
A Review of Childhood Acute Myeloid Leukemia: Diagnosis and Novel Treatment
Abstract
:1. Introduction
2. Well Known Subtypes of Childhood AML
2.1. Core-Binding Factor AML
2.2. KMT2Ar (KMT2A/11q23 Rearrangement)
2.3. Normal Karyotypes
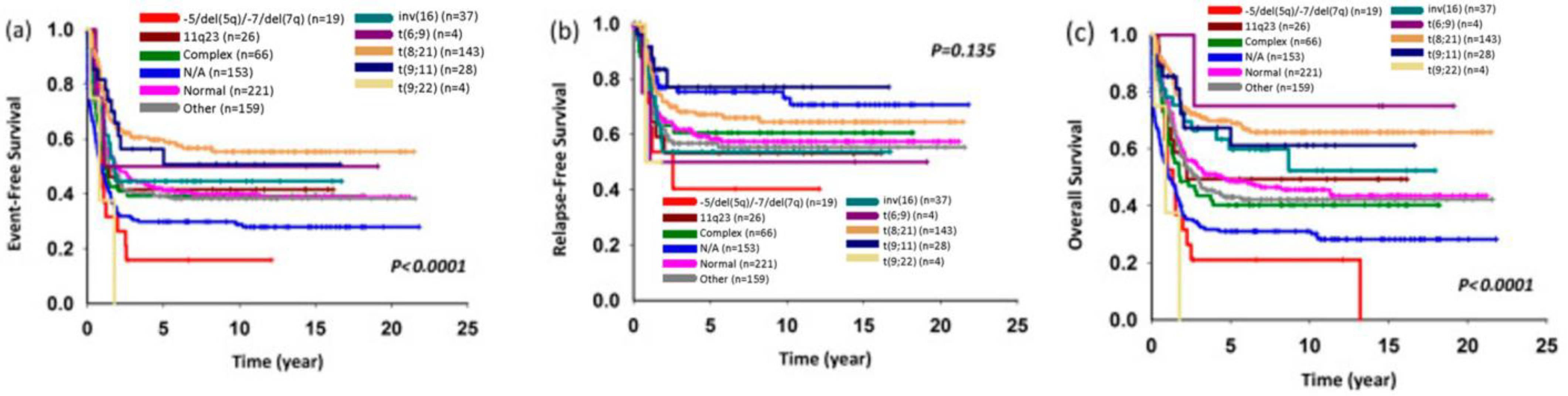
2.4. Unbalanced Cytogenetic Abnormalities in Childhood AML
2.5. NUP98 11p15/NUP09 Rearrangement
2.6. Acute Promyelocytic Leukemia (APL)
3. Current Treatment Regimens and Future Treatments
3.1. Target Therapies
3.1.1. Gemtuzumab Ozogamicin
3.1.2. CD123 Target Therapy
3.1.3. FLT3 Inhibitors
3.1.4. BCL-2 Inhibitors
3.1.5. AKT/MAPK/STAT Inhibitors
3.1.6. Menin-KMT2a Inhibitors
3.1.7. Anti-CD47 Antibody
3.2. Chimeric Antigen Receptor (CAR) T-Cell Therapy
3.3. Checkpoint Inhibitors
3.4. Epigenic Therapies
3.4.1. Hypomethylating Agents (HMAs)
3.4.2. Histone Deacetylase (HDAC) Inhibitors
3.5. TP53 Stabilizers
3.6. Repurposing of Chemotherapy Drugs
3.6.1. CPX-351
3.6.2. Oxidative Phosphorylation Inhibitors: Atovaquone Repurpose
3.7. Treatment for Acute Promyelocytic Leukemia (APL)
4. Hematopoietic Stem Cell Transplantation (HSCT) Guidelines for AML
4.1. Hematopoietic Stem Cell Transplant (HSCT) Recommendations
4.2. Unfavorable-Risk Molecular Abnormality
4.3. Unfavorable-Risk Cytogenetic Abnormality
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Rooij, J.D.E.; Zwaan, C.M.; Van den Heuvel-Eibrink, M. Pediatric AML: From Biology to Clinical Management. J. Clin. Med. 2015, 4, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Creutzig, U.; van den Heuvel-Eibrink, M.M.; Gibson, B.; Dworzak, M.N.; Adachi, S.; de Bont, E.; Harbott, J.; Hasle, H.; Johnston, D.; Kinoshita, A.; et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: Recommendations from an international expert panel. Blood 2012, 120, 3187–3205. [Google Scholar] [CrossRef] [PubMed]
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Conneely, S.E.; Stevens, A.M. Acute Myeloid Leukemia in Children: Emerging Paradigms in Genetics and New Approaches to Therapy. Curr. Oncol. Rep. 2021, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Rubnitz, J.E. Current Management of Childhood Acute Myeloid Leukemia. Paediatr. Drugs 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Yang, Y.L.; Jaing, T.H.; Chen, S.H.; Liu, H.C.; Hung, I.J.; Lin, D.T.; Yang, C.P.; Peng, C.T.; Lin, K.H.; Hsiao, C.C.; et al. Treatment outcomes of pediatric acute myeloid leukemia: A retrospective analysis from 1996 to 2019 in Taiwan. Sci. Rep. 2021, 11, 5893. [Google Scholar] [CrossRef]
- Obszański, P.; Kozłowska, A.; Wańcowiat, J.; Twardowska, J.; Lejman, M.; Zawitkowska, J. Molecular-Targeted Therapy of Pediatric Acute Myeloid Leukemia. Molecules 2022, 27, 3911. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef]
- Quan, X.; Deng, J. Core binding factor acute myeloid leukemia: Advances in the heterogeneity of. Mol. Clin. Oncol. 2020, 13, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Quessada, J.; Cuccuini, W.; Saultier, P.; Loosveld, M.; Harrison, C.J.; Lafage-Pochitaloff, M. Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge. Genes 2021, 12, 924. [Google Scholar] [CrossRef] [PubMed]
