
Development of Potential Multi-Target Inhibitors for Human Cholinesterases and Beta-Secretase 1: A Computational Approach
 ,
,  ,
,  , ,
, ,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Pharmacophore Models Building and Validation
2.2. Hierarchical Virtual Screening
2.3. Prediction of Toxicological and Physicochemical Parameters and Evaluation of Interaction Maps
2.4. Molecular Dynamics (MD)
3. Discussion
3.1. Pharmacophore Model Building and Validation
3.2. Hierarchical Virtual Screening
3.3. Prediction of Toxicological and Physicochemical Parameters Predictions and Evaluation of Interaction Maps
3.4. Molecular Dynamics (MD)
4. Materials and Methods
4.1. Pharmacophore Model Building and Validation
4.1.1. Dataset
4.1.2. Pharmacophore Model Building and Validation
4.2. Hierarchical Virtual Screening
4.3. Prediction of the Toxicological and Physicochemical Parameters and Evaluation of Interaction Maps
4.4. Molecular Dynamics (MD)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yusufzai, S.K.; Khan, M.S.; Sulaiman, O.; Osman, H.; Lamjin, D.N. Molecular Docking Studies of Coumarin Hybrids as Potential Acetylcholinesterase, Butyrylcholinesterase, Monoamine Oxidase A/B and β-Amyloid Inhibitors for Alzheimer’s Disease. Chem. Cent. J. 2018, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Chen, Y.G. Research Progress in the Pathogenesis of Alzheimer’s Disease. Chin. Med. J. 2018, 131, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Sakata, R.P. Docking Molecular, Síntese e Estudo Biológico de Potenciais Inibidores Da Beta-Secretase (BACE-1) e Da Acetilcolinesterase (ACHE); Universidade Estadual de Campinas: São Paulo, Brazil, 2018. [Google Scholar]
- Sağlık, B.N.; Ilgın, S.; Özkay, Y. Synthesis of New Donepezil Analogues and Investigation of Their Effects on Cholinesterase Enzymes. Eur. J. Med. Chem. 2016, 124, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- De Falco, A.; Cukierman, D.S.; Hauser-Davis, R.A.; Rey, N.A. Doença de Alzheimer: Hipóteses Etiológicas e Perspectivas de Tratamento. Quim. Nova 2016, 39, 63–80. [Google Scholar] [CrossRef]
- Najafi, Z.; Mahdavi, M.; Saeedi, M.; Karimpour-Razkenari, E.; Asatouri, R.; Vafadarnejad, F.; Moghadam, F.H.; Khanavi, M.; Sharifzadeh, M.; Akbarzadeh, T. Novel Tacrine-1,2,3-Triazole Hybrids: In Vitro, in Vivo Biological Evaluation and Docking Study of Cholinesterase Inhibitors. Eur. J. Med. Chem. 2017, 125, 1200–1212. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Gong, C.X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Beach, T.G.; Kuo, Y.M.; Spiegel, K.; Emmerling, M.R.; Sue, L.I.; Kokjohn, K.; Roher, A.E. The Cholinergic Deficit Coincides with Aβ Deposition at the Earliest Histopathologic Stages of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2000, 59, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Potter, P.E.; Rauschkolb, P.K.; Pandya, Y.; Sue, L.I.; Sabbagh, M.N.; Walker, D.G.; Beach, T.G. Pre- and Post-Synaptic Cortical Cholinergic Deficits Are Proportional to Amyloid Plaque Presence and Density at Preclinical Stages of Alzheimer’s Disease. Acta Neuropathol. 2011, 122, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Perry, R.H.; Blessed, G.; Tomlinson, B.E. Changes in Brain Cholinesterases in Senile Dementia of Alzheimer Type. Neuropathol. Appl. Neurobiol. 1978, 4, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.R.; Lehár, J.; Keith, C.T. Multi-Target Therapeutics: When the Whole Is Greater than the Sum of the Parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef] [PubMed]
- Denholm, R.; Morris, R.; Payne, R. Polypharmacy Patterns in the Last Year of Life in Patients with Dementia. Eur. J. Clin. Pharmacol. 2019, 75, 1583–1591. [Google Scholar] [CrossRef]
- Dias, K.S.T.; Viegas, C. Multi-Target Directed Drugs: A Modern Approach for Design of New Drugs for the Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 239–255. [Google Scholar] [CrossRef]
- Morphy, R.; Kay, C.; Rankovic, Z. From Magic Bullets to Designed Multiple Ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Borsari, C.; Trader, D.J.; Tait, A.; Costi, M.P. Designing Chimeric Molecules for Drug Discovery by Leveraging Chemical Biology. J. Med. Chem. 2020, 63, 1908–1928. [Google Scholar] [CrossRef]
