
The FDA’s New Guideline “Generally Accepted Scientific Knowledge” (GASK): An Opportunity to Expedite the Approval of Biosimilars


Abstract
:1. Introduction
- Monoclonal antibodies for in vivo use.
- Proteins intended for therapeutic use, including cytokines (e.g., interferons), enzymes (e.g., thrombolytics), and other novel proteins, except for those that are specifically assigned to CBER (e.g., vaccines and blood products). This category includes therapeutic proteins derived from plants, animals, or microorganisms and recombinant versions of these products.
- Non-antigen-specific immunomodulators (e.g., cytokines, growth factors, chemokines, etc.) that are intended to treat disease by inhibiting or modifying a pre-existing immune response; and proteins or peptides intended to act in antigen-specific fashion to treat or prevent autoimmune diseases by inhibiting or modifying pre-existing immune responses.
- Growth factors, cytokines, and monoclonal antibodies intended to mobilize, stimulate, decrease, or otherwise alter the production of cells in vivo. This category includes growth factors, cytokines, and monoclonal antibodies, as well as non-biological agents, administered as mobilizing agents for their direct therapeutic effect on the recipient, as well as growth factors, cytokines, and monoclonal antibodies administered to subsequently harvest the mobilized, stimulated, decreased or otherwise altered cells for use in a human cellular or tissue-based product (HCT/P).
2. The GASK Application to Biosimilars
3. Regulatory History
- March 2010: The BPCIA of 2009 was signed into law, creating a new class of biological drugs, namely biosimilars. It was amended to extend the exclusivity to twelve years from the initial eight years at the time of the approval of BPCIA. This change came at the protest of “big pharma,” claiming that the cost of developing new biological drugs has reached billion-dollar ranges. That extended times were needed to recover their investments. The EU allows exclusivity of ten years. In 2022, a new law, the Inflation Reduction ACT (IRA) [11], allowed the CMS (the agency responsible for Medicare and Medicaid patients) to reduce the price of biological drugs by 35% if they had a monopoly of 12 years, if there are no biosimilars and none expected within 12 months, and if these drugs are among the top ten highest reimbursed drugs. Surprisingly, the associations responsible for promoting biosimilars also opposed this bill, stating that reducing the price of reference products would reduce the incentive for biosimilar development efforts.
- February 2012: The FDA released draft guidance on biosimilar product development, outlining the scientific and regulatory considerations for demonstrating biosimilarity to a reference product.
- August 2014: Reference Product Exclusivity for Biological Products; Draft Guidance for Industry.
- April 2015: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product; Guidance for Industry.
- April 2015: Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product; Guidance for Industry.
- December 2016: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product; Guidance for Industry.
- January 2017: The FDA issued final guidance on interchangeability, providing recommendations on demonstrating that a biosimilar can be substituted for its reference product without the involvement of the prescribing healthcare provider.
- March 2018: The FDA released a Biosimilar Action Plan outlining the FDA’s commitment to promoting competition and access to biosimilars through various initiatives, including educational outreach, regulatory clarity, and market competition.
- May 2018: The Citizen Petition by the author to the FDA asked for a comprehensive review of the biosimilar guidelines to bring more rationality and scientific understanding of the testing required to approve biosimilars [12]. The FDA responded six months after the filing on 18 November 2018 [13], stating that it needed more time to review the requests made in the Petition, even though a reply is expected within 120–180 days [14]. The FDA has adopted several suggestions in the Petition:
- This Petition suggested that the FDA adopt scientific principles to conclude the arguments for removing animal toxicology testing. The BPCIA has been amended as of the end of 2022, removing the term “animal toxicology” and replacing it with “nonclinical testing” [15],
- The Petition had suggested removing the flawed statistical testing models. The FDA withdrew pivotal guidance, i.e., Statistical Approaches to Evaluate Analytical Similarity [16,17,18], and issued a new guideline [19] in May 2019: Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations Guidance for Industry. This guideline removed the tier 1 testing that was challenged in the Citizen Petition as non-scientific [20]. The statistical issue related to testing critical quality attributes, such as protein content and potency, requires the 90% confidence interval of biosimilar attributes to fall within 1.5 times the standard deviation in the reference product. The contradiction occurred because these attributes also meet the release specification that is fixed and different.
- The FDA adopted several suggestions in the Petition, including beginning an extensive teaching program about biosimilars [21] for all stakeholders. This action by the FDA has brought about significant change in the perception of the safety of biosimilars. Most recently, the FDA has changed the labeling requirements of interchangeable biosimilars to remove them from being listed as an interchangeable biosimilar, proposing that the interchangeable status is a legal designation, not related to quality matters; this change will significantly impact the perception of the safety and efficacy of the two classes of biosimilars in the US, a situation that needs a change, as discussed below.
- The denied recommendations included removing the four-letter suffix system to identify biological products, not just biologicals. An argument that this differentiation is needed to monitor post-market data is irrelevant since the registration numbers assigned to products are sufficient to meet this requirement. However, having gone this far, it is unlikely that there will be any change concerning this ruling by the FDA, and perhaps it is unnecessary to question it.
- Other recommendations to be considered include allowing approved non-US comparators to use essentially the same dossier as the US-licensed products and to declare to the public that biosimilars have “no clinically meaningful difference” from the originator product.
