

Regulatory Mechanism on Anti-Glycolytic and Anti-Metastatic Activities Induced by Strobilanthes crispus in Breast Cancer, In Vitro
 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
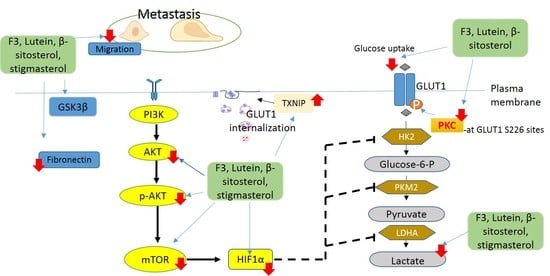
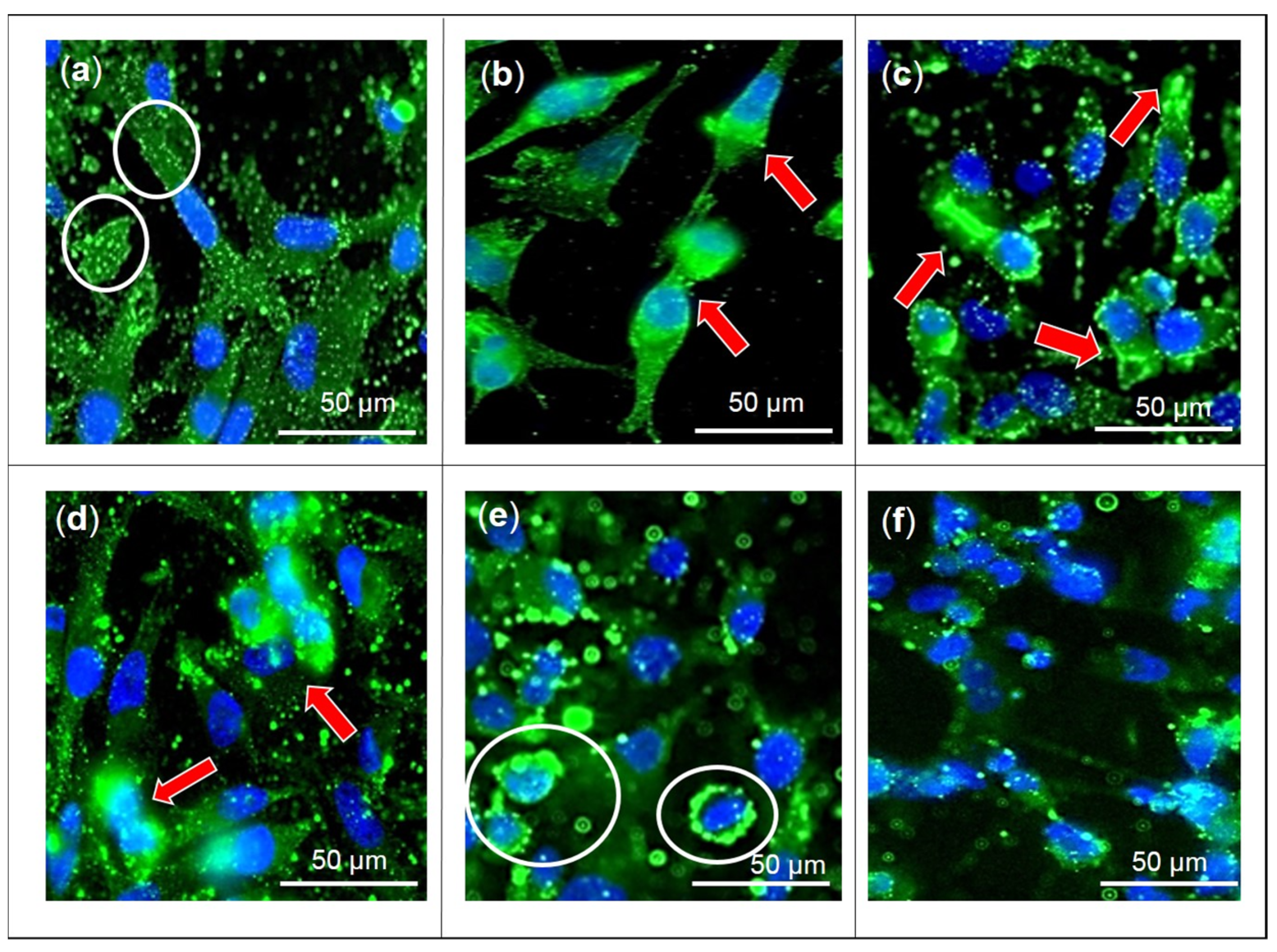
2.1. Effects of F3, Lutein, β-sitosterol, and Stigmasterol on GLUT1 Protein Expression and Localization
2.2. Effects of F3, Lutein, β-sitosterol, and Stigmasterol on PKC Activity
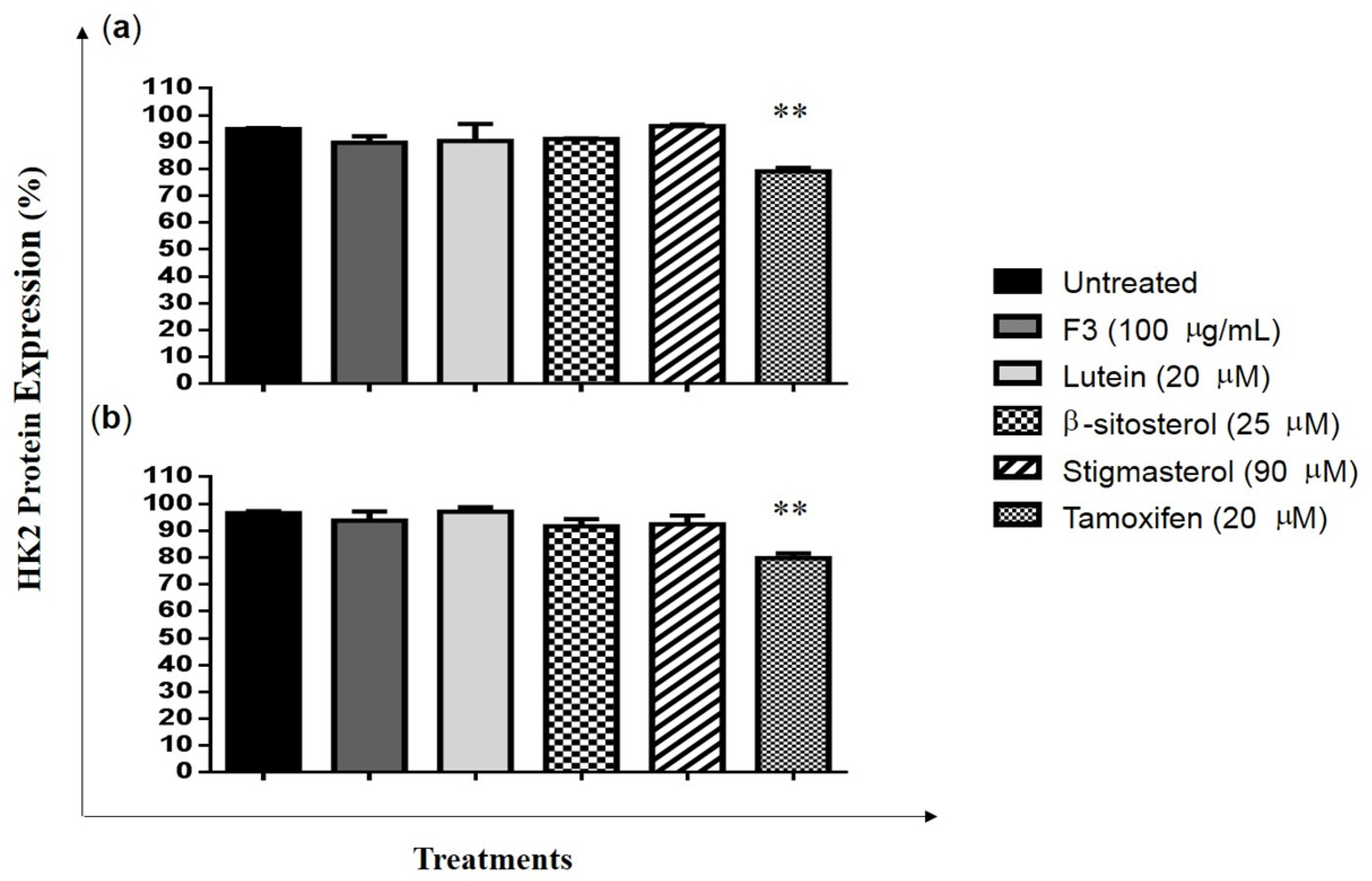
2.3. Effects of F3, Lutein, β-sitosterol, and Stigmasterol on TXNIP and HK2 Protein Expressions
2.4. Effects of F3, Lutein, β-sitosterol, and Stigmasterol on Glucose Metabolism Regulatory Proteins
2.5. Effects of F3, Lutein, β-sitosterol, and Stigmasterol on Metastatic-Related Activities
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Immunofluorescence Analysis
4.3. Cell Lysis and Western Blot Analysis
4.4. Flow Cytometric Analysis
4.5. PKC Activity Analysis
4.6. Migration Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GLUT | glucose transporter |
| TXNIP | Thioredoxin Interacting Protein |
| PKC | protein kinase c |
| AKT | Ak strain transforming |
| pAKT | phosphorylated Ak strain transforming |
| mTOR | mammalian target of rapamycin |
| HIF1α | Hypoxia-inducible factor 1-alpha |
| HK2 | hexokinase 2 |
| PKM2 | pyruvate kinase type M2 |
| LDH | lactate dehydrogenase |
| MCT | monocarboxylate transporters |
| ROS | reactive oxygen species |
| EMT | epithelial-mesenchymal transitions |
| ECM | extracellular matrix |
| CTC | circulating tumor cells |
| TNBC | triple negative breast cancer cells |
| NMU | N-Methyl-N-nitrosourea |
| GSK3β | Glycogen synthase kinase-3 beta |
| PDK1 | Pyruvate dehydrogenase kinase 1 |
| VDAC | voltage-dependent anion channel |
References
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Posener, K.; Negelein, E. Über den stoffwechsel der carcinomzelle (On metabolism of tumors). Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.; Schmidt, M.; Seeger, A.; Serras, A.F.; Vaupel, P.; Schmidberger, H. GLUT-1 expression is largely unrelated to both hypoxia and the Warburg phenotype in squamous cell carcinomas of the vulva. BMC Cancer 2014, 14, 760. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN-and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.; Ojo, D.; Yan, J.; Tang, D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015, 356, 184–191. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell. Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.N.; Koo, K.H.; Sung, J.Y.; Yun, U.J.; Kim, H. Anoikis resistance: An essential prerequisite for tumor metastasis. Int. J. Cell Biol. 2012, 2012, 306879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.J.; Semenza, G.L.; Zhang, H.F. Hypoxia-inducible factor 1 and breast cancer metastasis. J. Zhejiang Univ. Sci. B 2015, 16, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. Recent Results Cancer Res. 2016, 207, 39–72. [Google Scholar]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Bos, R.; Van der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; Van Diest, P.J.; Van der Wall, E. Levels of hypoxia-inducible factor-1α independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 2003, 97, 1573–1581. [Google Scholar] [CrossRef]
- Gruber, G.; Greiner, R.H.; Hlushchuk, R.; Aebersold, D.M.; Altermatt, H.J.; Berclaz, G.; Djonov, V. Hypoxia-inducible factor 1 alpha in high-risk breast cancer: An independent prognostic parameter? Breast Cancer Res. 2004, 6, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef] [Green Version]
- Al-Henhena, N.; Khalifa, S.A.; Ying, R.P.Y.; Ismail, S.; Hamadi, R.; Shawter, A.N.; Idris, A.M.; Azizan, A.; Al-Wajeeh, N.S.; Abdulla, M.A.; et al. Evaluation of chemopreventive potential of Strobilanthes crispus against colon cancer formation in vitro and in vivo. BMC Complement. Altern. Med. 2015, 15, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemzadeh, A.; Jaafar, H.Z.; Rahmat, A. Phytochemical constituents and biological activities of different extracts of Strobilanthes crispus (L.) Bremek leaves grown in different locations of Malaysia. BMC Complement. Altern. Med. 2015, 15, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaacob, N.S.; Hamzah, N.; Nik Mohamed Kamal, N.N.; Zainal Abidin, S.A.; Lai, C.S.; Navaratnam, V.; Norazmi, M.N. Anticancer activity of a sub-fraction of dichloromethane extract of Strobilanthes crispus on human breast and prostate cancer cells in vitro. BMC Complement. Altern. Med. 2010, 10, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaacob, N.S.; Nik Mohamed Kamal, N.N.; Wong, K.K.; Norazmi, M.N. Cell cycle modulation of MCF-7 and MDA-MB-231 by a sub-fraction of Strobilanthes crispus and its combination with tamoxifen. Asian Pac. J. Cancer Prev. 2015, 16, 8135–8140. [Google Scholar] [CrossRef] [Green Version]
- Yankuzo, H.M.; Baraya, Y.S.; Mustapha, Z.; Wong, K.K.; Yaacob, N.S. Immunomodulatory effects of a bioactive fraction of Strobilanthes crispus in NMU-induced rat mammary tumor model. J. Ethnopharmacol. 2018, 213, 31–37. [Google Scholar] [CrossRef]
- Baraya, Y.S.; Wong, K.K.; Yaacob, N.S. Strobilanthes crispus inhibits migration, invasion and metastasis in breast cancer. J. Ethnopharmacol. 2019, 233, 13–21. [Google Scholar] [CrossRef]