- Sun, D.; Zhao, Y.; Zhang, S.; Zhang, L.; Liu, B.; Ouyang, L. Dual-Target Kinase Drug Design: Current Strategies and Future Directions in Cancer Therapy. Eur. J. Med. Chem. 2020, 188, 112025. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y. Pharmacophore Modeling and Applications in Drug Discovery: Challenges and Recent Advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, E.; Do Amaral, A.T. Virtual Screening of Bioactive Compounds: Concepts and Aplications. Quim. Nova 2018, 41, 662–677. [Google Scholar] [CrossRef]
- Verli, H. Dinâmica Molecular. In Bioinformática da Biologia à Flexibilidade Molecular; SBBq: São Paulo, Brazil, 2014; pp. 173–187. [Google Scholar]
- de Almeida, R.B.M.; Conceição, R.S.; da Silva, K.S.; Santos Junior, M.C.; Branco, A.; Botura, M.B. Ocotea Daphnifolia: Phytochemical Investigation, in Vitro Dual Cholinesterase Inhibition, and Molecular Docking Studies. Braz. J. Pharm. Sci. 2021, 57, e18310. [Google Scholar] [CrossRef]
- Shelat, A.A.; Guy, R.K. Scaffold Composition and Biological Relevance of Screening Libraries Anang A Shelat & R Kiplin Guy. Nat. Chem. Biol. 2007, 3, 442–446. [Google Scholar]
- Empereur-Mot, C.; Guillemain, H.; Latouche, A.; Zagury, J.F.; Viallon, V.; Montes, M. Predictiveness Curves in Virtual Screening. J. Cheminform. 2015, 7, 52. [Google Scholar] [CrossRef]
- Mascarenhas, A.M.S.; de Almeida, R.B.M.; de Araujo Neto, M.F.; Mendes, G.O.; da Cruz, J.N.; dos Santos, C.B.R.; Botura, M.B.; Leite, F.H.A. Pharmacophore-Based Virtual Screening and Molecular Docking to Identify Promising Dual Inhibitors of Human Acetylcholinesterase and Butyrylcholinesterase. J. Biomol. Struct. Dyn. 2020, 39, 6021–6030. [Google Scholar] [CrossRef] [PubMed]
- Domingues, B.F. 3D-Pharma: Uma Ferramenta Para Triagem Virtual Baseada Em Fingerprints de Farmacóforos; Universidade Federal de Minas Gerais: Belo Horizonte, Brazil, 2013. [Google Scholar]
- Mendes, G.O.; Pita, S.S.d.R.; Carvalho, P.B.d.; Silva, M.P.d.; Taranto, A.G.; Leite, F.H.A. Molecular Multi-Target Approach for Human Acetylcholinesterase, Butyrylcholinesterase and β-Secretase 1: Next Generation for Alzheimer’s Disease Treatment. Pharmaceuticals 2023, 16, 880. [Google Scholar] [CrossRef] [PubMed]
- do Bomfim, M.R.; Barbosa, D.B.; de Carvalho, P.B.; da Silva, A.M.; de Oliveira, T.A.; Taranto, A.G.; Leite, F.H.A. Identification of Potential Human Beta-Secretase 1 Inhibitors by Hierarchical Virtual Screening and Molecular Dynamics. J. Biomol. Struct. Dyn. 2022, 41, 4560–4574. [Google Scholar] [CrossRef]
- Mortelmans, K.; Mortelmans, K.; Zeiger, E. The Ames Salmonella/Microsome Mutagenicity Assay The Ames Salmonella/Microsome Mutagenicity Assay. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2016, 5107, 29–60. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Hein, M.; Zilian, D.; Sotriffer, C.A. Docking Compared to 3D-Pharmacophores: The Scoring Function Challenge. Drug Discov. Today Technol. 2010, 7, e229–e236. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next Generation 3D Pharmacophore Modeling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef]
- Tran-Nguyen, V.K.; Da Silva, F.; Bret, G.; Rognan, D. All in One: Cavity Detection, Druggability Estimate, Cavity-Based Pharmacophore Perception, and Virtual Screening. J. Chem. Inf. Model. 2019, 59, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine Tools: An Online Service for Analyzing and Clustering Small Molecules. Nucleic Acids Res. 2011, 39, 486–491. [Google Scholar] [CrossRef]
- de Carvalho Gallo, J.C.; de Mattos Oliveira, L.; Araújo, J.S.C.; Santana, I.B.; dos Santos Junior, M.C. Virtual Screening to Identify Leishmania Braziliensis N-Myristoyltransferase Inhibitors: Pharmacophore Models, Docking, and Molecular Dynamics. J. Mol. Model. 2018, 24, 260. [Google Scholar] [CrossRef]
- Dorfman, R.J.; Smith, K.M.; Masek, B.B.; Clark, R.D. A Knowledge-Based Approach to Generating Diverse but Energetically Representative Ensembles of Ligand Conformers. J. Comput. Aided Mol. Des. 2008, 22, 681–691. [Google Scholar] [CrossRef]
- Xie, H.; Qiu, K.; Xie, X. 3D QSAR Studies, Pharmacophore Modeling and Virtual Screening on a Series of Steroidal Aromatase Inhibitors. Int. J. Mol. Sci. 2014, 15, 20927–20947. [Google Scholar] [CrossRef]
- Seidel, T.; Ibis, G.; Bendix, F.; Wolber, G. Strategies for 3D Pharmacophore-Based Virtual Screening. Drug Discov. Today Technol. 2010, 7, e221–e228. [Google Scholar] [CrossRef]