- The final recommendations are to encourage the development of in vitro immunogenicity testing methods to reduce test subject exposure on ethical grounds, to revise the perspective on the clinical relevance of the protocols and statistical methods used to establish PK/PD similarity, to end clinical efficacy testing in patients, and to remove the interchangeable status of biosimilars. In vitro immunogenicity tests assess the potential of a biological drug to induce an immune response in patients. Common techniques include enzyme-linked immunosorbent assays (ELISAs) [22] and cell-based assays [23] that measure the presence of anti-drug antibodies (ADAs) and neutralizing antibodies (NAbs). Furthermore, cell-based assays like the competitive ligand-binding assay (CLBA) are employed to assess the impact of ADAs on a drug’s efficacy. One notable example is the anti-TNF (tumor necrosis factor) biological drug infliximab [24]. Another drug that has undergone in vitro immunogenicity testing is adalimumab [25], and another is anti-TNF biologic [26]. Furthermore, in the context of cytokines, interferon-beta [27] and erythropoietin [28] have also been subjects of in vitro immunogenicity studies.
- May 2018: The FDA issued a letter to the United States Pharmacopoeia (USP) asking it not to create monographs for biological drugs [29]. “Because USP’s proposed revisions would aggravate existing concerns that a monograph could impede or delay the licensure of biosimilars and other biological products, the FDA strongly encourages USP to withdraw its proposal. The FDA welcomes future interaction with USP on these issues to ensure that biological product monographs do not create an unnecessary barrier to the availability of biosimilars and other biological products to patients. For example, we see opportunities for optional methodological standards that could encourage innovation and product development”. One of the FDA’s concerns was that using the analytical methodologies provided by the originator product companies might result in complex intellectual property issues. The USP has dropped its plan to create monographs of biological drugs.
- June 2018: The FDA withdraws Draft Guidance for Industry: Statistical Approaches to Evaluate Analytical Similarity, as recommended in the May 2018 Citizen Petition, as it required quality attribute comparisons based on a 90% confidence limit within 1.5× standard deviation of the reference product. This analysis conflicted with the release specification allowance and presented no scientific basis. The FDA also switched from “comparison” to “assessment” to give the evaluation a broader meaning.
- May 2019: The FDA issues Considerations in Demonstrating Interchangeability with a Reference Product; Guidance for Industry, a final guideline. The FDA can allow interchangeability without requiring the proposed three switching and alternating studies with the reference product to secure interchangeable status. The FDA cannot remove the interchangeable classification, unlike the May 2018 Citizen Petition. In February 2020, the FDA added clarification for allowing fewer conditions than the reference product in case it is dictated by the intellectual property or the choice of biosimilar developer. In 2023, the FDA issued a new guideline on labeling biosimilars, removing the mention of the interchangeable status of biosimilar in the prescribing information, asserting that this status is a legal description, not a quality classification.
- July 2021: The FDA released a Biosimilars Action Plan Progress Report, providing an update on the FDA’s efforts to enhance the biosimilars market and increase competition. The plan includes in silico testing and many other AI/ML-based technologies that biosimilar developers could adopt. The FDA issued a Biosimilars Action Plan [30] that encourages using in silico methods; the FDA has also removed the requirement for the immunogenic testing of products that have not impacted pharmacokinetics, such as insulin, and will allow the same for other products.
- December 2022: The FDA Modernization Act 2 amends the BPCIA, removing the term “animal toxicology” and replacing it with “nonclinical” after scientific evidence was presented against animal testing [31]. The scientific basis of this amendment came from the understanding that animals may not have the receptors required for biological drugs to act; the receptor binding leads to PD, leading to pharmacology that results in clinical and toxicological properties. This amendment required legislative action since the term “animal toxicology” was embedded in the requirement for biosimilars.
- December 2022: the FDA publishes its recommendations on using pharmacodynamic markers to support the efficacy of biosimilars [32], as well as a pivotal paper in January 2023 to establish the role of PD markers [33] in the waiving of clinical efficacy testing. The FDA conducted clinical trials to “discover” PD biomarkers, but these studies were limited to proteomics testing and did not differentiate between a PD biomarker and a pharmacology biomarker. The FDA also concluded that the PD biomarkers used to gain a waiver for clinical efficacy testing need not correlate with clinical efficacy, a weak point in justifying the use of PD biomarkers.
- November 2022 and July 2023: The US Senate received a bill to remove the interchangeability status of biosimilars based on scientific evidence that such studies can never fail and thus are tantamount to human abuse. This bill is opposed by the originator companies and even some biosimilar companies that have the additional resources to secure the interchangeable status to help them promote their product as superior to another biosimilar that does not have this status.
- August 2023: The CMS makes its first choice regarding drugs approved for price reduction [34], including three therapeutic proteins, Stelara (ustekinumab), Enbrel (etanercept), and FlaspPen and Novolog Pen (insulin aspart), because there were no biosimilars approved for these drugs; however, it is anticipated that by the time this selection becomes effective, the licensing of biosimilar will likely remove them from the list [35].
- September 2023: A Citizen Petition filed by the author suggests that the FDA consolidate its biosimilar guidance to follow the advice given in the May 2018 Citizen Petition by the author, with updates on how to approve efficacy testing waivers and other regulatory changes within the power of the FDA. This Petition identified the changes made by the FDA as suggested in the May 2018 petition and recommended additional recommendations that are detailed later in this paper.
- September 2023: The FDA calls a meeting to understand the scientific evidence for removing the clinical efficacy testing of biosimilars [36]. The MHRA (the UK Regulatory Agency) declared that it no longer requires patient efficacy testing based on a lack of scientific validity.