- Yaacob, N.S.; Yankuzo, H.M.; Devaraj, S.; Wong, J.K.M.; Lai, C.S. Anti-tumor action, clinical biochemistry profile and phytochemical constituents of a pharmacologically active fraction of S. crispus in NMU-induced rat mammary tumour model. PLoS ONE 2015, 10, e0126426. [Google Scholar] [CrossRef]
- Baraya, Y.S.; Yankuzo, H.M.; Wong, K.K.; Yaacob, N.S. Strobilanthes crispus bioactive subfraction inhibits tumor progression and improves hematological and morphological parameters in mouse mammary carcinoma model. J. Ethnopharmacol. 2021, 267, 113522. [Google Scholar] [CrossRef]
- Fadzelly, A.M.; Asmah, R.; Fauziah, O. Effects of Strobilanthes crispus tea aqueous extracts on glucose and lipid profile in normal and streptozotocin-induced hyperglycemic rats. Plant Foods Hum. Nutr. 2006, 61, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.N.; Yaacob, N.S.; Safuwan, N.A.; Fauzi, A.N. Antiglycolytic activities of Strobilanthes crispus active fraction and its bioactive components on triple-negative breast cancer cells in vitro. Anticancer Agents Med. Chem. 2022, 22, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Laudański, P.; Swiatecka, J.; Kovalchuk, O.; Wołczyński, S. Expression of GLUT1 gene in breast cancer cell lines MCF-7 and MDA-MB-231. Ginekol. Pol. 2003, 74, 782–785. [Google Scholar] [PubMed]
- Domoto, T.; Pyko, I.V.; Furuta, T.; Miyashita, K.; Uehara, M.; Shimasaki, T.; Nakada, M.; Minamoto, T. Glycogen synthase kinase-3β is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016, 107, 1363–1372. [Google Scholar] [CrossRef]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szablewski, L. Expression of glucose transporters in diseases. In Human Glucose Transporters in Health and Diseases; Szablewski, L., Ed.; Cambridge Scholars Publishing: New Castle, UK, 2019; pp. 63–227. [Google Scholar]
- Venturelli, L.; Nappini, S.; Bulfoni, M.; Gianfranceschi, G.; Dal Zilio, S.; Coceano, G.; Del Ben, F.; Turetta, M.; Scoles, G.; Vaccari, L.; et al. Glucose is a key driver for GLUT1-mediated nanoparticles internalization in breast cancer cells. Sci. Rep. 2016, 6, 21629. [Google Scholar] [CrossRef] [Green Version]
- Amaro, A.; Angelini, G.; Mirisola, V.; Esposito, A.I.; Reverberi, D.; Matis, S.; Maffei, M.; Giaretti, W.; Viale, M.; Gangemi, R.; et al. A highly invasive subpopulation of MDA-MB-231 breast cancer cells shows accelerated growth, differential chemoresistance, features of apocrine tumors and reduced tumorigenicity in vivo. Oncotarget 2016, 7, 68803. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Kim, H.; Nam, K.; Shin, I. Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple-negative breast cancer cells. BMB Rep. 2017, 50, 132. [Google Scholar] [CrossRef]