- Rizzi, A.; Fioni, A. Virtual Screening Using PLS Discriminant Analysis and ROC Curve Approach: An Application Study on PDE4 Inhibitors. J. Chem. Inf. Model. 2008, 48, 1686–1692. [Google Scholar] [CrossRef]
- Metz, C.E. Basic Principles of ROC Analysis. Semin. Nucl. Med. 1978, 8, 283–298. [Google Scholar] [CrossRef]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the Performance of 3D Virtual Screening Protocols: RMSD Comparisons, Enrichment Assessments, and Decoy Selection—What Can We Learn from Earlier Mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Grover, S.; Dhanjal, J.K.; Goyal, S.; Tyagi, C.; Grover, A. Molecular Modelling Studies on Flavonoid Derivatives as Dual Site Inhibitors of Human Acetyl Cholinesterase Using 3D-QSAR, Pharmacophore and High Throughput Screening Approaches. Med. Chem. Res. 2014, 23, 2122–2132. [Google Scholar] [CrossRef]
- Gupta, S.; Mohan, C.G. Dual Binding Site and Selective Acetylcholinesterase Inhibitors Derived from Integrated Pharmacophore Models and Sequential Virtual Screening. Biomed Res. Int. 2014, 2014, 291214. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yu, H.; Sheng, R.; Li, J.; Hu, Y. Identification of Pharmacophore Model, Synthesis and Biological Evaluation of N-Phenyl-1-Arylamide and N-Phenylbenzenesulfonamide Derivatives as BACE 1 Inhibitors. Bioorg. Med. Chem. 2008, 16, 10190–10197. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Thangapandian, S.; Sakkiah, S.; Lee, K.W. Potent Bace-1 Inhibitor Design Using Pharmacophore Modeling, in Silico Screening and Molecular Docking Studies. BMC Bioinform. 2011, 12, S28. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Irwin, J. Docking Screens: Right for the Right Reasons? Curr. Top. Med. Chem. 2009, 9, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.H.; Wu, J.W.; Liu, H.L.; Zhao, J.H.; Liu, K.T.; Chuang, C.K.; Lin, H.Y.; Tsai, W.B.; Ho, Y. The Discovery of Potential Acetylcholinesterase Inhibitors: A Combination of Pharmacophore Modeling, Virtual Screening, and Molecular Docking Studies. J. Biomed. Sci. 2011, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.; Lee, X.Y.; De Raeymaeker, J.; Tame, J.R.; Zhang, K.Y.; De Maeyer, M.; Voet, A.R. Pharmacophore Modeling: Advances, Limitations, And Current Utility in Drug Discovery. J. Recept. Ligand Channel Res. 2014, 7, 81–92. [Google Scholar] [CrossRef]
- Kumar, V.; Saha, A.; Roy, K. In Silico Modeling for Dual Inhibition of Acetylcholinesterase (AChE) and Butyrylcholinesterase (BuChE) Enzymes in Alzheimer’s Disease; Elsevier Ltd.: Amsterdam, The Netherlands, 2020; Volume 88, ISBN 9133283710. [Google Scholar]
- dos Santos, K.L.B.; Cruz, J.N.; Silva, L.B.; Ramos, R.S.; Neto, M.F.A.; Lobato, C.C.; Ota, S.S.B.; Leite, F.H.A.; Borges, R.S.; da Silva, C.H.T.P.; et al. Identification of Novel Chemical Entities for Adenosine Receptor Type 2a Using Molecular Modeling Approaches. Molecules 2020, 25, 1245. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA-J. Am. Med. Assoc. 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Gopi Mohan, C.; Gandhi, T.; Garg, D.; Shinde, R. Computer-Assisted Methods in Chemical Toxicity Prediction. Mini-Rev. Med. Chem. 2007, 7, 499–507. [Google Scholar] [CrossRef]
- Barbezan, A.B.; Martins, R.; Bueno, J.B.; Villavicencio, A.L.C.H. Ames Test to Detect Mutagenicity of 2-Alkylcyclobutanones: A Review. J. Food Sci. 2017, 82, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Hosea, N.A.; Jones, H.M. Predicting Pharmacokinetic Profiles Using in Silico Derived Parameters. Mol. Pharm. 2013, 10, 1207–1215. [Google Scholar] [CrossRef]
- Boobis, A.; Gundert-Remy, U.; Kremers, P.; Macheras, P.; Pelkonen, O. In Silico Prediction of ADME and Pharmacokinetics: Report of an Expert Meeting Organised by COST B15. Eur. J. Pharm. Sci. 2002, 17, 183–193. [Google Scholar] [CrossRef]
- Chhabra, N.; Aseri, M.; Padmanabhan, D. A Review of Drug Isomerism and Its Significance. Int. J. Appl. Basic Med. Res. 2013, 3, 16. [Google Scholar] [CrossRef]
- Institute, N.C. NCIthesaurus: Silodosin. Available online: https://ncit.nci.nih.gov/ncitbrowser/pages/concept_details.jsf?dictionary=NCI_Thesaurus&version=20.09d&code=C81372&ns=NCI_Thesaurus&type=properties&key=null&b=1&n=0&vse=null (accessed on 1 October 2020).
