazolines as cholinesterase inhibitors: Molecular docking and MM/GBSA studies. Comput. Biol. Chem. 2018, 74, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Lee, J.; Park, B.-G.; Park, I.; Pae, A.N.; Roh, E.J. Novel quinazoline-urea analogues as modulators for Aβ-induced mitochondrial dysfunction: Design, synthesis, and molecular docking study. Eur. J. Med. Chem. 2014, 84, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Nam, J.H.; Kim, J.H.; Kim, H.J.; Onnis, V.; Balboni, G.; Lee, K.-T.; Park, J.H.; Catto, M.; Carotti, A. 3,4-Dihydroquinazoline derivatives inhibit the activities of cholinesterase enzymes. Bioorganic Med. Chem. Lett. 2017, 27, 1179–1185. [Google Scholar] [CrossRef]
- Mohamed, T.; Rao, P.P. 2,4-Disubstituted quinazolines as amyloid-β aggregation inhibitors with dual cholinesterase inhibition and antioxidant properties: Development and structure-activity relationship (SAR) studies. Eur. J. Med. Chem. 2017, 126, 823–843. [Google Scholar] [CrossRef]
- Le-Nhat-Thuy, G.; Thi, N.N.; Pham-The, H.; Thi, T.A.D.; Thi, H.N.; Thi, T.H.N.; Hoang, S.N.; Van Nguyen, T. Synthesis and biological evaluation of novel quinazoline-triazole hybrid compounds with potential use in Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2020, 30, 127404. [Google Scholar] [CrossRef] [PubMed]
- Obniska, J.; Rapacz, A.; Rybka, S.; Góra, M.; Żmudzki, P.; Kamiński, K. Synthesis and Anticonvulsant Properties of New 3,3-Diphenyl-2,5-dioxo-pyrrolidin-1-yl-acetamides and 3,3-Diphenyl-propionamides. Arch. Pharm. 2017, 350, 1600368. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, M.; Lamie, P.; Ahmed, O. Synthesis of new quinolone derivatives linked to benzothiazole or benzoxazole moieties as anticancer and anti-oxidant agents. Med. Chem. 2016, 6, 652–657. [Google Scholar] [CrossRef] [Green Version]
- Sales, E.M.; Figueroa-Villar, J.D. Recent studies about synthesis and biological activity of quinolones and derivatives: A Review. World J. Pharm. Pharm. Sci. 2016, 5, 253–268. [Google Scholar]
- Boteva, A.; Krasnykh, O. The methods of synthesis, modification, and biological activity of 4-quinolones. Chem. Heterocycl. Compd. 2009, 45, 757–785. [Google Scholar] [CrossRef]
- Larsson, E.A.; Jansson, A.; Ng, F.M.; Then, S.W.; Panicker, R.; Liu, B.; Sangthongpitag, K.; Pendharkar, V.; Tai, S.J.; Hill, J. Fragment-based ligand design of novel potent inhibitors of tankyrases. J. Med. Chem. 2013, 56, 4497–4508. [Google Scholar] [CrossRef]
- Mermer, A.; Demirbaş, N.; Şirin, Y.; Uslu, H.; Özdemir, Z.; Demirbaş, A. Conventional and microwave prompted synthesis, antioxidant, anticholinesterase activity screening and molecular docking studies of new quinolone-triazole hybrids. Bioorganic Chem. 2018, 78, 236–248. [Google Scholar] [CrossRef]