- Balliet, R.M.; Capparelli, C.; Guido, C.; Pestell, T.G.; Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Pestell, R.G.; Howell, A.; et al. Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production, promoting breast cancer tumor growth: Understanding the aging and cancer connection. Cell Cycle 2011, 10, 4065–4073. [Google Scholar] [CrossRef] [Green Version]
- Pliszka, M.; Szablewski, L. Glucose transporters as a target for anticancer therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [PubMed]
- Gwak, H.; Haegeman, G.; Tsang, B.K.; Song, Y.S. Cancer-specific interruption of glucose metabolism by resveratrol is mediated through inhibition of Akt/GLUT1 axis in ovarian cancer cells. Mol. Carcinog. 2015, 54, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Tsujihara, K.; Hongu, M.; Saito, K.; Inamasu, M.; Arakawa, K.; Oku, A.; Matsumoto, M. Na+-Glucose Contransporter Inhibitors as Antidiabetics. I. Synthesis and Pharmacological Properties of 4′-Dehydroxyphlorizin Derivatives Based on a New Concept. Chem. Pharm. Bull. 1996, 44, 1174–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blodgett, D.M.; De Zutter, J.K.; Levine, K.B.; Karim, P.; Carruthers, A. Structural basis of GLUT1 inhibition by cytoplasmic ATP. J. Gen. Physiol. 2007, 130, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Antonescu, C.N.; McGraw, T.E.; Klip, A. Reciprocal regulation of endocytosis and metabolism. Cold Spring Harb. Perspect. 2014, 6, a016964. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Aliereza, B.; Faramarz, I.B. Regulation of glucose transport by hypoxia. Am. J. Kidney Dis. 1999, 34, 189–202. [Google Scholar] [CrossRef]
- Li, Z.Y.; Shi, Y.L.; Liang, G.X.; Yang, J.; Zhuang, S.K.; Lin, J.B.; Ghodbane, A.; Tam, M.S.; Liang, Z.J.; Zha, Z.G.; et al. Visualization of GLUT1 trafficking in live cancer cells by the use of a dual-fluorescence reporter. ACS Omega 2020, 5, 15911–15921. [Google Scholar] [CrossRef]
- Wieman, H.L.; Horn, S.R.; Jacobs, S.R.; Altman, B.J.; Kornbluth, S.; Rathmell, J.C. An essential role for the Glut1 PDZ-binding motif in growth factor regulation of Glut1 degradation and trafficking. Biochem. J. 2009, 418, 345–367. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.E.; Ma, J.; Sacharidou, A.; Mi, W.; Salato, V.K.; Nguyen, N.; Jiang, Y.; Pascual, J.M.; North, P.E.; Shaul, P.W.; et al. A protein kinase C phosphorylation motif in GLUT1 affects glucose transport and is mutated in GLUT1 deficiency syndrome. Mol. Cell 2015, 58, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Leidal, A.M.; Ye, J.; Ronen, S.M.; Debnath, J. Autophagy-dependent shuttling of TBC1D5 controls plasma membrane translocation of GLUT1 and glucose uptake. Mol. Cell 2017, 67, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Langemeyer, L.; Fröhlich, F.; Ungermann, C. Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol. 2018, 28, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Witters, L.A.; Vater, C.A.; Lienhard, G.E. Phosphorylation of the glucose transporter in vitro and in vivo by protein kinase C. Nature 1985, 315, 777–778. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Rathmell, J.C. PKCs sweeten cell metabolism by phosphorylation of Glut1. Mol. Cell 2015, 58, 711–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yu, Q.; Chng, W.J. TXNIP (VDUP-1, TBP-2): A major redox regulator commonly suppressed in cancer by epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2011, 43, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Yu, F.X.; Luo, Y.; Hagen, T. Oncogenic activation of the PI3K/Akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell. Signal. 2016, 28, 377–383. [Google Scholar] [CrossRef]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT mediates acute influx of glucose in response to insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. Pi3k-pkb/akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Clarke, J.F.; Young, P.W.; Yonezawa, K.; Kasuga, M.; Holman, G.D. Inhibition of the translocation of GLUT1 and GLUT4 in 3T3-L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem. J. 1994, 300, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Samih, N.; Hovsepian, S.; Aouani, A.; Lombardo, D.; Fayet, G. Glut-1 translocation in FRTL-5 thyroid cells: Role of phosphatidylinositol 3-kinase and N-glycosylation. Endocrinology 2000, 141, 4146–4155. [Google Scholar] [CrossRef]
- Kim, D.I.; Lim, S.K.; Park, M.J.; Han, H.J.; Kim, G.Y.; Park, S.H. The involvement of phosphatidylinositol 3-kinase/Akt signaling in high glucose-induced downregulation of GLUT-1 expression in ARPE cells. Life Sci. 2007, 80, 626–632. [Google Scholar] [CrossRef]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Moten, A.; Lin, H.K. Akt: A new activation mechanism. Cell Res. 2014, 24, 785–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Zhu, X.; Wu, H.; Jiang, K.; Zhao, G.; Shaukat, A.; Deng, G.; Qiu, C. Targeting the ROS/PI3K/AKT/HIF-1α/HK2 axis of breast cancer cells: Combined administration of Polydatin and 2-Deoxy-d-glucose. J. Cell Mol. Med. 2019, 23, 3711–3723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Tan, Z.; Peng, C.; Yi, W. HK2 is associated with the Warburg effect and proliferation in liver cancer: Targets for effective therapy with glycyrrhizin. Mol. Med. Rep. 2021, 23, 343. [Google Scholar] [CrossRef] [PubMed]