- Degoey, D.A.; Chen, H.J.; Cox, P.B.; Wendt, M.D. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem. 2018, 61, 2636–2651. [Google Scholar] [CrossRef]
- Türkeş, C.; Arslan, M.; Demir, Y.; Çoçaj, L.; Rifati Nixha, A.; Beydemir, Ş. Synthesis, Biological Evaluation and in Silico Studies of Novel N-Substituted Phthalazine Sulfonamide Compounds as Potent Carbonic Anhydrase and Acetylcholinesterase Inhibitors. Bioorg. Chem. 2019, 89, 103004. [Google Scholar] [CrossRef]
- Brahmachari, G.; Choo, C.; Ambure, P.; Roy, K. In Vitro Evaluation and in Silico Screening of Synthetic Acetylcholinesterase Inhibitors Bearing Functionalized Piperidine Pharmacophores. Bioorg. Med. Chem. 2015, 23, 4567–4575. [Google Scholar] [CrossRef]
- Senol, F.S.; Ślusarczyk, S.; Matkowski, A.; Pérez-Garrido, A.; Girón-Rodríguez, F.; Cerón-Carrasco, J.P.; den-Haan, H.; Peña-García, J.; Pérez-Sánchez, H.; Domaradzki, K.; et al. Selective In Vitro and In Silico Butyrylcholinesterase Inhibitory Activity of Diterpenes and Rosmarinic Acid Isolated from Perovskia Atriplicifolia Benth. and Salvia glutinosa L. Phytochemistry 2017, 133, 33–44. [Google Scholar] [CrossRef]
- Dubey, S.K.; Lakshmi, K.K.; Krishna, K.V.; Agrawal, M.; Singhvi, G.; Saha, R.N.; Saraf, S.; Saraf, S.; Shukla, R.; Alexander, A. Insulin Mediated Novel Therapies for the Treatment of Alzheimer’s Disease. Life Sci. 2020, 249, 117540. [Google Scholar] [CrossRef]
- Wajid, S.; Khatoon, A.; Khan, M.A.; Zafar, H.; Kanwal, S.; Atta-ur-Rahman; Choudhary, M.I.; Basha, F.Z. Microwave-Assisted Organic Synthesis, Structure–Activity Relationship, Kinetics and Molecular Docking Studies of Non-Cytotoxic Benzamide Derivatives as Selective Butyrylcholinesterase Inhibitors. Bioorg. Med. Chem. 2019, 27, 4030–4040. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; Correa-Basurto, J.; Gutiérrez, A.; Vitorica, J.; Rosales-Hernández, M.C. Asp32 and Asp228 Determine the Selective Inhibition of BACE1 as Shown by Docking and Molecular Dynamics Simulations. Eur. J. Med. Chem. 2016, 124, 1142–1154. [Google Scholar] [CrossRef]
- Semighini, E.P. In Silico Design of Beta-Secretase Inhibitors in Alzheimer’s Disease. Chem. Biol. Drug Des. 2015, 86, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Ali, M.Y.; Choi, R.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Kinetics and Molecular Docking Studies of Fucosterol and Fucoxanthin, BACE1 Inhibitors from Brown Algae Undaria Pinnatifida and Ecklonia Stolonifera. Food Chem. Toxicol. 2016, 89, 104–111. [Google Scholar] [CrossRef]
- Winneroski, L.L.; Erickson, J.A.; Green, S.J.; Lopez, J.E.; Stout, S.L.; Porter, W.J.; Timm, D.E.; Audia, J.E.; Barberis, M.; Beck, J.P.; et al. Preparation and Biological Evaluation of BACE1 Inhibitors: Leveraging Trans-Cyclopropyl Moieties as Ligand Efficient Conformational Constraints. Bioorg. Med. Chem. 2020, 28, 115194. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Pan, Y.; Jennifer, L.M.; Zhan, C.-G. Active Site Gating and Substrate Specificity of Butyrylcholinesterase and Acetylcholinesterase: Insights from Molecular Dynamics Simulations. J. Phys. Chem. B 2011, 115, 8797–8805. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. Encycl. Life Sci. 2010. [Google Scholar] [CrossRef]
- Niu, C.; Xu, Y.; Xu, Y.; Luo, X.; Duan, W.; Silman, I.; Sussman, J.L.; Zhu, W.; Chen, K.; Shen, J.; et al. Dynamic Mechanism of E2020 Binding to Acetylcholinesterase: A Steered Molecular Dynamics Simulation. J. Phys. Chem. B 2005, 109, 23730–23738. [Google Scholar] [CrossRef]
- Delogu, G.L.; Matos, M.J.; Fanti, M.; Era, B.; Medda, R.; Pieroni, E.; Fais, A.; Kumar, A.; Pintus, F. 2-Phenylbenzofuran Derivatives as Butyrylcholinesterase Inhibitors: Synthesis, Biological Activity and Molecular Modeling. Bioorg. Med. Chem. Lett. 2016, 26, 2308–2313. [Google Scholar] [CrossRef]
- Dhanjal, J.K.; Goyal, S.; Sharma, S.; Hamid, R.; Grover, A. Mechanistic Insights into Mode of Action of Potent Natural Antagonists of BACE-1 for Checking Alzheimer’s Plaque Pathology. Biochem. Biophys. Res. Commun. 2014, 443, 1054–1059. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Luo, X.; Silman, I.; Sussman, J.L.; Chen, K.; Jiang, H. How Does Huperzine A Enter and Leave the Binding Gorge of Acetylcholinesterase? Steered Molecular Dynamics Simulations. J. Am. Chem. Soc. 2003, 125, 11340–11349. [Google Scholar] [CrossRef]