- Mantoani, S.P.; Chierrito, T.P.; Vilela, A.F.; Cardoso, C.L.; Martínez, A.; Carvalho, I. Novel triazole-quinoline derivatives as selective dual binding site acetylcholinesterase inhibitors. Molecules 2016, 21, 193. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Wu, P.; Yang, R.; Gao, L.; Li, C.; Wang, D.; Wu, S.; Liu, A.-L.; Du, G.-H. Inhibition of acetylcholinesterase by two genistein derivatives: Kinetic analysis, molecular docking and molecular dynamics simulation. Acta Pharm. Sin. B 2014, 4, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Alhomida, A.S.; Al-Rajhi, A.A.; Kamal, M.A.; Al-Jafari, A.A. Kinetic analysis of the toxicological effect of tacrine (Cognex®) on human retinal acetylcholinesterase activity. Toxicology 2000, 147, 33–39. [Google Scholar] [CrossRef]
- Saeedi, M.; Ansari, S.; Mahdavi, M.; Sabourian, R.; Akbarzadeh, T.; Foroumadi, A.; Shafiee, A. Synthesis of Novel 1,2,3-Triazole-dihydro[3,2-c]chromenones as Acetylcholinesterase Inhibitors. Synth. Commun. 2015, 45, 2311–2318. [Google Scholar] [CrossRef]
- Bagheri, S.M.; Khoobi, M.; Nadri, H.; Moradi, A.; Emami, S.; Jalili-Baleh, L.; Jafarpour, F.; Homayouni Moghadam, F.; Foroumadi, A.; Shafiee, A. Synthesis and anticholinergic activity of 4-hydroxycoumarin derivatives containing substituted benzyl-1,2,3-triazole moiety. Chem. Biol. Drug Des. 2015, 86, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.C.; Gonçalves, G.A.; Vanzolini, K.L.; Merlo, A.A.; Gauer, B.; Holzschuh, M.; Andrade, S.; Piedade, M.; Garcia, S.C.; Carvalho, I.; et al. Combining the pharmacophore features of coumarins and 1,4-substituted 1,2,3-triazoles to design new acetylcholinesterase inhibitors: Fast and easy generation of 4-methylcoumarins/1,2,3-triazoles conjugates via click chemistry. J. Braz. Chem. Soc. 2016, 27, 1541–1550. [Google Scholar] [CrossRef]
- Saeedi, M.; Safavi, M.; Karimpour-Razkenari, E.; Mahdavi, M.; Edraki, N.; Moghadam, F.H.; Khanavi, M.; Akbarzadeh, T. Synthesis of novel chromenones linked to 1,2,3-triazole ring system: Investigation of biological activities against Alzheimer’s disease. Bioorganic Chem. 2017, 70, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Moradi, A.; Faraji, L.; Nadri, H.; Hasanpour, Z.; Moghadam, F.H.; Pakseresht, B.; Golshani, M.; Moghimi, S.; Ramazani, A.; Firoozpour, L.; et al. Synthesis, docking study, and biological evaluation of novel umbellipherone/hymecromone derivatives as acetylcholinesterase/butyrylcholinesterase inhibitors. Med. Chem. Res. 2018, 27, 1741–1747. [Google Scholar] [CrossRef]
- Sepehri, N.; Mohammadi-Khanaposhtani, M.; Asemanipoor, N.; Hosseini, S.; Biglar, M.; Larijani, B.; Mahdavi, M.; Hamedifar, H.; Taslimi, P.; Sadeghian, N. Synthesis, characterization, molecular docking, and biological activities of coumarin—1,2,3-triazole-acetamide hybrid derivatives. Archiv der Pharmazie 2020, 353, 2000109. [Google Scholar] [CrossRef]
- Rastegari, A.; Nadri, H.; Mahdavi, M.; Moradi, A.; Mirfazli, S.S.; Edraki, N.; Moghadam, F.H.; Larijani, B.; Akbarzadeh, T.; Saeedi, M. Design, synthesis and anti-Alzheimer’s activity of novel 1,2,3-triazole-chromenone carboxamide derivatives. Bioorganic Chem. 2019, 83, 391–401. [Google Scholar] [CrossRef]
- Singh, A.; Sharma, S.; Arora, S.; Attri, S.; Kaur, P.; Gulati, H.K.; Bhagat, K.; Kumar, N.; Singh, H.; Singh, J.V.; et al. New coumarin-benzotriazole based hybrid molecules as inhibitors of acetylcholinesterase and amyloid aggregation. Bioorganic Med. Chem. Lett. 2020, 30, 127477. [Google Scholar] [CrossRef]