- De Cerqueira Cesar, M.; Wilson, J.E. Further studies on the coupling of mitochondrially bound hexokinase to intramitochondrially compartmented ATP, generated by oxidative phosphorylation. Arch. Biochem. Biophys. 1998, 350, 109–117. [Google Scholar] [CrossRef]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef]
- Ahuatzi, D.; Riera, A.; Pela, R.; Herrero, P.; Moreno, F. Hxk2 regulates the phosphorylation state of Mig1 and therefore its nucleocytoplasmic distribution. J. Biol. Chem. 2007, 282, 4485–4493. [Google Scholar] [CrossRef] [PubMed]
- Tan, V.P.; Miyamoto, S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy 2015, 11, 963–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neary, C.L.; Pastorino, J.G. Akt inhibition promotes hexokinase 2 redistribution and glucose uptake in cancer cells. J. Cell. Physiol. 2013, 228, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 3b disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Harwood, F.C.; Klein Geltink, R.I.; O’Hara, B.P.; Cardone, M.; Janke, L.; Finkelstein, D.; Entin, I.; Paul, L.; Houghton, P.J.; Grosveld, G.C. ETV7 is an essential component of a rapamycin-insensitive mTOR complex in cancer. Sci. Adv. 2018, 4, eaar3938. [Google Scholar] [CrossRef] [Green Version]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 2001, 11, 293–299. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Singh, D.; Arora, R.; Kaur, P.; Singh, B.; Mannan, R.; Arora, S. Overexpression of hypoxia-inducible factor and metabolic pathways: Possible targets of cancer. Cell Biosci. 2017, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The transcription factor HIF-1α plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Jóźwiak, P.; Krześlak, A.; Bryś, M.; Lipińska, A. Glucose-dependent glucose transporter 1 expression and its impact on viability of thyroid cancer cells. Oncol. Rep. 2015, 33, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Riemann, A.; Rauschner, M.; Gießelmann, M.; Reime, S.; Haupt, V.; Thews, O. Extracellular acidosis modulates the expression of epithelial-mesenchymal transition (EMT) markers and adhesion of epithelial and tumor cells. Neoplasia 2019, 21, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Liu, X.; Wang, M.; Wang, P.; Yang, J.; Zhang, S. Lutein inhibits proliferation, invasion and migration of hypoxic breast cancer cells via downregulation of HES1. Int. J. Oncol. 2018, 52, 2119–2129. [Google Scholar] [CrossRef] [Green Version]