- Lu, X.; Yang, H.; Li, Q.; Chen, Y.; Li, Q.; Zhou, Y.; Feng, F.; Liu, W.; Guo, Q.; Sun, H. Expansion of the Scaffold Diversity for the Development of Highly Selective Butyrylcholinesterase (BChE) Inhibitors: Discovery of New Hits through the Pharmacophore Model Generation, Virtual Screening and Molecular Dynamics Simulation. Bioorg. Chem. 2019, 85, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Beck, J.M.; Xia, S.; Zhang, J.; Hadad, C.M. Butyrylcholinesterase and G116H, G116S, G117H, G117N, E197Q and G117H/E197Q Mutants: A Molecular Dynamics Study. Chem. Biol. Interact. 2010, 187, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Dhanabalan, A.K.; Kesherwani, M.; Velmurugan, D.; Gunasekaran, K. Identification of New BACE1 Inhibitors Using Pharmacophore and Molecular Dynamics Simulations Approach. J. Mol. Graph. Model. 2017, 76, 56–69. [Google Scholar] [CrossRef]
- Manoharan, P.; Ghoshal, N. Fragment-Based Virtual Screening Approach and Molecular Dynamics Simulation Studies for Identification of BACE1 Inhibitor Leads. J. Biomol. Struct. Dyn. 2018, 36, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, X.H.; Guan, J.; Ge, S.; Wu, M.B.; Lin, J.P.; Yang, L.R. Advancement of Multi-Target Drug Discoveries and Promising Applications in the Field of Alzheimer’s Disease. Eur. J. Med. Chem. 2019, 169, 200–223. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-Mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Cuya, T.; Baptista, L.; Celmar Costa França, T. A Molecular Dynamics Study of Components of the Ginger (Zingiber Officinale) Extract inside Human Acetylcholinesterase: Implications for Alzheimer Disease. J. Biomol. Struct. Dyn. 2018, 36, 3843–3855. [Google Scholar] [CrossRef]
- Jiang, C.S.; Ge, Y.X.; Cheng, Z.Q.; Wang, Y.Y.; Tao, H.R.; Zhu, K.; Zhang, H. Discovery of New Selective Butyrylcholinesterase (BCHE) Inhibitors with Anti-Aβ Aggregation Activity: Structure-Based Virtual Screening, Hit Optimization and Biological Evaluation. Molecules 2019, 24, 2568. [Google Scholar] [CrossRef] [PubMed]
- Digiacomo, M.; Chen, Z.; Wang, S.; Lapucci, A.; Macchia, M.; Yang, X.; Chu, J.; Han, Y.; Pi, R.; Rapposelli, S. Synthesis and Pharmacological Evaluation of Multifunctional Tacrine Derivatives against Several Disease Pathways of AD. Bioorg. Med. Chem. Lett. 2015, 25, 807–810. [Google Scholar] [CrossRef]
- González-Naranjo, P.; Pérez-Macias, N.; Pérez, C.; Roca, C.; Vaca, G.; Girón, R.; Sánchez-Robles, E.; Martín-Fontelles, M.I.; de Ceballos, M.L.; Martin-Requero, A.; et al. Indazolylketones as New Multitarget Cannabinoid Drugs. Eur. J. Med. Chem. 2019, 166, 90–107. [Google Scholar] [CrossRef]
- Lee, S.; Youn, K.; Lim, G.T.; Lee, J.; Jun, M. In Silico Docking and in Vitro Approaches towards BACE1 and Cholinesterases Inhibitory Effect of Citrus Flavanones. Molecules 2018, 23, 1509. [Google Scholar] [CrossRef]
- Mohamed, T.; Yeung, J.C.K.; Vasefi, M.S.; Beazely, M.A.; Rao, P.P.N. Development and Evaluation of Multifunctional Agents for Potential Treatment of Alzheimer’s Disease: Application to a Pyrimidine-2,4-Diamine Template. Bioorg. Med. Chem. Lett. 2012, 22, 4707–4712. [Google Scholar] [CrossRef] [PubMed]
- Nuthakki, V.K.; Sharma, A.; Kumar, A.; Bharate, S.B. Identification of Embelin, a 3-Undecyl-1,4-Benzoquinone from Embelia Ribes as a Multitargeted Anti-Alzheimer Agent. Drug Dev. Res. 2019, 80, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Viayna, E.; Gómez, T.; Galdeano, C.; Ramírez, L.; Ratia, M.; Badia, A.; Clos, M.V.; Verdaguer, E.; Junyent, F.; Camins, A.; et al. Novel Huprine Derivatives with Inhibitory Activity toward β-Amyloid Aggregation and Formation as Disease-Modifying Anti-Alzheimer Drug Candidates. ChemMedChem 2010, 5, 1855–1870. [Google Scholar] [CrossRef]
- Jannat, S.; Balupuri, A.; Ali, M.Y.; Hong, S.S.; Choi, C.W.; Choi, Y.-H.; Ku, J.-M.; Kim, W.J.; Leem, J.Y.; Kim, J.E.; et al. Inhibition of β-site amyloid precursor protein cleaving enzyme 1 and cholinesterases by pterosins via a specific structure—Activity relationship with a strong BBB permeability. Exp. Mol. Med. 2019, 51, 1–18. [Google Scholar] [CrossRef]
- Chemaxon MarvinSketch Version 19.9.0. Available online: https://www.chemaxon.com (accessed on 1 May 2019).