- Solh, M.; Yohe, S.; Weisdorf, D.; Ustun, C. Core-binding factor acute myeloid leukemia: Heterogeneity, monitoring, and therapy. Am. J. Hematol. 2014, 89, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Rau, R.E. Beyond KIT in CBF-AML: Chromatin and cohesin. Blood 2016, 127, 2370–2371. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Sinha, C.; Cunningham, L.C.; Liu, P.P. Core Binding Factor Acute Myeloid Leukemia: New Prognostic Categories and Therapeutic Opportunities. Semin. Hematol. 2015, 52, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Surapally, S.; Tenen, D.G.; Pulikkan, J.A. Emerging therapies for inv(16) AML. Blood 2021, 137, 2579–2584. [Google Scholar] [CrossRef]
- Marcucci, G.; Geyer, S.; Laumann, K.; Zhao, W.; Bucci, D.; Uy, G.L.; Blum, W.; Eisfeld, A.K.; Pardee, T.S.; Wang, E.S.; et al. Combination of dasatinib with chemotherapy in previously untreated core binding factor acute myeloid leukemia: CALGB 10801. Blood Adv. 2020, 4, 696–705. [Google Scholar] [CrossRef]
- Klein, K.; Kaspers, G.; Harrison, C.J.; Beverloo, H.B.; Reedijk, A.; Bongers, M.; Cloos, J.; Pession, A.; Reinhardt, D.; Zimmerman, M.; et al. Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Münster Study Group. J. Clin. Oncol. 2015, 33, 4247–4258. [Google Scholar] [CrossRef]
- Harrison, C.J.; Hills, R.K.; Moorman, A.V.; Grimwade, D.J.; Hann, I.; Webb, D.K.; Wheatley, K.; de Graaf, S.S.; van den Berg, E.; Burnett, A.K.; et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J. Clin. Oncol. 2010, 28, 2674–2681. [Google Scholar] [CrossRef]
- Brown, P.A. Neonatal Leukemia. Clin. Perinatol. 2021, 48, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Zarka, J.; Sasaki, K.; Qiao, W.; Pak, D.; Ning, J.; Short, N.J.; Haddad, F.; Tang, Z.; Patel, K.P.; et al. Predictors of outcomes in adults with acute myeloid leukemia and KMT2A rearrangements. Blood Cancer J. 2021, 11, 162. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Kaspers, G.J.L. How I treat pediatric acute myeloid leukemia. Blood 2021, 138, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Hernández Sánchez, A.; González, T.; Sobas, M.A.; Sträng, E.; Gastone, C.; Abáigar, M.; Valk, P.J.; Villaverde Ramiro, A.; Benner, A.; Metzeler, K.H.; et al. Rearrangements Involving 11q23/KMT2A: Mutational Landscape and Prognostic Implications—Results of the Harmony Alliance AML Database. Blood 2022, 140, 551–554. [Google Scholar] [CrossRef]
- Pollard, J.A.; Guest, E.; Alonzo, T.A.; Gerbing, R.B.; Loken, M.R.; Brodersen, L.E.; Kolb, E.A.; Aplenc, R.; Meshinchi, S.; Raimondi, S.C.; et al. Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2021, 39, 3149–3160. [Google Scholar] [CrossRef]
- Tregnago, C.; Benetton, M.; Da Ros, A.; Borella, G.; Longo, G.; Polato, K.; Francescato, S.; Biffi, A.; Pigazzi, M. Novel Compounds Synergize With Venetoclax to Target KMT2A-Rearranged Pediatric Acute Myeloid Leukemia. Front. Pharmacol. 2021, 12, 820191. [Google Scholar] [CrossRef]