- September 2023: The FDA issues a new guideline, namely Labeling for Biosimilar and Interchangeable Biosimilar Products [37], allowing the removal of identification if it is approved as an interchangeable biosimilar, stating that the interchangeable status is a legal description and not a quality differentiation, and thus stakeholders do not need this information, leaving it available only in the Purple Book.
4. Patent Dance
5. Bridging Studies
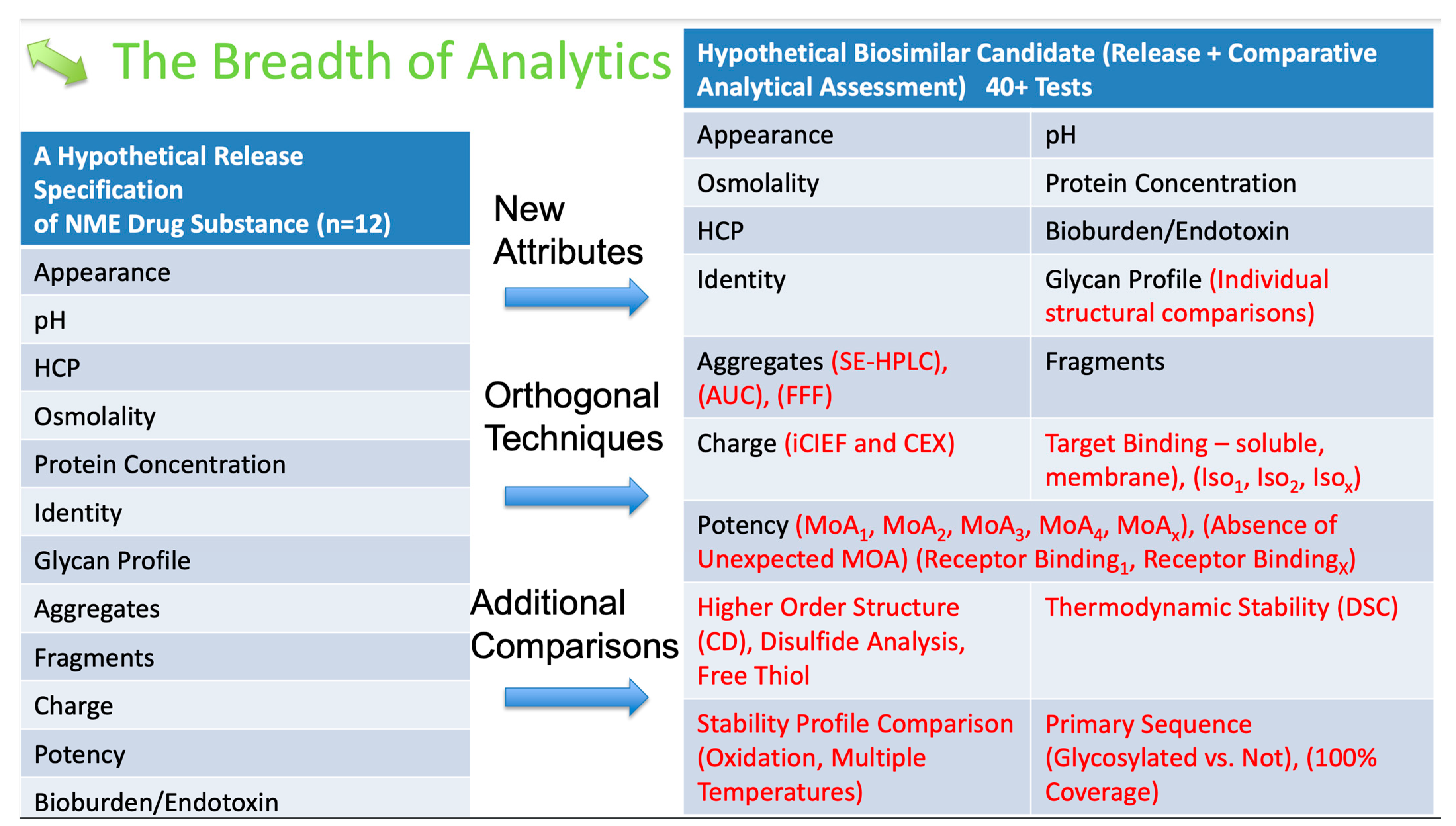
6. Analytical Assessment
6.1. US Pharmacopoeia
6.2. Immunogenicity Study
- Let developers present in vitro tests instead of clinical immunogenicity testing where required.
- Continue its internal development in finding and prescribing testing modalities that reduce the need for clinical immunogenicity testing.
7. Clinical Pharmacology
8. Clinical Efficacy Studies (CES)
“(cc) a clinical study or studies (including the assessment of immunogenicity and pharmacokinetics or pharmacodynamics) that are sufficient to demonstrate safety, purity, and potency in 1 or more appropriate conditions of use for which the reference product is licensed and intended to be used and for which licensure is sought for the biological product.”
“(cc) a clinical study or studies (including the assessment of immunogenicity and pharmacokinetics or pharmacodynamics) that are sufficient to demonstrate safety purity.”
8.1. CES Alternatives
- The analysis and validation of biomarkers will require extensive research, which should not be expected of biosimilar developers.
- The cost of identifying and validating a biomarker may exceed the CES, making it an improbable alternative.
- Biosimilars use different biosimilars; some may be easier to match than others.