- TriposInc SYBYL-X 2.0, version; Discovery Software for Computacional Chemistry and Molecular Modelling; Tripos: Saint Louis, MO, USA, 2011.
- Clark, M.; Cramer, R.D.; Van Opdenbosch, N. Validation of the General Purpose Tripos 5.2 Force Field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative Partial Equalization of Orbital Electronegativity—A Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Tripos GALAHAD Manual; Tripos: Saint Louis, MO, USA, 2011.
- Cereto-Massagué, A.; Guasch, L.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. DecoyFinder: An Easy-to-Use Python GUI Application for Building Target-Specific Decoy Sets. Bioinformatics 2012, 28, 1661–1662. [Google Scholar] [CrossRef] [PubMed]
- Tripos UNITY Manual; Tripos: Saint Louis, MO, USA, 2011.
- Gigliarano, C.; Figini, S.; Muliere, P. Making Classifier Performance Comparisons When ROC Curves Intersect. Comput. Stat. Data Anal. 2014, 77, 300–312. [Google Scholar] [CrossRef]
- Systat Software SigmaPlot 2019; Systat: San Jose, CA, USA, 2019.
- Lätti, S.; Niinivehmas, S.; Pentikäinen, O.T. Rocker: Open Source, Easy-to-Use Tool for AUC and Enrichment Calculations and ROC Visualization. J. Cheminform. 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Gary, E.N.; Shiomi, K.; Rosenberry, T.L. Structures of Human Acetylcholinesterase Bound to Dihydrotanshinone i and Territrem B Show Peripheral Site Flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. [Google Scholar] [CrossRef]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.Y. Crystal Structures of Human Cholinesterases in Complex with Huprine W and Tacrine: Elements of Specificity for Anti-Alzheimer’s Drugs Targeting Acetyl- and Butyryl-Cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Domínguez, J.L.; Christopeit, T.; Villaverde, M.C.; Gossas, T.; Otero, J.M.; Nyström, S.; Baraznenok, V.; Lindström, E.; Danielson, U.H.; Sussman, F. Effect of the Protonation State of the Titratable Residues on the Inhibitor Affinity to BACE-1. Biochemistry 2010, 49, 7255–7263. [Google Scholar] [CrossRef]