- Park, J.-Y.; Shin, S.; Park, K.C.; Jeong, E.; Park, J.H. Synthesis and in vitro Assay of New Triazole Linked Decursinol Derivatives Showing Inhibitory Activity against Cholinesterase for Alzheimer’s Disease Therapeutics. J. Korean Chem. Soc. 2016, 60, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Jalili-Baleh, L.; Forootanfar, H.; Küçükkılınç, T.T.; Nadri, H.; Abdolahi, Z.; Ameri, A.; Jafari, M.; Ayazgok, B.; Baeeri, M.; Rahimifard, M.; et al. Design, synthesis and evaluation of novel multi-target-directed ligands for treatment of Alzheimer’s disease based on coumarin and lipoic acid scaffolds. Eur. J. Med. Chem. 2018, 152, 600–614. [Google Scholar] [CrossRef]
- Jalili-Baleh, L.; Nadri, H.; Forootanfar, H.; Samzadeh-Kermani, A.; Küçükkılınç, T.T.; Ayazgok, B.; Rahimifard, M.; Baeeri, M.; Doostmohammadi, M.; Firoozpour, L.; et al. Novel 3-phenylcoumarin–lipoic acid conjugates as multi-functional agents for potential treatment of Alzheimer’s disease. Bioorganic Chem. 2018, 79, 223–234. [Google Scholar] [CrossRef]
- Chekir, S.; Debbabi, M.; Regazzetti, A.; Dargère, D.; Laprévote, O.; Jannet, H.B.; Gharbi, R. Design, synthesis and biological evaluation of novel 1,2,3-triazole linked coumarinopyrazole conjugates as potent anticholinesterase, anti-5-lipoxygenase, anti-tyrosinase and anti-cancer agents. Bioorganic Chem. 2018, 80, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Askarani, H.K.; Iraji, A.; Rastegari, A.; Bukhari, S.N.A.; Firuzi, O.; Akbarzadeh, T.; Saeedi, M. Design and synthesis of multi-target directed 1,2,3-triazole-dimethylaminoacryloyl-chromenone derivatives with potential use in Alzheimer’s disease. BMC Chem. 2020, 14, 1–13. [Google Scholar] [CrossRef]
- Roussaki, M.; Kontogiorgis, C.A.; Hadjipavlou-Litina, D.; Hamilakis, S.; Detsi, A. A novel synthesis of 3-aryl coumarins and evaluation of their antioxidant and lipoxygenase inhibitory activity. Bioorganic Med. Chem. Lett. 2010, 20, 3889–3892. [Google Scholar] [CrossRef]
- Kiani, A.; Jalili-baleh, L.; Abdollahi, Z.; Nadri, H.; Foroumadi, A.; Sadat Ebrahimi, S.E.; Khoobi, M. Cholinesterase inhibition activity and docking simulation study of coumarin mannich base derivatives. J. Sci. Islam. Repub. Iran 2019, 30, 5–12. [Google Scholar]
- Pourabdi, L.; Küçükkılınç, T.T.; Khoshtale, F.; Ayazgök, B.; Nadri, H.; Farokhi Alashti, F.; Forootanfar, H.; Akbari, T.; Shafiei, M.; Foroumadi, A. Synthesis of New 3-Arylcoumarins Bearing N-Benzyl Triazole Moiety: Dual Lipoxygenase and Butyrylcholinesterase Inhibitors With Anti-Amyloid Aggregation and Neuroprotective Properties Against Alzheimer’s Disease. Front. Chem. 2022, 9, 810233. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Malaguti, M.; Barbalace, M.C.; Hrelia, S. Bioactivity of olive oil phenols in neuroprotection. Int. J. Mol. Sci. 2017, 18, 2230. [Google Scholar] [CrossRef] [Green Version]
- Atochin, D.N.; Chernysheva, G.A.; Smolyakova, V.I.; Osipenko, A.N.; Logvinov, S.V.; Zhdankina, A.A.; Sysolyatin, S.V.; Kryukov, Y.A.; Anfinogenova, Y.; Plotnikova, T.M. Neuroprotective effects of p-tyrosol after the global cerebral ischemia in rats. Phytomedicine 2016, 23, 784–792. [Google Scholar] [CrossRef]