- The lack of correlation between a biomarker profile and clinical response leads to uncertainty about whether meeting the biomarker profile is clinically meaningful.
- The nonlinearity of the biomarker profile cannot be established; thus, it will be impossible to conclude whether a failed matching is a study failure, and the same will be true if a study passes.
8.2. Known Biomarkers
8.3. Receptor Binding
8.4. Pharmacokinetics
9. Interchangeable Biosimilars
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- U.S. Food and Drug Administration. Notice of Availability, Generally Accepted Scientific Knowledge in Applications for Drug and Biological Products: Nonclinical Information; Draft Guidance for Industry; Availability. Fed. Regist. 2023, 88, 33887–33888. [Google Scholar]
- Available online: https://www.govinfo.gov/app/details/BILLS-117s5002cps (accessed on 22 September 2023).
- Available online: https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/role-pharmacodynamic-biomarkers-biosimilar-drug-development (accessed on 15 July 2023).
- Available online: https://www.fda.gov/media/78946/download (accessed on 22 September 2023).
- Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information (accessed on 22 September 2023).
- Available online: https://drugs.ncats.io (accessed on 22 September 2023).
- Available online: https://www.fda.gov/about-fda/histories-fda-regulated-products/history-biologics-regulation (accessed on 22 September 2023).
- Available online: https://www.fda.gov/combination-products/jurisdictional-information/transfer-therapeutic-biological-products-center-drug-evaluation-and-research (accessed on 22 September 2023).
- Available online: https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/biosimilars-guidances (accessed on 22 September 2023).
- Available online: https://www.fda.gov/media/119272/download#:~:text=FDA%20interprets%20section%207002(e)%20of%20the%20BPCI%20Act%20and,product%20or%20a%20proposed%20interchangeable (accessed on 22 September 2023).
- Niazi, S.K. The Inflation Reduction Act: A boon for the generic and biosimilar industry. J. Clin. Pharm. Ther. 2022, 47, 1738–1751. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.regulations.gov/document/FDA-2018-P-1876-0001 (accessed on 22 September 2023).
- Available online: https://www.regulations.gov/document/FDA-2018-P-1876-0007 (accessed on 22 September 2023).
- Available online: https://www.fda.gov/files/about%20fda/published/The-FDA-Docket-Frequently-Asked-Questions.pdf (accessed on 22 September 2023).
- Available online: https://www.science.org/doi/10.1126/science.add4664 (accessed on 22 September 2023).
- Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-withdraws-draft-guidance-industry-statistical-approaches-evaluate-analytical-similarity (accessed on 22 September 2023).
- Available online: https://www.fda.gov/media/159261/download (accessed on 22 September 2023).
- Available online: https://www.biospace.com/article/fda-withdraws-draft-guidance-on-biosimilar-development/ (accessed on 22 September 2023).
- Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analytical-assessment-and-other-quality (accessed on 22 September 2023).
- Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-withdraws-draft-guidance-industry-statistical-approaches-evaluate-analytical-similarity#:~:text=After%20considering%20public%20comments%20that,could%20impact%20the%20cost%20and (accessed on 22 September 2023).
- Available online: https://www.fda.gov/drugs/biosimilars/multimedia-education-materials (accessed on 22 September 2023).
- Gupta, S.; Devanarayan, V.; Finco, D.; Gunn, G.R., 3rd; Kirshner, S.; Richards, S.; Rup, B.; Song, A.; Subramanyam, M. Recommendations for the Validation of Cell-Based Assays Used for the Detection of Neutralizing Antibody Immune Responses Elicited Against Biological Therapeutics. J. Pharm. Biomed. Anal. 2016, 117, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Jiskoot, W.; EMEA. Immunogenicity of therapeutic proteins: Clinical implications and future prospects. Clin. Ther. 2011, 33, 764–777. [Google Scholar] [CrossRef]
- Vande Casteele, N.; Ferrante, M.; Van Assche, G.; Ballet, V.; Compernolle, G.; Van Steen, K.; Simoens, S.; Rutgeerts, P.; Gils, A.; Vermeire, S. Trough Concentrations of Infliximab Guide Dosing for Patients with Inflammatory Bowel Disease. Gastroenterology 2017, 152, 812–814. [Google Scholar] [CrossRef]
- Bartelds, G.M.; Wijbrandts, C.A.; Nurmohamed, M.T.; Stapel, S.; Lems, W.F.; Aarden, L.; Dijkmans, B.A.; Tak, P.P.; Wolbink, G.J. Clinical Response to Adalimumab: Relationship to Anti-Adalimumab Antibodies and Serum Adalimumab Concentrations in Rheumatoid Arthritis. Ann. Rheum. Dis. 2011, 70, 142–149. [Google Scholar] [CrossRef]
- Mok, C.C.; van der Kleij, D.; Wolbink, G.J. Drug Levels, Anti-drug Antibodies, and Clinical Efficacy of the Anti-TNFα Biologics in Rheumatic Diseases. Clin. Rheumatol. 2016, 35, 1423–1431. [Google Scholar] [CrossRef]
- Zare, N.; Zarkesh-Esfahani, S.H.; Gharagozloo, M.; Shaygannejad, V. Antibodies to interferon beta in patients with multiple sclerosis receiving CinnoVex, rebif, and betaferon. J. Korean Med. Sci. 2013, 28, 1801–1806. [Google Scholar] [CrossRef]
- van Schie, K.A.; Hart, M.H.; de Groot, E.R.; Kruithof, S.; Aarden, L.A.; Wolbink, G.J.; Rispens, T. The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann. Rheum. Dis. 2015, 74, 311–314. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/media/112103/download (accessed on 22 September 2023).