- Stark, B.; Jeison, M.; Gabay, L.G.; Mardoukh, J.; Luria, D.; Bar-Am, I.; Avrahami, G.; Kapeliushnik, Y.; Sthoeger, D.; Herzel, G.; et al. Classical and molecular cytogenetic abnormalities and outcome of childhood acute myeloid leukaemia: Report from a referral centre in Israel. Br. J. Haematol. 2004, 126, 320–337. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Meshinchi, S.; Radich, J.P.; Veerman, A.J.; Huismans, D.R.; Munske, L.; Podleschny, M.; Hählen, K.; Pieters, R.; Zimmermann, M.; et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: Prognostic significance and relation to cellular drug resistance. Blood 2003, 102, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; McIntyre, E.; Rau, R.; Meshinchi, S.; Lacayo, N.; Dahl, G.; Alonzo, T.A.; Chang, M.; Arceci, R.J.; Small, D. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 2007, 110, 979–985. [Google Scholar] [CrossRef]
- Shimada, A.; Taki, T.; Tabuchi, K.; Taketani, T.; Hanada, R.; Tawa, A.; Tsuchida, M.; Horibe, K.; Tsukimoto, I.; Hayashi, Y. Tandem duplications of MLL and FLT3 are correlated with poor prognoses in pediatric acute myeloid leukemia: A study of the Japanese childhood AML Cooperative Study Group. Pediatr. Blood Cancer 2008, 50, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Manola, K.N. Cytogenetics of pediatric acute myeloid leukemia. Eur. J. Haematol. 2009, 83, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.S.; Dickerson, K.E.; Nakano, T.A.; Wlodarski, M. Monosomy 7 Predisposition Syndromes Overview. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Galaverna, F.; Ruggeri, A.; Locatelli, F. Myelodysplastic syndromes in children. Curr. Opin. Oncol. 2018, 30, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hosono, N. Genetic abnormalities and pathophysiology of MDS. Int. J. Clin. Oncol. 2019, 24, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Laursen, A.C.; Sandahl, J.D.; Kjeldsen, E.; Abrahamsson, J.; Asdahl, P.; Ha, S.Y.; Heldrup, J.; Jahnukainen, K.; Jónsson, Ó.G.; Lausen, B.; et al. Trisomy 8 in pediatric acute myeloid leukemia: A NOPHO-AML study. Genes Chromosomes Cancer 2016, 55, 719–726. [Google Scholar] [CrossRef]
- Schmoellerl, J.; Barbosa, I.A.M.; Eder, T.; Brandstoetter, T.; Schmidt, L.; Maurer, B.; Troester, S.; Pham, H.T.T.; Sagarajit, M.; Ebner, J.; et al. CDK6 is an essential direct target of NUP98 fusion proteins in acute myeloid leukemia. Blood 2020, 136, 387–400. [Google Scholar] [CrossRef]
- Heikamp, E.B.; Henrich, J.A.; Perner, F.; Wong, E.M.; Hatton, C.; Wen, Y.; Barwe, S.P.; Gopalakrishnapillai, A.; Xu, H.; Uckelmann, H.J.; et al. The menin-MLL1 interaction is a molecular dependency in NUP98-rearranged AML. Blood 2022, 139, 894–906. [Google Scholar] [CrossRef]
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.; Barragán, E.; Cervera, J. Acute Promyelocytic Leukemia: A Constellation of Molecular Events around a Single. Cancers 2020, 12, 624. [Google Scholar] [CrossRef]
- Conneely, S.E.; Stevens, A.M. Advances in Pediatric Acute Promyelocytic Leukemia. Children 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.M. Acute Promyelocytic Leukemia: A Summary. J. Adv. Pract. Oncol. 2018, 9, 178–187. [Google Scholar]
- Mrózek, K.; Bloomfield, C.D. Clinical significance of the most common chromosome translocations in adult acute myeloid leukemia. J. Natl. Cancer Inst. Monogr. 2008, 2008, 52–57. [Google Scholar] [CrossRef] [PubMed]