- Cremonini, A.L.; Caffa, I.; Cea, M.; Nencioni, A.; Odetti, P.; Monacelli, F. Nutrients in the prevention of Alzheimer’s disease. Oxidative Med. Cell. Longev. 2019, 2019, 9874159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Yamamoto, F.; Arai, T.; Yang, J.; Sakai, Y.; Itoh, M.; Mamada, N.; Sekiguchi, M.; Yamada, D.; Saitoh, A. Tyrosol reduces amyloid-β oligomer neurotoxicity and alleviates synaptic, oxidative, and cognitive disturbances in Alzheimer’s disease model mice. J. Alzheimer’s Dis. 2019, 70, 937–952. [Google Scholar] [CrossRef]
- Huo, J.; Hu, H.; Zhang, M.; Hu, X.; Chen, M.; Chen, D.; Liu, J.; Xiao, G.; Wang, Y.; Wen, Z. A mini review of the synthesis of poly-1,2,3-triazole-based functional materials. RSC Adv. 2017, 7, 2281–2287. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-C.; Zhang, J.; Rodrigues, M.C.; Ding, D.-J.; Longo, J.P.F.; Azevedo, R.B.; Muehlmann, L.A.; Jiang, C.-S. Synthesis and evaluation of novel 1,2,3-triazole-based acetylcholinesterase inhibitors with neuroprotective activity. Bioorganic Med. Chem. Lett. 2016, 26, 3881–3885. [Google Scholar] [CrossRef]
- Bousada, G.M.; de Sousa, B.L.; Furlani, G.; Agrizzi, A.P.; Ferreira, P.G.; Leite, J.P.V.; Mendes, T.A.d.O.; Varejão, E.V.; Pilau, E.J.; Dos Santos, M.H. Tyrosol 1,2,3-triazole analogues as new acetylcholinesterase (AChE) inhibitors. Comput. Biol. Chem. 2020, 88, 107359. [Google Scholar] [CrossRef]
- Sousa, B.L.d.; Leite, J.P.; Mendes, T.A.; Varejão, E.V.; Chaves, A.; Silva, J.G.d.; Agrizzi, A.P.; Ferreira, P.G.; Pilau, E.J.; Silva, E. Inhibition of acetylcholinesterase by coumarin-linked amino acids synthetized via triazole associated with molecule partition coefficient. J. Braz. Chem. Soc. 2021, 32, 652–664. [Google Scholar] [CrossRef]
- Najafi, Z.; Mahdavi, M.; Saeedi, M.; Karimpour-Razkenari, E.; Asatouri, R.; Vafadarnejad, F.; Moghadam, F.H.; Khanavi, M.; Sharifzadeh, M.; Akbarzadeh, T. Novel tacrine-1,2,3-triazole hybrids: In vitro, in vivo biological evaluation and docking study of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 125, 1200–1212. [Google Scholar] [CrossRef]
- Wu, G.; Gao, Y.; Kang, D.; Huang, B.; Huo, Z.; Liu, H.; Poongavanam, V.; Zhan, P.; Liu, X. Design, synthesis and biological evaluation of tacrine-1,2,3-triazole derivatives as potent cholinesterase inhibitors. MedChemComm 2018, 9, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roldan-Pena, J.M.; Romero-Real, V.; Hicke, J.; Maya, I.; Franconetti, A.; Lagunes, I.; Padron, J.M.; Petralla, S.; Poeta, E.; Naldi, M. Tacrine-O-protected phenolics heterodimers as multitarget-directed ligands against Alzheimer’s disease: Selective subnanomolar BuChE inhibitors. Eur. J. Med. Chem. 2019, 181, 111550. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Anand, A.; Kumar, K.; Kumar, V. Recent developments in biological aspects of chalcones: The odyssey continues. Expert Opin. Drug Discov. 2019, 14, 249–288. [Google Scholar] [CrossRef]