- Awad, A.B.; Williams, H.; Fink, C.S. Phytosterols reduce in vitro metastatic ability of MDA-MB-231 human breast cancer cells. Nutr. Cancer 2001, 40, 157–164. [Google Scholar] [CrossRef]
- Li, K.; Yuan, D.; Yan, R.; Meng, L.; Zhang, Y.; Zhu, K. Stigmasterol exhibits potent antitumor effects in human gastric cancer cells mediated via inhibition of cell migration, cell cycle arrest, mitochondrial mediated apoptosis and inhibition of JAK/STAT signalling pathway. J. BUON 2018, 23, 1420–1425. [Google Scholar]
- Bae, H.; Song, G.; Lim, W. Stigmasterol causes ovarian cancer cell apoptosis by inducing endoplasmic reticulum and mitochondrial dysfunction. Pharmaceutics 2020, 12, 488. [Google Scholar] [CrossRef]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and associated signaling pathways involved in cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Quintayo, M.A.; Munro, A.F.; Thomas, J.; Kunkler, I.H.; Jack, W.; Kerr, G.R.; Dixon, J.M.; Chetty, U.; Bartlett, J. GSK3β and cyclin D1 expression predicts outcome in early breast cancer patients. Breast Cancer Res. Treat. 2012, 136, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wolgamott, L.; Tcherkezian, J.; Vallabhapurapu, S.; Yu, Y.; Roux, P.P.; Yoon, S.O. Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene 2014, 33, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-mesenchymal transition and breast cancer. J. Clin. Med. 2016, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay, G.V.; Zhao, N.; Den Hollander, P.; Toneff, M.J.; Joseph, R.; Pietila, M.; Taube, J.H.; Sarkar, T.R.; Ramirez-Pena, E.; Werden, S.J.; et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Wierzbicka-Patynowski, I.; Schwarzbauer, J.E. The ins and outs of fibronectin matrix assembly. J. Cell. Sci. 2003, 116, 3269–3276. [Google Scholar] [CrossRef] [Green Version]
- Rick, J.W.; Chandra, A.; Dalle Ore, C.; Nguyen, A.T.; Yagnik, G.; Aghi, M.K. Fibronectin in malignancy: Cancer-specific alterations, protumoral effects, and therapeutic implications. Semin. Oncol. 2019, 46, 284–290. [Google Scholar] [CrossRef]