- McNeer, N.A.; Philip, J.; Geiger, H.; Ries, R.E.; Lavallée, V.P.; Walsh, M.; Shah, M.; Arora, K.; Emde, A.K.; Robine, N.; et al. Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 2019, 33, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, D.; Chi, S.; Uchiyama, S.; Nakamura, H.; Guo, Y.M.; Yamauchi, N.; Yuda, J.; Minami, Y. Molecular Classification and Overcoming Therapy Resistance for Acute Myeloid Leukemia with Adverse Genetic Factors. Int. J. Mol. Sci. 2022, 23, 5950. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, R.; Chauhan, L.; Alonzo, T.A.; Wang, Y.C.; Elmasry, A.; Loken, M.R.; Pollard, J.; Aplenc, R.; Raimondi, S.; Hirsch, B.A.; et al. ABCB1 SNP predicts outcome in patients with acute myeloid leukemia treated with Gemtuzumab ozogamicin: A report from Children’s Oncology Group AAML0531 Trial. Blood Cancer J. 2019, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Zhao, J.; Song, Y.; Liu, D. Gilteritinib: A novel FLT3 inhibitor for acute myeloid leukemia. Biomark. Res. 2019, 7, 19. [Google Scholar] [CrossRef]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef]
- Röllig, C.; Serve, H.; Hüttmann, A.; Noppeney, R.; Müller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Arana Yi, C.; Cortes, J.E.; Levis, M.; Faderl, S.; Garcia-Manero, G.; Jabbour, E.; Konopleva, M.; O’Brien, S.; Estrov, Z.; et al. Final report of phase II study of sorafenib, cytarabine and idarubicin for initial therapy in younger patients with acute myeloid leukemia. Leukemia 2014, 28, 1543–1545. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.A.; Alonzo, T.A.; Brown, P.A.; Gerbing, R.B.; Fox, E.; Choi, J.K.; Fisher, B.T.; Hirsch, B.A.; Kahwash, S.; Levine, J.E. Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ AML improves event-free survival and reduces relapse risk: A report from the Children’s Oncology Group Protocol AAML1031. Blood 2019, 134, 292. [Google Scholar] [CrossRef]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Röllig, C.; Wollmer, E.; Wäsch, R.; Bornhäuser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia with FLT3-Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Stone, R.M.; Deangelo, D.J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E.J.; Schiller, G.J.; et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010, 28, 4339–4345. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Maloney, K.W.; Treece, A.L.; Gore, L.; Franklin, A.K. Single-center pediatric experience with venetoclax and azacitidine as treatment for myelodysplastic syndrome and acute myeloid leukemia. Pediatr. Blood Cancer 2020, 67, e28398. [Google Scholar] [CrossRef]
- Karol, S.E.; Alexander, T.B.; Budhraja, A.; Pounds, S.B.; Canavera, K.; Wang, L.; Wolf, J.; Klco, J.M.; Mead, P.E.; Das Gupta, S.; et al. Venetoclax in combination with cytarabine with or without idarubicin in children with relapsed or refractory acute myeloid leukaemia: A phase 1, dose-escalation study. Lancet Oncol. 2020, 21, 551–560. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [CrossRef]