- Jones, G.; Willet, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully Automated Protein-Ligand Interaction Profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Mermelstein, D.J.; McCammon, J.A.; Walker, R.C. PH-Dependent Conformational Dynamics of Beta-Secretase 1: A Molecular Dynamics Study. J. Mol. Recognit. 2019, 32, e2765. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Energy (kcal/mol) | Pareto | Sterics | HBond | Mol_qry |
|---|---|---|---|---|---|
| 01 * | 1450.77 | 0 | 876.2 | 126.8 | 41.05 |
| 02 | 62.98 | 0 | 796.1 | 128.0 | 32.26 |
| 03 * | 8.71 × 109 | 0 | 813.6 | 134.4 | 39.55 |
| 04 * | 154.37 | 0 | 766.5 | 129.7 | 31.13 |
| 05 | 17.39 | 0 | 686.0 | 123.4 | 36.62 |
| 06 | 60.38 | 0 | 719.8 | 126.2 | 33.27 |
| 07 | 27.95 | 0 | 799.4 | 125.9 | 20.92 |
| 08 | 26.64 | 0 | 791.3 | 118.6 | 35.18 |
| 09 | 17.97 | 0 | 785.6 | 120.7 | 28.20 |
| 10 * | 1343.38 | 0 | 811.9 | 118.9 | 36.97 |
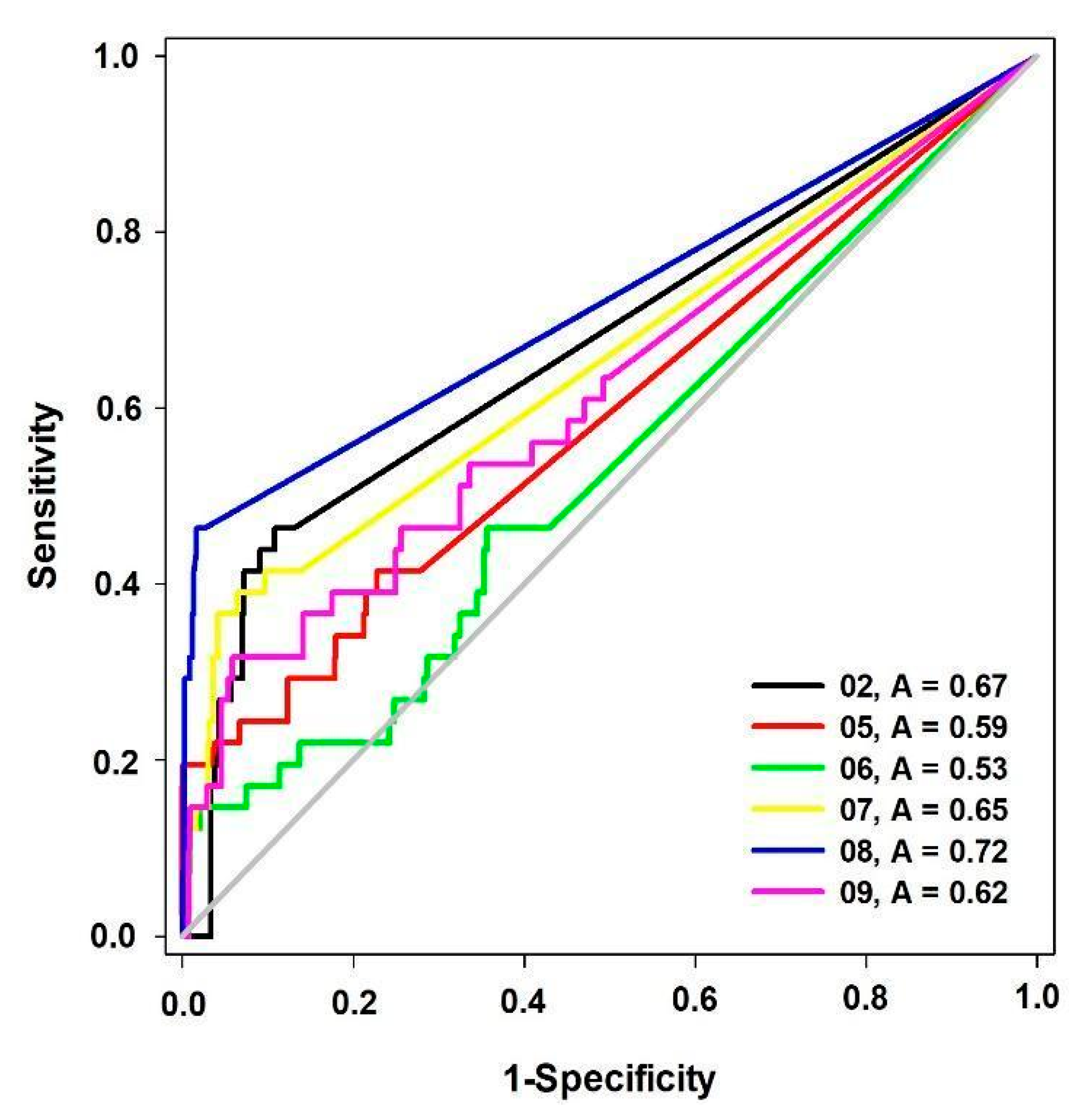
| Model | BEDROC (α = 20) |
|---|---|
| 02 | 0.24 |
| 05 | 0.25 |
| 06 | 0.17 |
| 07 | 0.33 |
| 08 | 0.75 |
| 09 | 0.22 |
| MW (g/mol) | cLogP | Rot. Bond | HBA | HBD | PSA (Å2) | |
|---|---|---|---|---|---|---|
| ZINC6063 | 495.542 | 3.0711 | 13 | 6 | 3 | 101 |
| ZINC1733 | 318.424 | 3.8045 | 5 | 4 | 1 | 42 |
| ZINC1958 | 697.924 * | 6.2240 * | 13 * | 10 | 2 | 139 |
| ZINC5368 | 509.647 * | 5.4016 * | 17 * | 6 | 2 | 106 |
| ZINC6214 | 557.054 * | 5.9325 * | 11 * | 7 | 3 | 113 |
| ZINC1219 | 480.948 | 4.7006 | 6 | 7 | 0 | 85 |
| ZINC1221 | 480.48 | 4.7006 | 6 | 7 | 0 | 85 |