- Wang, J.P.; Hielscher, A. Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic targeting. J. Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.C.; Li, J.R.; Wang, J.D.; Chang, C.Y.; Wu, C.C.; Chen, W.Y.; Kuan, Y.H.; Liao, S.L.; Lu, H.C.; Chen, C.J. Fibronectin promotes cell growth and migration in human renal cell carcinoma cells. Int. J. Mol. Sci. 2019, 20, 2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousif, N.G. Fibronectin promotes migration and invasion of ovarian cancer cells through up-regulation of FAK–PI3K/Akt pathway. Cell Biol. Int. 2014, 3, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Thant, A.A.; Nawa, A.; Kikkawa, F.; Ichigotani, Y.; Zhang, Y.; Sein, T.T.; Amin, A.R.; Hamaguchi, M. Fibronectin activates matrix metalloproteinase-9 secretion via the MEK1-MAPK and the PI3K-Akt pathways in ovarian cancer cells. Clin. Exp. Metastasis 2000, 18, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, P.; Enns, A.; Haier, J. Role of tumor cell adhesion and migration in organ-specific metastasis formation. Oncology 2004, 27, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.Z.; Milam, B.M.; Strouch, M.; Pelling, J.C.; Bentrem, D.J. Apigenin inhibits the GLUT-1 glucose transporter and the phosphoinositide 3-kinase/Akt pathway in human pancreatic cancer cells. Pancreas 2008, 37, 426–431. [Google Scholar] [CrossRef]
- Bao, Y.Y.; Zhou, S.H.; Lu, Z.J.; Fan, J.; Huang, Y.P. Inhibiting GLUT-1 expression and PI3K/Akt signaling using apigenin improves the radiosensitivity of laryngeal carcinoma in vivo. Oncol. Rep. 2015, 34, 1805–1814. [Google Scholar] [CrossRef] [Green Version]
- Gundimeda, U.; Chen, Z.H.; Gopalakrishna, R. Tamoxifen modulates protein kinase C via oxidative stress in estrogen receptor-negative breast cancer cells. J. Biol. Chem. 1996, 271, 13504–13514. [Google Scholar] [CrossRef] [Green Version]
- Nguyen Hoang, A.T.; Hoe, K.L.; Lee, S.J. CSNK1G2 differently sensitizes tamoxifen-induced decrease in PI3K/AKT/mTOR/S6K and ERK signaling according to the estrogen receptor existence in breast cancer cells. PLoS ONE 2021, 16, e0246264. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, S.N.H.; Safuwan, N.A.M.; Yaacob, N.S.; Fauzi, A.N. Regulatory Mechanism on Anti-Glycolytic and Anti-Metastatic Activities Induced by Strobilanthes crispus in Breast Cancer, In Vitro. Pharmaceuticals 2023, 16, 153. https://doi.org/10.3390/ph16020153
Muhammad SNH, Safuwan NAM, Yaacob NS, Fauzi AN. Regulatory Mechanism on Anti-Glycolytic and Anti-Metastatic Activities Induced by Strobilanthes crispus in Breast Cancer, In Vitro. Pharmaceuticals. 2023; 16(2):153. https://doi.org/10.3390/ph16020153
Chicago/Turabian StyleMuhammad, Siti Nur Hasyila, Nur Arnida Mohd Safuwan, Nik Soriani Yaacob, and Agustine Nengsih Fauzi. 2023. "Regulatory Mechanism on Anti-Glycolytic and Anti-Metastatic Activities Induced by Strobilanthes crispus in Breast Cancer, In Vitro" Pharmaceuticals 16, no. 2: 153. https://doi.org/10.3390/ph16020153