- Verstovsek, S.; Odenike, O.; Singer, J.W.; Granston, T.; Al-Fayoumi, S.; Deeg, H.J. Phase 1/2 study of pacritinib, a next generation JAK2/FLT3 inhibitor, in myelofibrosis or other myeloid malignancies. J. Hematol. Oncol. 2016, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Fareed, A.; Inam, N.; Faraz, F. Breakthrough treatment choice for Acute Myeloid Leukemia in pediatric and adult patients: Revumenib, an oral selective inhibitor of KMTA2Ar. Rare Tumors 2023, 15, 20363613231183785. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Al Malki, M.M.; Asch, A.S.; Wang, E.S.; Jurcic, J.G.; Bradley, T.J.; Flinn, I.W.; Pollyea, D.A.; Kambhampati, S.; Tanaka, T.N.; et al. Magrolimab in Combination With Azacitidine in Patients with Higher-Risk Myelodysplastic Syndromes: Final Results of a Phase Ib Study. J. Clin. Oncol. 2023, 41, 2815–2826. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed]
- Tabata, R.; Chi, S.; Yuda, J.; Minami, Y. Emerging Immunotherapy for Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 1944. [Google Scholar] [CrossRef]
- Albinger, N.; Pfeifer, R.; Nitsche, M.; Mertlitz, S.; Campe, J.; Stein, K.; Kreyenberg, H.; Schubert, R.; Quadflieg, M.; Schneider, D.; et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 2022, 12, 61. [Google Scholar] [CrossRef]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef]
- Loff, S.; Dietrich, J.; Meyer, J.E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Gründer, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly Switchable Universal CAR-T Cells for Treatment of CD123-Positive Leukemia. Mol. Ther. Oncolytics 2020, 17, 408–420. [Google Scholar] [CrossRef]
- Willier, S.; Rothämel, P.; Hastreiter, M.; Wilhelm, J.; Stenger, D.; Blaeschke, F.; Rohlfs, M.; Kaeuferle, T.; Schmid, I.; Albert, M.H.; et al. CLEC12A and CD33 coexpression as a preferential target for pediatric AML combinatorial immunotherapy. Blood 2021, 137, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, K. Targeting Immune Signaling Checkpoints in Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Ganser, A. Treatment of Relapsed Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2020, 21, 66. [Google Scholar] [CrossRef] [PubMed]
- Cruijsen, M.; Lübbert, M.; Wijermans, P.; Huls, G. Clinical Results of Hypomethylating Agents in AML Treatment. J. Clin. Med. 2014, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Santos, F.P.; Garcia-Manero, G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011, 25, 226–235. [Google Scholar] [CrossRef]
- San José-Enériz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef]
- Craddock, C.F.; Houlton, A.E.; Quek, L.S.; Ferguson, P.; Gbandi, E.; Roberts, C.; Metzner, M.; Garcia-Martin, N.; Kennedy, A.; Hamblin, A.; et al. Outcome of Azacitidine Therapy in Acute Myeloid Leukemia Is not Improved by Concurrent Vorinostat Therapy but Is Predicted by a Diagnostic Molecular Signature. Clin. Cancer Res. 2017, 23, 6430–6440. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Walker, A.R.; Schlenk, R.F.; Sierra, J.; Medeiros, B.C.; Ocio, E.M.; Röllig, C.; Strickland, S.A.; Thol, F.; Valera, S.Z.; et al. Safety and efficacy of oral panobinostat plus chemotherapy in patients aged 65 years or younger with high-risk acute myeloid leukemia. Leuk. Res. 2019, 85, 106197. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2017, 292, 19607. [Google Scholar] [CrossRef] [PubMed]