- Available online: https://www.fda.gov/media/114574/download (accessed on 22 September 2023).
- Niazi, S.K. End animal testing for biosimilar approval. Science 2022, 377, 162–163. [Google Scholar] [CrossRef]
- Florian, J.; Sun, Q.; Schrieber, S.J.; White, R.; Shubow, S.; Johnson-Williams, B.E.; Sheikhy, M.; Harrison, N.R.; Parker, V.J.; Wang, Y.M.; et al. Pharmacodynamic Biomarkers for Biosimilar Development and Approval: A Workshop Summary. Clin. Pharmacol. Ther. 2023, 113, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.; Racz, R.; Burkhart, K.; Florian, J.; Ford, K.; Iveth Garcia, M.; Geiger, R.M.; Howard, K.E.; Hyland, P.L.; Ismaiel, O.A.; et al. New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Front. Med. 2023, 9, 1109541. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.hhs.gov/about/news/2023/08/29/hhs-selects-the-first-drugs-for-medicare-drug-price-negotiation.html (accessed on 22 September 2023).
- Available online: https://www.centerforbiosimilars.com/view/biorationality-first-price-reductions-under-the-ira-3-biologics-with-no-biosimilars (accessed on 22 September 2023).
- Available online: https://www.fda.gov/drugs/news-events-human-drugs/fda-workshop-increasing-efficiency-biosimilar-development-programs-09192022 (accessed on 22 September 2023).
- Available online: https://www.federalregister.gov/documents/2023/09/18/2023-20141/labeling-for-biosimilar-and-interchangeable-biosimilar-products-draft-guidance-for-industry (accessed on 22 September 2023).
- Sarpatwari, A.; Gluck, A.R.; Curfman, G.D. The Supreme Court Ruling in Sandoz v Amgen: A Victory for Follow-on Biologics. JAMA Intern. Med. 2018, 178, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.bigmoleculewatch.com/bpcia-patent-litigations/ (accessed on 22 September 2023).
- Available online: https://www.congress.gov/search?q=%7B%22search%22%3A%5B%22Biosimilars%22%5D%2C%22type%22%3A%22bills%22%2C%22bill-status%22%3A%5B%22law%22%5D%7D (accessed on 22 September 2023).
- Available online: https://www.congress.gov/search?q=%7B%22search%22%3A%5B%22Biosimilars%22%5D%2C%22type%22%3A%22bills%22%7D (accessed on 22 September 2023).
- Available online: https://www.congress.gov/search?q=%7B%22search%22%3A%5B%22Biosimilars+patent%22%5D%2C%22type%22%3A%22bills%22%7D (accessed on 22 September 2023).
- Available online: https://www.congress.gov/search?q=%7B%22search%22%3A%5B%22Biosimilars+patent%22%5D%2C%22type%22%3A%22bills%22%2C%22bill-status%22%3A%22law%22%7D (accessed on 22 September 2023).
- Available online: https://www.congress.gov/bill/116th-congress/house-bill/4850?q=%7B%22search%22%3A%22S.659+-+Biologic+Patent+Transparency+Act%22%7D&s=2&r=16 (accessed on 22 September 2023).
- Available online: https://www.congress.gov/bill/116th-congress/house-bill/133 (accessed on 22 September 2023).
- Available online: https://www.cbo.gov/publication/55203 (accessed on 22 September 2023).
- Webster, C.J.; Woollett, G.R. A ‘Global Reference’ Comparator for Biosimilar Development. BioDrugs 2017, 31, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.; Associate Director for Science & Biosimilar Strategy; Chair of Emerging Technology Team; Office of Biotechnology Products; Office of Pharmaceutical Quality. CDER|US FDA FDA Workshop: Increasing the Efficiency of Biosimilar Development Programs. 19 September 2022. Available online: https://www.fda.gov/media/163283/download (accessed on 22 September 2023).
- Available online: https://www.raps.org/news-and-articles/news-articles/2019/6/fda-usp-clash-over-biologics-monographs (accessed on 22 September 2023).
- FDA: Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. Available online: https://www.fda.gov/media/71489/download (accessed on 22 September 2023).
- Yie, J.; Dey, M.; Su, J.; Sergi, J.; Zhang, Y.; Le, T.H.; Kashi, S.; Gurney, K. Development of a robust functional cell-based assay for replacing the rabbit blood sugar bioidentity test of insulin glargine drug substance. J. Pharm. Biomed. Anal. 2020, 186, 113328. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-immunogenicity-considerations-biosimilar-and-interchangeable-insulin-products (accessed on 22 September 2023).
- Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q5e-comparability-biotechnologicalbiological-products-subject-changes-their-manufacturing-process (accessed on 22 September 2023).
- Available online: http://www.fda.gov/BiologicsBloodVaccines/ScienceResearch/BiologicsResearchAreas/ucm246804.htm (accessed on 22 September 2023).