- Rani, A.; Singh, A.; Kaur, J.; Singh, G.; Bhatti, R.; Gumede, N.; Kisten, P.; Singh, P.; Kumar, V. 1H-1,2,3-triazole grafted tacrine-chalcone conjugates as potential cholinesterase inhibitors with the evaluation of their behavioral tests and oxidative stress in mice brain cells. Bioorganic Chem. 2021, 114, 105053. [Google Scholar] [CrossRef] [PubMed]
- Najafi, Z.; Mahdavi, M.; Saeedi, M.; Karimpour-Razkenari, E.; Edraki, N.; Sharifzadeh, M.; Khanavi, M.; Akbarzadeh, T. Novel tacrine-coumarin hybrids linked to 1,2,3-triazole as anti-Alzheimer’s compounds: In vitro and in vivo biological evaluation and docking study. Bioorganic Chem. 2019, 83, 303–316. [Google Scholar] [CrossRef]
- Gulati, H.K.; Choudhary, S.; Kumar, N.; Ahmed, A.; Bhagat, K.; Singh, J.V.; Singh, A.; Kumar, A.; Bedi, P.M.S.; Singh, H. Design, Synthesis, biological investigations and molecular interactions of triazole linked tacrine glycoconjugates as Acetylcholinesterase inhibitors with reduced hepatotoxicity. Bioorganic Chem. 2022, 118, 105479. [Google Scholar] [CrossRef]
- Arslan, T.; Çakır, N.; Keleş, T.; Biyiklioglu, Z.; Senturk, M. Triazole substituted metal-free, metallo-phthalocyanines and their water soluble derivatives as potential cholinesterases inhibitors: Design, synthesis and in vitro inhibition study. Bioorganic Chem. 2019, 90, 103100. [Google Scholar] [CrossRef] [PubMed]
- Kantekin, H.; Yalazan, H.; Barut, B.; Güngör, Ö.; Ünlüer, D.; Demirbaş, Ü.; Özel, A.; Durmuş, M. Dual-purpose both peripheral and non-peripheral triazole substituted ZnII, MgII and PbII phthalocyanines: Synthesis, characterization, photophysicochemical and acetylcholinesterase inhibitory properties. Polyhedron 2021, 208, 115416. [Google Scholar] [CrossRef]
- Çelik, F.; Türkan, F.; Aras, A.; Atalar, M.N.; Karaman, H.S.; Ünver, Y.; Kahriman, N. Synthesis of novel 1,2,3 triazole derivatives and assessment of their potential cholinesterases, glutathione S-transferase enzymes inhibitory properties: An in vitro and in silico study. Bioorganic Chem. 2021, 107, 104606. [Google Scholar] [CrossRef] [PubMed]
- Faraji, L.; Shahkarami, S.; Nadri, H.; Moradi, A.; Saeedi, M.; Foroumadi, A.; Ramazani, A.; Haririan, I.; Ganjali, M.R.; Shafiee, A. Synthesis of novel benzimidazole and benzothiazole derivatives bearing a 1,2,3-triazole ring system and their acetylcholinesterase inhibitory activity. J. Chem. Res. 2017, 41, 30–35. [Google Scholar] [CrossRef]
- Akrami, H.; Mirjalili, B.F.; Khoobi, M.; Moradi, A.; Nadri, H.; Emami, S.; Foroumadi, A.; Vosooghi, M.; Shafiee, A. 9H-Carbazole Derivatives Containing the N-Benzyl-1,2,3-triazole Moiety as New Acetylcholinesterase Inhibitors. Arch. Pharm. 2015, 348, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.; Pourmousavi, S.A.; Mahdavi, M.; Taslimi, P. Synthesis, and in vitro biological evaluations of novel naphthoquinone conjugated to aryl triazole acetamide derivatives as potential anti-Alzheimer agents. J. Mol. Struct. 2022, 1255, 132229. [Google Scholar] [CrossRef]
- Jain, A.; Piplani, P. Design, synthesis and biological evaluation of triazole-oxadiazole conjugates for the management of cognitive dysfunction. Bioorganic Chem. 2020, 103, 104151. [Google Scholar] [CrossRef]