- Rangel, L.P.; Ferretti, G.D.S.; Costa, C.L.; Andrade, S.M.M.V.; Carvalho, R.S.; Costa, D.C.F.; Silva, J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019, 294, 3670–3682. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients with Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Absalon, M.J.; Alonzo, T.A.; Gerbing, R.B.; Leger, K.J.; Hirsch, B.A.; Pollard, J.; Razzouk, B.I.; Aplenc, R.; Kolb, E.A. Phase I/II Study of CPX-351 Followed by Fludarabine, Cytarabine, and Granulocyte-Colony Stimulating Factor for Children with Relapsed Acute Myeloid Leukemia: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2020, 38, 2170–2177. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.M.; Schafer, E.S.; Li, M.; Terrell, M.; Rashid, R.; Paek, H.; Bernhardt, M.B.; Weisnicht, A.; Smith, W.T.; Keogh, N.J.; et al. Repurposing Atovaquone as a Therapeutic against Acute Myeloid Leukemia (AML): Combination with Conventional Chemotherapy Is Feasible and Well Tolerated. Cancers 2023, 15, 1344. [Google Scholar] [CrossRef]
- Stevens, A.M.; Xiang, M.; Heppler, L.N.; Tošić, I.; Jiang, K.; Munoz, J.O.; Gaikwad, A.S.; Horton, T.M.; Long, X.; Narayanan, P.; et al. Atovaquone is active against AML by upregulating the integrated stress pathway and suppressing oxidative phosphorylation. Blood Adv. 2019, 3, 4215–4227. [Google Scholar] [CrossRef]
- Taga, T.; Tomizawa, D.; Takahashi, H.; Adachi, S. Acute myeloid leukemia in children: Current status and future directions. Pediatr. Int. 2016, 58, 71–80. [Google Scholar] [CrossRef]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef]
- Lo Coco, F.; Ammatuna, E.; Noguera, N. Treatment of acute promyelocytic leukemia with gemtuzumab ozogamicin. Clin. Adv. Hematol. Oncol. 2006, 4, 57–62, 76–77. [Google Scholar] [PubMed]
- Martino, O.D.; Welch, J.S. Retinoic Acid Receptors in Acute Myeloid Leukemia Therapy. Cancers 2019, 11, 1915. [Google Scholar] [CrossRef]
- Elgarten, C.W.; Aplenc, R. Pediatric acute myeloid leukemia: Updates on biology, risk stratification, and therapy. Curr. Opin. Pediatr. 2020, 32, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Sulis, M.L.; Chewning, J.H.; Pollard, J.A.; Cooper, T.; Gamis, A.; Shenoy, S.; Kutny, M.; Horan, J.; Meshinchi, S.; et al. Hematopoietic Cell Transplantation in the Treatment of Pediatric Acute Myelogenous Leukemia and Myelodysplastic Syndromes: Guidelines from the American Society of Transplantation and Cellular Therapy. Transplant. Cell Ther. 2022, 28, 530–545. [Google Scholar] [CrossRef]
- Gale, R.E.; Green, C.; Allen, C.; Mead, A.J.; Burnett, A.K.; Hills, R.K.; Linch, D.C.; Medical Research Council Adult Leukaemia Working Party. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008, 111, 2776–2784. [Google Scholar] [CrossRef] [PubMed]