- van Beers, M.M.C.; Sauerborn, M.; Gilli, F.; Brinks, V.; Schellekens, H.; Jiskoot, W. Oxidized and Aggregated Recombinant Human Interferon Beta is Immunogenic in Human Interferon Beta Transgenic Mice. Pharm. Res. 2011, 28, 2393. [Google Scholar] [CrossRef]
- Koren, E.; De Groot, A.S.; Jawa, V.; Beck, K.D.; Boone, T. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin. Immunol. 2007, 124, 26–32. [Google Scholar] [CrossRef]
- Sharma, B. Immunogenicity of therapeutic proteins. Part 3: Impact of manufacturing changes. Biotechnol. Adv. 2007, 25, 325–331. [Google Scholar] [CrossRef]
- Kringelum, J.V.; Undegaard, C.; Lund, O.; Nielsen, M. Reliable B cell epitope predictions: Impacts of method development and improved benchmarking. PLoS Comput. Biol. 2012, 8, e1002829. [Google Scholar] [CrossRef]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef]
- Mire-Sluis, A.R.; Barrett, Y.C.; Devanarayan, V.; Koren, E.; Liu, H.; Maia, M.; Parish, T.; Scott, G.; Shankar, G.; Shores, E.; et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J. Immunol. Methods 2004, 289, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Ebenhoch, R.; Schiele, F.; Nar, H. Anti-GARP Antibodies Inhibit Release of TGF-β by Regulatory T Cells via Different Modes of Action, but Do Not Influence Their Function In Vitro. Immunohorizons 2023, 7, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S. Scientific Rationale for Waiving Clinical Efficacy Testing of Biosimilars. Drug Des. Dev. Ther. 2022, 16, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Oh, W. Recombinant Protein Products Division, National Institute of Food and Drug Safety Evaluation (NIFDS). Ministry of Food and Drug Safety (MFDS). Available online: https://www.fda.gov/media/172197/download (accessed on 22 September 2023).
- EMA Biosimilar EPAR Program. Available online: https://www.ema.europa.eu/en/search/search/field_ema_web_categories%253Aname_field/Human/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar?search_api_views_fulltext=epar%20biosimilar (accessed on 22 September 2023).
- FDA. Biosimilars Approval Documents. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 22 September 2023).
- Clinical Trial Database. Available online: https://clinicaltrials.gov/ct2/results?term=biosimilar&age_v=&gndr=&type=&rslt=With&Search=Apply (accessed on 22 September 2023).
- Available online: https://pubmed.ncbi.nlm.nih.gov/?term=biosimilars+&filter=pubt.clinicaltrial&sort=pubdate&size=200 (accessed on 22 September 2023).
- Woodcock. Available online: https://www.biopharmadive.com/news/fdas-woodcock-the-clinical-trial-system-is-broken/542698/ (accessed on 22 September 2023).
- US Congress. Available online: https://www.govinfo.gov/content/pkg/STATUTE−98/pdf/STATUTE−98-Pg1585.pdf (accessed on 22 September 2023).
- Available online: https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence (accessed on 22 September 2023).
- Available online: https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products (accessed on 22 September 2023).
- Available online: https://www.centerforbiosimilars.com/view/part-3-biorationality-fda-webinar-on-biosimilars-efficacy-testing-marks-major-step-forward (accessed on 22 September 2023).
- van der Graaf, P.H. Innovations in Biosimilars. Clin. Pharmacol. Ther. 2023, 113, 1–195. [Google Scholar]
- Available online: https://www.fda.gov/drugs/news-events-human-drugs/pharmacodynamic-biomarkers-their-role-biosimilar-product-development (accessed on 22 September 2023).
- Reff, M.E.; Carner, K.; Chambers, K.S.; Chinn, P.C.; Leonard, J.E.; Raab, R.; Newman, R.A.; Hanna, N.; Anderson, D.R. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994, 83, 435–445. [Google Scholar] [CrossRef]
- Holgate, S.T.; Chuchalin, A.G.; Hébert, J.; Lötvall, J.; Persson, G.B.; Chung, K.F.; Bousquet, J.; Kerstjens, H.A.; Fox, H.; Thirlwell, J.; et al. Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clin. Exp. Allergy 2004, 34, 632–638. [Google Scholar] [CrossRef]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; FitzGerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W.; et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J. Novotny WBevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2004, 333, 328–335. [Google Scholar] [CrossRef]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) anti-CD2 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M.; Tuddenham, E.G. The hemophilias—From royal genes to gene therapy. N. Engl. J. Med. 2001, 344, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Hall, C.; Marsh, J.C.; Elebute, M.; Bombara, M.P.; Petro, B.E.; Cullen, M.J.; Richards, S.J.; Rollins, S.A.; Mojcik, C.F.; et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2006, 355, 1233–1243. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef]