- Mohammadi-Khanaposhtani, M.; Saeedi, M.; Zafarghandi, N.S.; Mahdavi, M.; Sabourian, R.; Razkenari, E.K.; Alinezhad, H.; Khanavi, M.; Foroumadi, A.; Shafiee, A. Potent acetylcholinesterase inhibitors: Design, synthesis, biological evaluation, and docking study of acridone linked to 1,2,3-triazole derivatives. Eur. J. Med. Chem. 2015, 92, 799–806. [Google Scholar] [CrossRef]
- Lan, T.T.; Anh, D.T.; Dung, D.T.M.; Huong, L.T.T.; Park, E.J.; Jeon, H.W.; Kang, J.S.; Thuan, N.T.; Han, S.-B.; Nam, N.-H. Design, synthesis, and bioevaluation of novel oxoindolin-2-one derivatives incorporating 1-benzyl-1H-1,2,3-triazole. Med. Chem. Res. 2020, 29, 396–408. [Google Scholar] [CrossRef]
- Marques, C.S.; López, Ó.; Bagetta, D.; Carreiro, E.P.; Petralla, S.; Bartolini, M.; Hoffmann, M.; Alcaro, S.; Monti, B.; Bolognesi, M.L. N-1,2,3-triazole-isatin derivatives for cholinesterase and β-amyloid aggregation inhibition: A comprehensive bioassay study. Bioorganic Chem. 2020, 98, 103753. [Google Scholar] [CrossRef]
- Saeedi, M.; Maleki, A.; Iraji, A.; Hariri, R.; Akbarzadeh, T.; Edraki, N.; Firuzi, O.; Mirfazli, S.S. Synthesis and bio-evaluation of new multifunctional methylindolinone-1,2,3-triazole hybrids as anti-Alzheimer’s agents. J. Mol. Struct. 2021, 1229, 129828. [Google Scholar] [CrossRef]
- Bhagat, K.; Singh, J.V.; Sharma, A.; Kaur, A.; Kumar, N.; Gulati, H.K.; Singh, A.; Singh, H.; Bedi, P.M.S. Novel series of triazole containing coumarin and isatin based hybrid molecules as acetylcholinesterase inhibitors. J. Mol. Struct. 2021, 1245, 131085. [Google Scholar] [CrossRef]
- Mehrazar, M.; Hassankalhori, M.; Toolabi, M.; Goli, F.; Moghimi, S.; Nadri, H.; Bukhari, S.N.A.; Firoozpour, L.; Foroumadi, A. Design and synthesis of benzodiazepine-1,2,3-triazole hybrid derivatives as selective butyrylcholinesterase inhibitors. Mol. Divers. 2020, 24, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tian, L.; Wu, L.; Chen, H.; Wang, N.; Liu, X.; Zhao, C.; Wu, Z.; Jiang, X.; Wu, Q. Discovery of novel β-carboline-1,2,3-triazole hybrids as AChE/GSK-3β dual inhibitors for Alzheimer’s disease treatment. Bioorganic Chem. 2022, 129, 106168. [Google Scholar] [CrossRef]
- Merde, İ.B.; Önel, G.T.; Türkmenoğlu, B.; Gürsoy, Ş.; Dilek, E. Novel(p-Tolyl)-3(2H)-Pyridazinone Derivatives Containing Substituted-1,2,3-Triazole Moiety as New Anti-Alzheimer Agents: Synthesis, In vitro and In silico Assays. FABAD J. Pharm. Sci. 2022, 47, 355–368. [Google Scholar]
- Park, J.Y.; Shin, S.; Kim, J.k.; Park, K.C.; Park, J.H. Synthesis of Benzoisoxazole Derivatives and Evaluation of Inhibitory Potency against Cholinesterase for Alzheimer’s Disease Therapeutics. Bull. Korean Chem. Soc. 2016, 37, 1464–1471. [Google Scholar] [CrossRef]
- Son, M.; Lee, H.; Jeon, C.; Kang, Y.; Park, C.; Lee, K.W.; Park, J.H. Tryptamine–Triazole Hybrid Compounds for Selective Butyrylcholinesterase Inhibition. Bull. Korean Chem. Soc. 2019, 40, 544–553. [Google Scholar] [CrossRef]
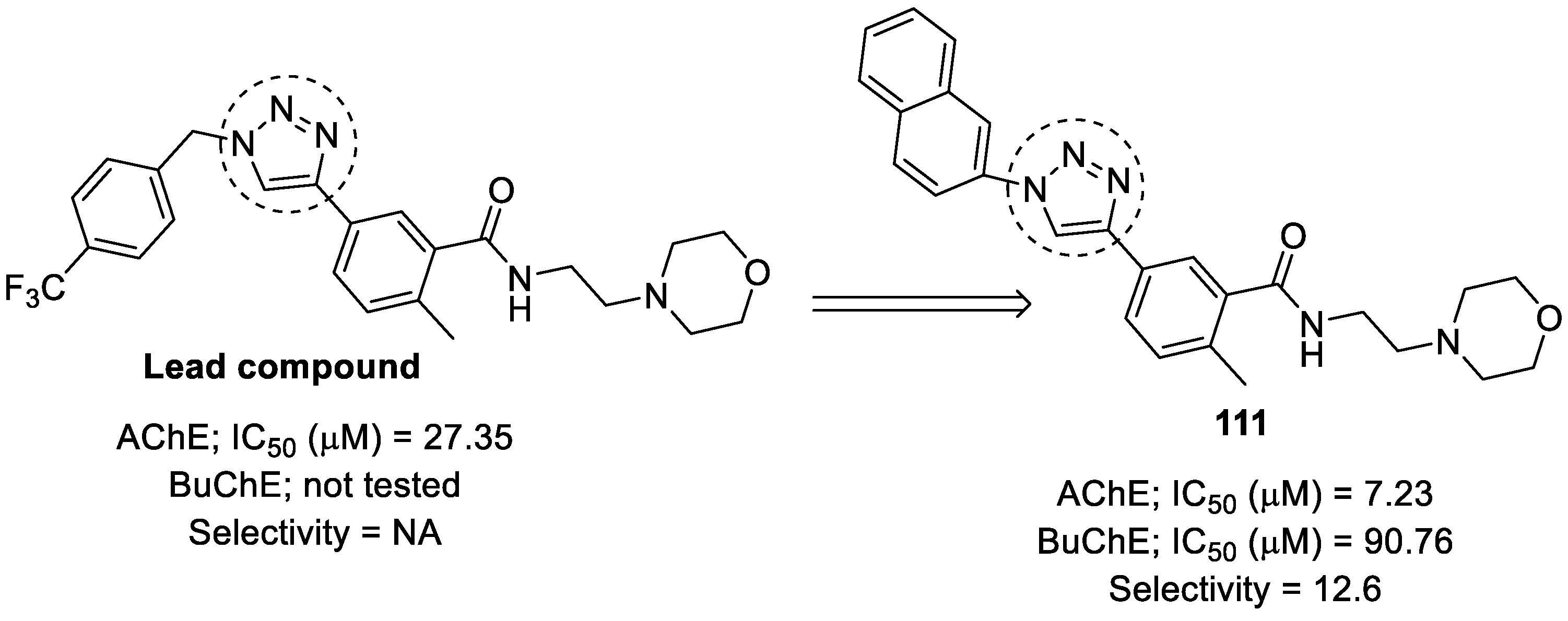
- Ouach, A.; Pin, F.; Bertrand, E.; Vercouillie, J.; Gulhan, Z.; Mothes, C.; Deloye, J.-B.; Guilloteau, D.; Suzenet, F.; Chalon, S. Design of α7 nicotinic acetylcholine receptor ligands using the (het) Aryl-1,2,3-triazole core: Synthesis, in vitro evaluation and SAR studies. Eur. J. Med. Chem. 2016, 107, 153–164. [Google Scholar] [CrossRef]