- Shouval, R.; Labopin, M.; Bomze, D.; Baerlocher, G.M.; Capria, S.; Blaise, D.; Hänel, M.; Forcade, E.; Huynh, A.; Saccardi, R.; et al. Risk stratification using FLT3 and NPM1 in acute myeloid leukemia patients autografted in first complete remission. Bone Marrow Transplant. 2020, 55, 2244–2253. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Alonzo, T.A.; Gerbing, R.B.; Ries, R.E.; Gibson, B.; Niktoreh, N.; Noort, S.; Van den Heuvel-Eibrink, M.M.; Zwaan, C.M.; Meshinchi, S. Distinct Co-Occurring Mutational Profiles in Acute Myeloid Leukemia Confers Prognostic Significance in Children and Young Adults with FLT3/ITD Mutations. Blood 2018, 132, 443. [Google Scholar] [CrossRef]
- Kobos, R.; Steinherz, P.G.; Kernan, N.A.; Prockop, S.E.; Scaradavou, A.; Small, T.N.; Shukla, N.; Khalaf, R.; O’Reilly, R.J.; Boulad, F. Allogeneic Hematopoietic Stem Cell Transplantation for Pediatric Patients with Treatment-Related Myelodysplastic Syndrome or Acute Myelogenous Leukemia. Biol. Blood Marrow Transplant. 2012, 18, 473–480. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2017 | 2021 | |
|---|---|---|
| Favorable | t(8;21)(q22;q22)/RUNX1-RUNX1T1 | t(8;21)(q22;q22)/RUNX1-RUNX1T1 |
| inv(16)(p13.1;q22)/CBFb-MYH11 | inv(16)(p13.1;q22)/t(16;16)(p13.1;q22)/CBFB-MYH11 | |
| t(16;16)(p13.1;q22)/CBFb-MYH11 | ||
| Mutated NPM1 without FLT3-ITD | NPM1 mutation with or without FLT3-ITD | |
| Biallelic mutations of CEBPA | CEBPA mutation with or without FLT3-ITD | |
| t(1;11)(q21;q23)/MLLT11-KMT2A | ||
| Unfavorable | t(6;11)(q27;q23)/MLLT4-KMT2A | inv(16)(p13.3q24.3)/CBFA2T3-GLIS2 |
| t(10;11)(p12;q23)/MLLT10-KMT2A | t(10;11)(p12;q23)/KMT2A-AF10 | |
| t(10;11)(p11.2;q23)/ABI1-KMT2A | t(10;11)(p11.2;q23)/KMT2A-ABI1 | |
| t(6;9)(p23;q34)/DEK-NUP214 | t(6;11)(q27;q23)/KMT2A-MLLT4 | |
| t(8;16)(p11;p13)/KAT6A-CREBBP | t(4;11)(q21;q23.3)/KMT2A-MLLT2 | |
| t(16;21)(q24;q22)/RUNX1-CBFA2T3 | t(11;12)(p15;p13)/NUP98-KDM5A | |
| t(5;11)(q35;p15.5)/NUP98-NSD1 | t(7;11)(p15.4;p15)/NUP98-HOXA9 | |
| inv(16)(p13.3q24.3)/CBFA2T3-GLIS2 | t(5;11)(q35;p15)/NUP98-NSD1 | |
| t(11;15)(p15;q35)/NUP98-KDM5A | t(6;9)(p23;q34)/DEK-NUP214 | |
| t(3;5)(q25;q34)/NPM1-MLF1 | t(8;16)(p11;p13)/KAT6A-CREBBP | |
| FLT3-ITD | t(16;21)(q24;q22)/RUNX1-CBFA2T3 | |
| Monosomy 7 | t(7;12)(q36;p13)/ETV6-HLXB | |
| t(3;21)(26.2;q22)/RUNX1-MECOM | ||
| t(16;21)(p11.2;q22.2)/FUS-ERG | ||
| FLT3-ITD without CEPBA or NPM1 mutation | ||
| inv(3)(q21.3q26.2)/t(3;3)(q21.3q26.2)/RPN1-MECOM | ||
| t(3;5)(q25;q34)/NPM1-MLF1 | ||
| t(10;11)(p12.3;q14.2)/PICALM-MLLT10 | ||
| −7, −5, 5q- | ||
| Intermediate or unknown | t(9;11)(p12;q23)/MLLT3-KMT2A | |
| Other KMT2A fusions | ||
| t(1;22)(p13;q13)/RBM15-MKL1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, S.; Lee, M.-E.; Lin, P.-C. A Review of Childhood Acute Myeloid Leukemia: Diagnosis and Novel Treatment. Pharmaceuticals 2023, 16, 1614. https://doi.org/10.3390/ph16111614
Tseng S, Lee M-E, Lin P-C. A Review of Childhood Acute Myeloid Leukemia: Diagnosis and Novel Treatment. Pharmaceuticals. 2023; 16(11):1614. https://doi.org/10.3390/ph16111614
Chicago/Turabian StyleTseng, Serena, Mu-En Lee, and Pei-Chin Lin. 2023. "A Review of Childhood Acute Myeloid Leukemia: Diagnosis and Novel Treatment" Pharmaceuticals 16, no. 11: 1614. https://doi.org/10.3390/ph16111614