| ZINC1223 | 480.948 | 4.7006 | 6 | 7 | 0 | 85 |
| ZINC6949 | 409.534 | 3.7990 | 8 | 7 | 0 | 65 |
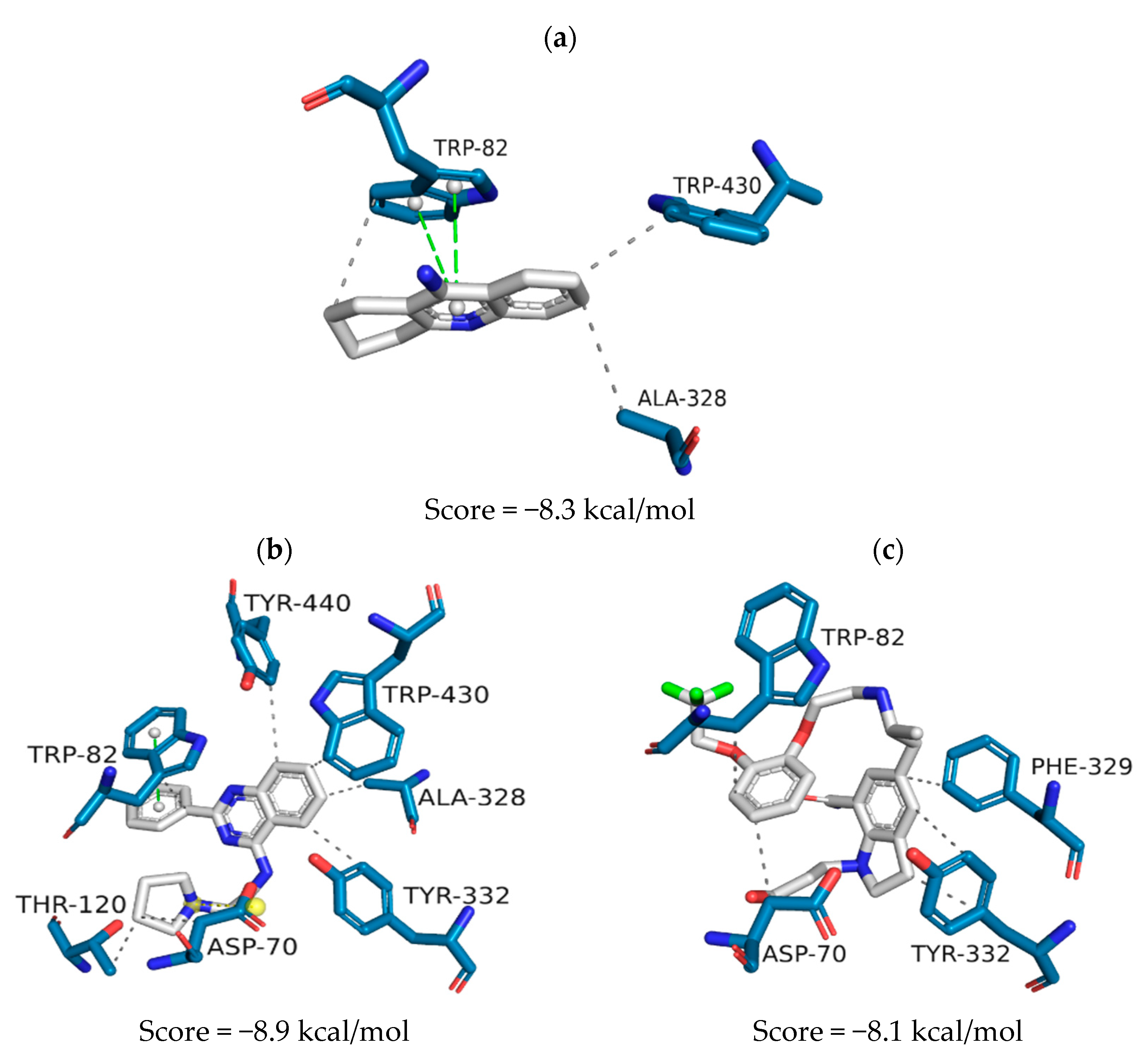
| QFIT | ScoreAChE (kcal/mol) | ScoreBuChE (kcal/mol) | ScoreBACE-1 | |
|---|---|---|---|---|
| ZINC1733 | 31.95 | −9.3 | −8.9 | 38.83 |
| ZINC6063 | 31.67 | −8.0 | −8.1 | 43.55 |
| System | EvdW (kJ/mol) | Eelec (kJ/mol) | GMM (kJ/mol) | Gpolar (kJ/mol) | Gnonpolar (kJ/mol) | ΔGbinding (kJ/mol) |
|---|---|---|---|---|---|---|
| AChE-ZN1733 | −120.938 | −1.557 | −122.495 | 66.866 | 3.569 | −67.980 |
| BuChE-ZN1733 | −144.441 | −17.825 | −162.266 | 96.478 | 3.516 | −80.487 |
| BACE-ZN1733 | −190.126 | −28.716 | −218.842 | 132.305 | 3.443 | −104.466 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, D.B.; do Bomfim, M.R.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G.; Cruz, J.N.; de Carvalho, P.B.; Campos, J.M.; Santos, C.B.R.; Leite, F.H.A. Development of Potential Multi-Target Inhibitors for Human Cholinesterases and Beta-Secretase 1: A Computational Approach. Pharmaceuticals 2023, 16, 1657. https://doi.org/10.3390/ph16121657
Barbosa DB, do Bomfim MR, de Oliveira TA, da Silva AM, Taranto AG, Cruz JN, de Carvalho PB, Campos JM, Santos CBR, Leite FHA. Development of Potential Multi-Target Inhibitors for Human Cholinesterases and Beta-Secretase 1: A Computational Approach. Pharmaceuticals. 2023; 16(12):1657. https://doi.org/10.3390/ph16121657
Chicago/Turabian StyleBarbosa, Deyse B., Mayra R. do Bomfim, Tiago A. de Oliveira, Alisson M. da Silva, Alex G. Taranto, Jorddy N. Cruz, Paulo B. de Carvalho, Joaquín M. Campos, Cleydson B. R. Santos, and Franco H. A. Leite. 2023. "Development of Potential Multi-Target Inhibitors for Human Cholinesterases and Beta-Secretase 1: A Computational Approach" Pharmaceuticals 16, no. 12: 1657. https://doi.org/10.3390/ph16121657