- Petrat, W.; Wattanapiromsakul, C.; Nualnoi, T.; Sabri, N.H.; Lee, V.S.L.; Lomlim, L. Cholinesterase inhibitory activity, kinetic and molecular docking studies of N-(1-substituted-1H-1,2,3-triazole-4-yl)-aralkylamide derivatives. Walailak J. Sci. Technol. (WJST) 2017, 14, 687–701. [Google Scholar]
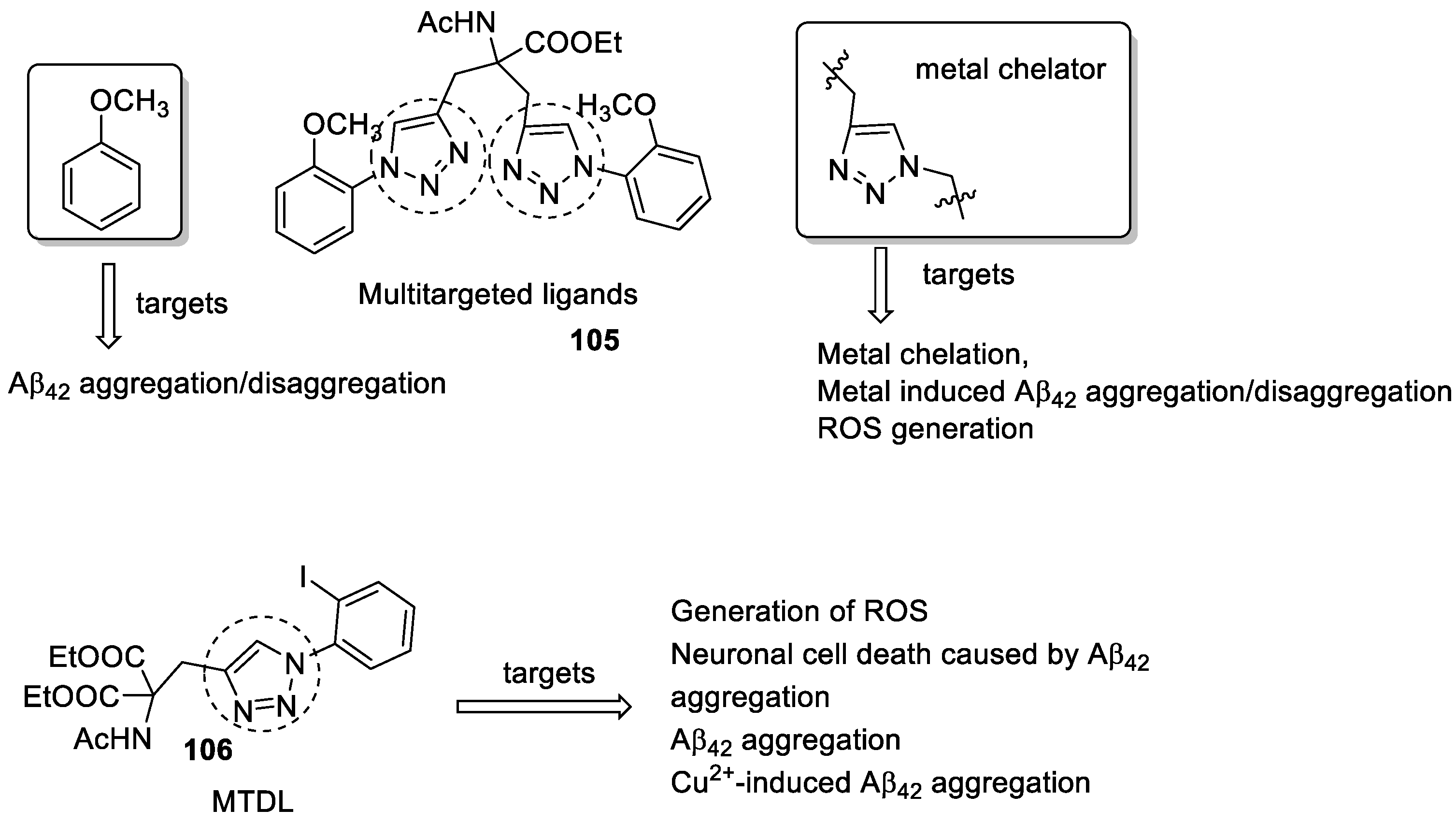
- Kaur, A.; Mann, S.; Kaur, A.; Priyadarshi, N.; Goyal, B.; Singhal, N.K.; Goyal, D. Multi-target-directed triazole derivatives as promising agents for the treatment of Alzheimer’s disease. Bioorganic Chem. 2019, 87, 572–584. [Google Scholar] [CrossRef]
- Kaur, A.; Narang, S.S.; Kaur, A.; Mann, S.; Priyadarshi, N.; Goyal, B.; Singhal, N.K.; Goyal, D. Multifunctional mono-triazole derivatives inhibit Aβ42 aggregation and Cu2+-mediated Aβ42 aggregation and protect against Aβ42-induced cytotoxicity. Chem. Res. Toxicol. 2019, 32, 1824–1839. [Google Scholar] [CrossRef] [PubMed]
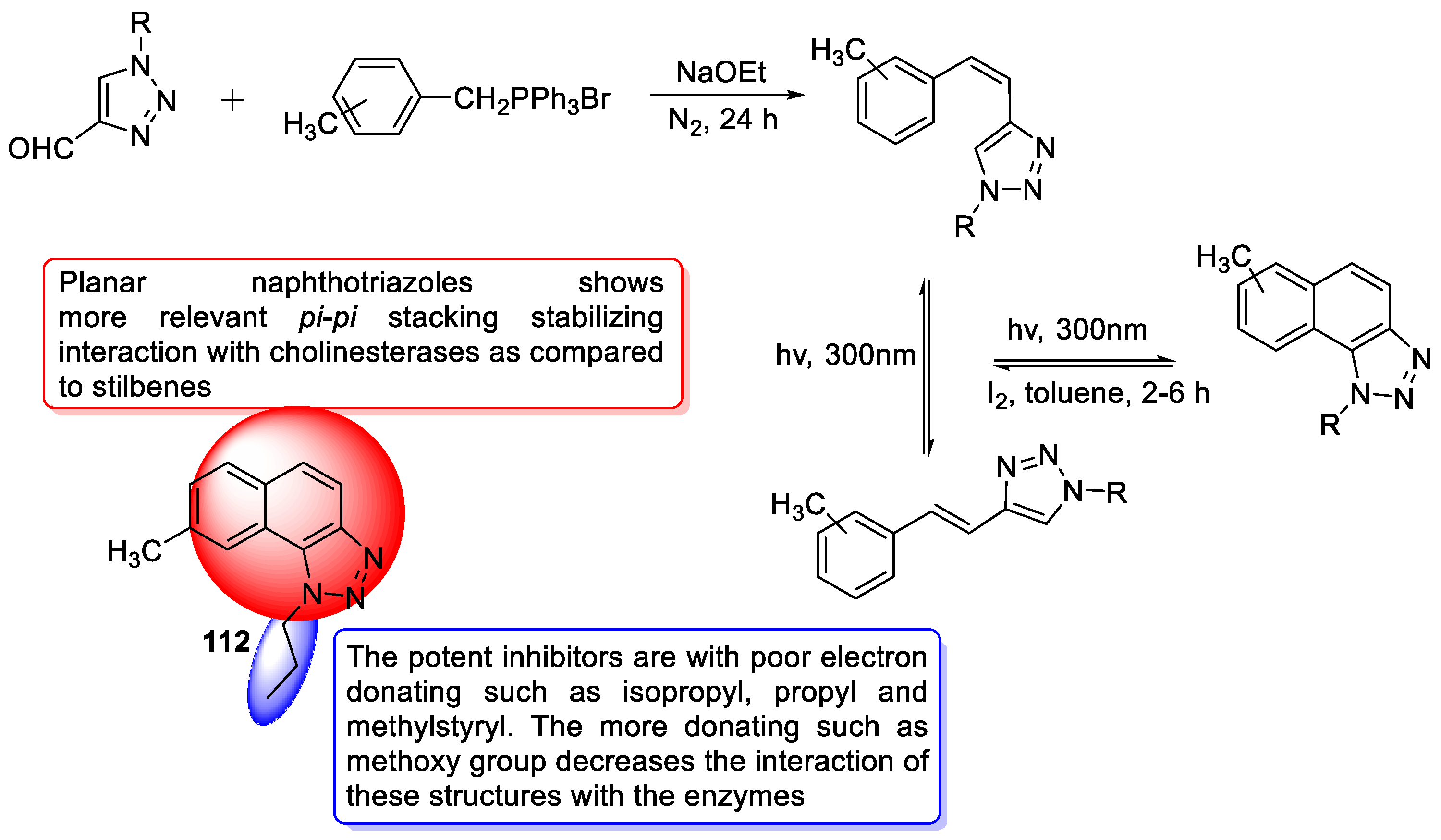
- Mlakić, M.; Faraho, I.; Odak, I.; Talić, S.; Vukovinski, A.; Raspudić, A.; Bosnar, M.; Zadravec, R.; Ratković, A.; Lasić, K. Synthesis, photochemistry and computational study of novel 1,2,3-triazole heterostilbenes: Expressed biological activity of their electrocyclization photoproducts. Bioorganic Chem. 2022, 121, 105701. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Odak, I.; Faraho, I.; Talić, S.; Bosnar, M.; Lasić, K.; Barić, D.; Škorić, I. New naphtho/thienobenzo-triazoles with interconnected anti-inflammatory and cholinesterase inhibitory activity. Eur. J. Med. Chem. 2022, 241, 114616. [Google Scholar] [CrossRef] [PubMed]
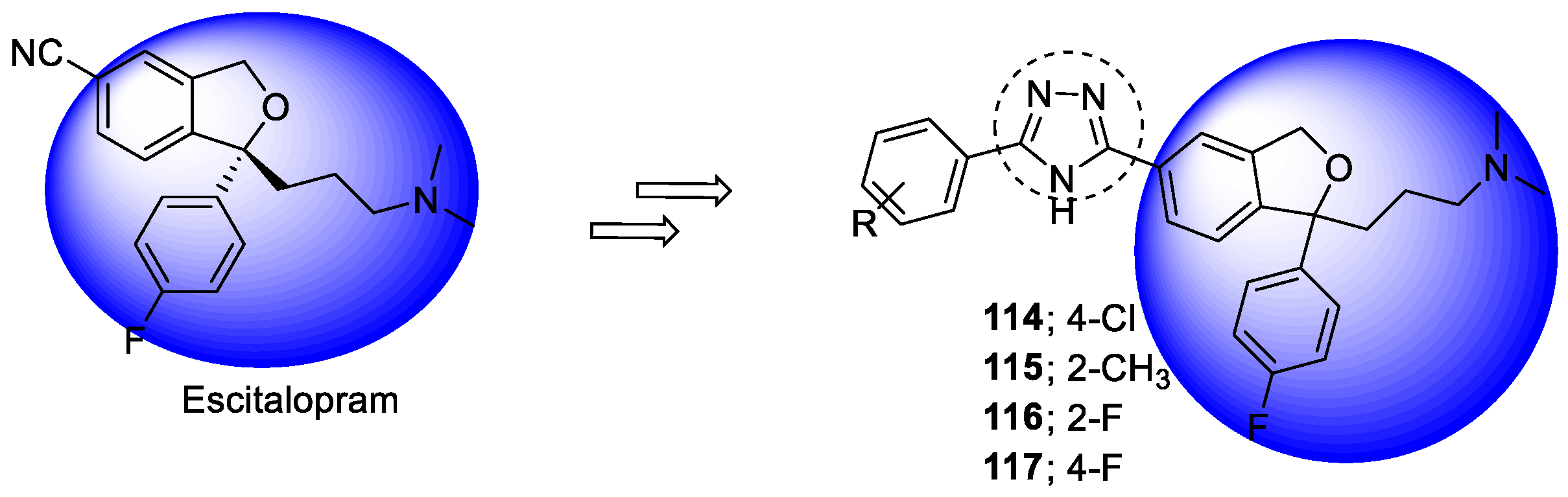
- Mehr un, N.; Munawar, M.A.; Chattha, F.A.; Kousar, S.; Munir, J.; Ismail, T.; Ashraf, M.; Khan, M.A. Synthesis of novel triazoles and a tetrazole of escitalopram as cholinesterase inhibitors. Bioorg. Med. Chem. 2015, 23, 6014–6024. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Almaz, Z. A series of 1,2,3-triazole compounds: Synthesis, characterization, and investigation of the cholinesterase inhibitory properties via in vitro and in silico studies. J. Mol. Struct. 2022, 1277, 134854. [Google Scholar] [CrossRef]
















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacological Activities | Chemical Structures |
|---|---|
| Antifungal |  |
| Antibiotic |  |
| Anticancer |  |
| Antiviral |  |
| Miscellaneous |  |
| S. No | Chemical Structure | Neuroprotective Activity | Structural Features | Ref. |
|---|---|---|---|---|
| 1 |  | 55% inhibition of AChE at 40 μg/mL. | Substitution of –Br group with a –CH3 or –Cl in the phenyl ring decreases activity. Introduction of –OCH3 group to the aryloxy moiety does not improve anti-AChE activity. | [81] |
| 2 |  | (IC50 = 0.18 μM-hAChE). | 2-Cl on phenyl ring significantly improves the activity, but changing its position to p or replacing it with p-Br/p-NO2 greatly reduces anti-AChE activity (~40–250-times). | [82] |
| 3 |  | 59.52% inhibition of AChE at 200 μmol/L. | The phenylethyl ring is important for exhibiting AChE inhibition | [83] |