- White, G.C.; Rosendaal, F.; Aledort, L.M.; Lusher, J.M.; Rothschild, C.; Ingerslev, J. Definitions in hemophilia: Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost. 2001, 85, 560. [Google Scholar]
- Konkle, B.A.; Stasyshyn, O.; Chowdary, P.; Bevan, D.H.; Mant, T.; Shima, M. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood 2015, 126, 1071–1079. [Google Scholar] [CrossRef]
- Connolly, S.J.; Crowther, M.; Eikelboom, J.W.; Gibson, C.M.; Curnutte, J.T.; Lawrence, J.H.; Yue, P.; Bronson, M.D.; Lu, G.; Conley, P.B.; et al. Full study report of andexanet alfa for bleeding associated with factor Xa inhibitors. N. Engl. J. Med. 2019, 380, 1326–1335. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.M.; Tesser, J.; Schiff, M.H.; Schechtman, J.; Burmester, G.-R.; Bennett, R.; Zrubek, J. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Krueger, G.G.; Langley, R.G.; Leonardi, C.; Yeilding, N.; Guzzo, C.; Wang, Y.; Dooley, L.T.; Lebwohl, M.; CNTO 1275 Psoriasis Study Group. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N. Engl. J. Med. 2007, 356, 580–592. [Google Scholar] [CrossRef]
- Deodhar, A.; Mease, P.J.; McInnes, I.B.; Baraliakos, X.; Reich, K.; Blauvelt, A.; Leonardi, C.; Porter, B.; Das Gupta, A.; Widmer, A.; et al. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: Integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res. Ther. 2019, 21, 111. [Google Scholar] [CrossRef]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.; Randazzo, B.; Wasfi, Y.; Shen, Y.K. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo- and active comparator–controlled VOYAGE 1 trial. J. Am. Acad. Dermatol. 2017, 76, 405–417. [Google Scholar]
- Nishimoto, N.; Hashimoto, J.; Miyasaka, N.; Yamamoto, K.; Kawai, S.; Takeuchi, T.; Murata, N.; van der Heijde, D.; Kishimoto, T. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): Evidence of clinical and radiographic benefit from an X-ray reader-blinded randomised controlled trial of tocilizumab. Ann. Rheum. Dis. 2007, 66, 1162–1167. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2013, 66, 311–328. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Stasch, J.P.; Schmidt, P.M.; Nedvetsky, P.I.; Nedvetskaya, T.Y.; Kumar, A.H.S.; Meurer, S.; Deile, M.; Taye, A.; Knorr, A.; Lapp, H.; et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J. Clin. Investig. 2011, 121, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.; Elliott, M.J.; Davis, D.; Potter, A.; Kalden, J.R.; Antoni, C.; Breedveld, F.C.; Smolen, J.S.; Eberl, G.; deWoody, K.; et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-α therapy in rheumatoid arthritis. J. Immunol. 1999, 163, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- de Jong, L.A.; Uges, D.R.; Franke, J.P.; Bischoff, R. Receptor-ligand binding assays: Technologies and applications. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 829, 1–25. [Google Scholar] [CrossRef]
- Dash, R.; Singh, S.K.; Chirmule, N.; Rathore, A.S. Assessment of Functional Characterization and Comparability of Biotherapeutics: A Review. AAPS J. 2021, 24, 15. [Google Scholar] [CrossRef]
- Available online: https://drugs.ncats.io/substances?facet=Development%20Status%2FUS%20Approved%20Rx&facet=Substance%20Class%2Fprotein&facet=Substance%20Form%2FPrincipal%20Form&page=1 (accessed on 22 September 2023).
- Niazi, S. Volume of distribution as a function of time. J. Pharm. Sci. 1976, 65, 452–454. [Google Scholar] [CrossRef]
- Wesolowski, C.A.; Wesolowski, M.J.; Babyn, P.S.; Wanasundara, S.N. Time Varying Apparent Volume of Distribution and Drug Half-Lives Following Intravenous Bolus Injections. PLoS ONE 2016, 11, e0158798. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/science-research/about-science-research-fda/modeling-simulation-fda (accessed on 22 September 2023).
- Available online: https://www.jdsupra.com/legalnews/first-interchangeable-fda-approval-1928445/ (accessed on 22 September 2023).
- Available online: https://www.fda.gov/media/162416/download (accessed on 22 September 2023).
- Available online: https://www.fda.gov/media/71336/download (accessed on 22 September 2023).
- Niazi, S.K. No two classes of biosimilars: Urgent advice to the US Congress and the FDA. J. Clin. Pharm. Ther. 2022, 47, 1352–1361. [Google Scholar] [CrossRef]
- Available online: https://www.lee.senate.gov/2023/7/lee-seeks-increased-competition-in-biological-drug-market (accessed on 22 September 2023).
- US Congress. BPCIA. Available online: https://www.congress.gov/bill/111th-congress/house-bill/3590 (accessed on 22 September 2023).