| 4 |  | (IC50 = 15.42 μM)—A dual inhibitor of AChE. Protected PC12 neurons against H2O2-induced cell death. | Removal of 8-OCH3 group and increasing the number of chlorine atoms at 3- and 4- positions of the phenyl ring decreases AChE inhibitory activity. Unsubstituted coumarin and the phenyl ring of 1,2,3-triazole display very weak activity. | [84] |
| 5 |  | IC50 = 3.4 and 1.1 μM—AChE and BuChE inhibition. | 2-substituted phenyl ring derivatives (Cl > F > NO2) are more active against AChE and BuChE than the unsubstituted derivatives. | [85] |
| 6 |  | Potent AChE inhibitor (Ki: 24.85 nM). | More selective toward AChE over BuChE. Acetamide group improves activity. | [86] |
| 7 |  | 30: selective AChE inhibitory activity (IC50 = 1.80 μM). 31: anti-BuChE activity (IC50 = 1.71 μM). Metal chelator. | 3,4-di-methylaryl group is important for AChE inhibition. Substitution with halogen decreases AChE inhibition but enhances anti-BuChE activity. | [87] |
| 8 |  | Mixed-type AChE inhibition (IC50 = 0.059 μΜ), copper-induced Aβ1–42 aggregation inhibition (34.26% at 50 μΜ). | A 3-carbon alkyl chain linker between coumarin and 1,2,3-triazole is optimal for activity. Selective AChEI, as it failed to inhibit the BuChE. | [88] |
| 9 |  | More potent BuChE inhibitor (IC50 = 5.89 ± 0.31 μM) than galantamine (IC50 = 9.4 ± 2.5 μM). | It is a decursinol hybrid which itself is not active against BuChE. Increase in activity of hybrids could be attributed to the 1,2,3-triazole–lipoic acid scaffold. | [89]. |
| 10 |  | Inhibits both AChE and BuChE (IC50 of 7.3 and 68.6 μM). | A 4-carbon chain-linker between 1,2,3-triazole and 3-hydroxycoumarin is optimal for inhibitory activity. | [90] |
| 11 |  | AChEI (IC50 = 16.4 μM)—Inhibitor of self-induced and AChE-induced Aβ1–42 aggregation (51.2 and 47.4%); about twofold stronger than donepezil (26 and 22.1%). Inhibits the formation of intracellular reactive oxygen species (ROS) in PC12 neuronal cells. Selective biometal chelator. | 3,4-OCH3 phenyl at the C-3 position of a coumarin ring and linked via a three-carbon-long chain to 1,2,3-triazole is the most potent compound of the series. | [91] |
| 12 |  | AChEI (IC50 = 18 μM) 5- LOX inhibitor. | The potent anti-AChE activity of this compound is attributed to the presence of three -Cl atoms on the phenyl ring. | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.A.; Akhtar, M.J.; Gogoi, U.; Meenakshi, D.U.; Das, A. An Overview of 1,2,3-triazole-Containing Hybrids and Their Potential Anticholinesterase Activities. Pharmaceuticals 2023, 16, 179. https://doi.org/10.3390/ph16020179
Khan SA, Akhtar MJ, Gogoi U, Meenakshi DU, Das A. An Overview of 1,2,3-triazole-Containing Hybrids and Their Potential Anticholinesterase Activities. Pharmaceuticals. 2023; 16(2):179. https://doi.org/10.3390/ph16020179
Chicago/Turabian StyleKhan, Shah Alam, Mohammad Jawaid Akhtar, Urvashee Gogoi, Dhanalekshmi Unnikrishnan Meenakshi, and Aparoop Das. 2023. "An Overview of 1,2,3-triazole-Containing Hybrids and Their Potential Anticholinesterase Activities" Pharmaceuticals 16, no. 2: 179. https://doi.org/10.3390/ph16020179