- Schreitmüller, T.; Barton, B.; Zharkov, A.; Bakalos, G. Comparative immunogenicity assessment of biosimilars. Future Oncol. 2019, 15, 319–329. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Number of Lawsuits (#Resolved) |
|---|---|
| Aflibercept | 1 |
| Denosumab | 1 |
| Natalizumab | 1 |
| Tocilizumab | 1 |
| Adalimumab | 5 (5) |
| Bevacizumab | 4 (4) |
| Epoetin alfa | 2 (2) |
| Etanercept | 2 (2) |
| Filgrastim | 5 (5) |
| Infliximab | 2 (2) |
| Pegfilgrastim | 6 (6) |
| Rituximab | 2 (2) |
| Trastuzumab | 5 (5) |
| Ustekinumab | 1 (1) |
| Therapeutic Protein Class | PD Marker | Receptor Binding Test |
|---|---|---|
| Anti-CD20 antibodies (e.g., Rituximab) [75] | CD20+ B-cell counts, immunoglobulin levels | CD20 binding assay |
| Anti-IgE therapies (e.g., Omalizumab) [76] | Serum-free IgE levels, basophil activation | IgE binding assay |
| Anti-IL-5 therapies (e.g., Mepolizumab, Reslizumab) [77] | Blood eosinophil count, asthma exacerbation rates | IL-5 receptor binding assay |
| Anti-PD-1 antibodies (e.g., Nivolumab, Pembrolizumab) [78] | Tumor responses, immune cell activation | PD-1 binding assay |
| Anti-VEGF agents (e.g., Bevacizumab, Aflibercept) [79] | Tumor response, normalization of tumor vasculature | VEGF binding assay |
| Bruton’s tyrosine kinase (BTK) inhibitors (e.g., Ibrutinib) [80] | B-cell counts, lymph node size, BTK phosphorylation | BTK binding assay |
| BTK inhibitors (e.g., Ibrutinib) [81] | B-cell counts, lymph node size, BTK phosphorylation | BTK binding assay |
| CD20-directed therapies (e.g., Rituximab) [82] | B-cell counts, tumor response in B-cell malignancies | CD20 binding assay |
| CD3-directed therapies (e.g., Blinatumomab) [83] | T-cell counts, tumor response in B-ALL | CD3 binding assay |
| CD38-directed therapies (e.g., Daratumumab) [84] | Plasma cell counts in multiple myeloma, immunoglobulin levels | CD38 binding assay |
| Coagulation factors (e.g., Factor VIII, Factor IX) [85] | Clotting times, bleeding episodes | N/A (Functional activity assays used) |
| Complement C5 inhibitors (e.g., Eculizumab) [86] | Hemolysis markers, renal function in aHUS | Complement C5 binding assay |
| CTLA-4 inhibitors (e.g., Ipilimumab) [87] | Tumor response in melanoma, immune cell activation | CTLA-4 binding assay |
| EGFR inhibitors (e.g., Cetuximab, Panitumumab) [88] | Tumor response, skin rash, EGFR phosphorylation levels | EGFR binding assay |
| Factor IX products (e.g., BeneFIX) [89] | Control and prevention of bleeding episodes in Hemophilia B | Factor IX binding assays |
| Factor VIII products (e.g., Advate) [90] | Control and prevention of bleeding episodes in Hemophilia A | Factor VIII binding assays |
| Factor Xa inhibitors (e.g., Andexanet alfa) [91] | Bleeding control, clotting times | Factor Xa binding assay |
| HER2/neu-directed therapies (e.g., Trastuzumab) [92] | Tumor response, cardiac monitoring, HER2 phosphorylation | HER2/neu binding assay |
| IL-1 inhibitors (e.g., Anakinra, Canakinumab) [93] | Inflammatory markers, clinical scores | IL-1 receptor binding assay |
| IL-1 inhibitors (e.g., Anakinra) [94] | Symptom relief in RA, systemic JIA; inflammatory markers | IL-1 receptor binding assay |
| IL-12/23 inhibitors (e.g., Ustekinumab) [95] | Cytokine levels (IL-12, IL-23), PASI score in psoriasis | IL-12/IL-23 p40 subunit binding assay |
| IL-17 inhibitors (e.g., Secukinumab, Ixekizumab) [96] | PASI score in psoriasis, inflammatory markers in spondylitis | IL-17 receptor binding assay |
| IL-23 inhibitors (e.g., Guselkumab) [97] | PASI score in psoriasis | IL-23 receptor binding assay |
| IL-5 inhibitors (e.g., Mepolizumab) [98] | Eosinophil counts, symptom control in severe asthma | IL-5 receptor binding assay |
| IL-6 inhibitors (e.g., Tocilizumab) [99] | CRP, IL-6 serum levels, clinical scores in diseases like RA | IL-6R binding assay |
| JAK inhibitors (e.g., Tofacitinib, Baricitinib) [98] | Inflammatory markers, clinical scores | JAK protein binding assay (biochemical assay) |
| mTOR inhibitors (e.g., Everolimus) [99] | Tumor response, organ transplant graft survival | mTOR binding assay |
| PCSK9 inhibitors (e.g., Evolocumab, Alirocumab) [100] | Serum LDL cholesterol levels | PCSK9 binding assay |
| PD-1 inhibitors (e.g., Pembrolizumab) [101] | Tumor response in various cancers | PD-1 receptor binding assay |
| PD-L1 inhibitors (e.g., Atezolizumab) [102] | Tumor responses, immune cell activation | PD-L1 binding assay |
| SGLT2 inhibitors (e.g., Dapagliflozin, Empagliflozin) [103] | Blood glucose levels, HbA1c levels | SGLT2 receptor binding assay |
| Soluble guanylate cyclase (sGC) stimulators [104] | Plasma cyclic GMP levels, vasodilatory responses | sGC binding assay |
| TNF-α inhibitors (e.g., Infliximab, Adalimumab) [105] | Inflammatory markers (CRP, ESR), clinical scores in diseases like RA | TNF-α binding assay |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K. The FDA’s New Guideline “Generally Accepted Scientific Knowledge” (GASK): An Opportunity to Expedite the Approval of Biosimilars. Pharmaceuticals 2023, 16, 1517. https://doi.org/10.3390/ph16111517
Niazi SK. The FDA’s New Guideline “Generally Accepted Scientific Knowledge” (GASK): An Opportunity to Expedite the Approval of Biosimilars. Pharmaceuticals. 2023; 16(11):1517. https://doi.org/10.3390/ph16111517
Chicago/Turabian StyleNiazi, Sarfaraz K. 2023. "The FDA’s New Guideline “Generally Accepted Scientific Knowledge” (GASK): An Opportunity to Expedite the Approval of Biosimilars" Pharmaceuticals 16, no. 11: 1517. https://doi.org/10.3390/ph16111517