
Neural–Cardiac Inflammasome Axis after Traumatic Brain Injury
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Inflammasome Signaling Proteins Are Elevated in the Cortex after TBI
2.2. Pyrin Is Elevated in the Brain of Mice after TBI
2.3. ASC Specks Are Elevated in the Brain of Mice after TBI
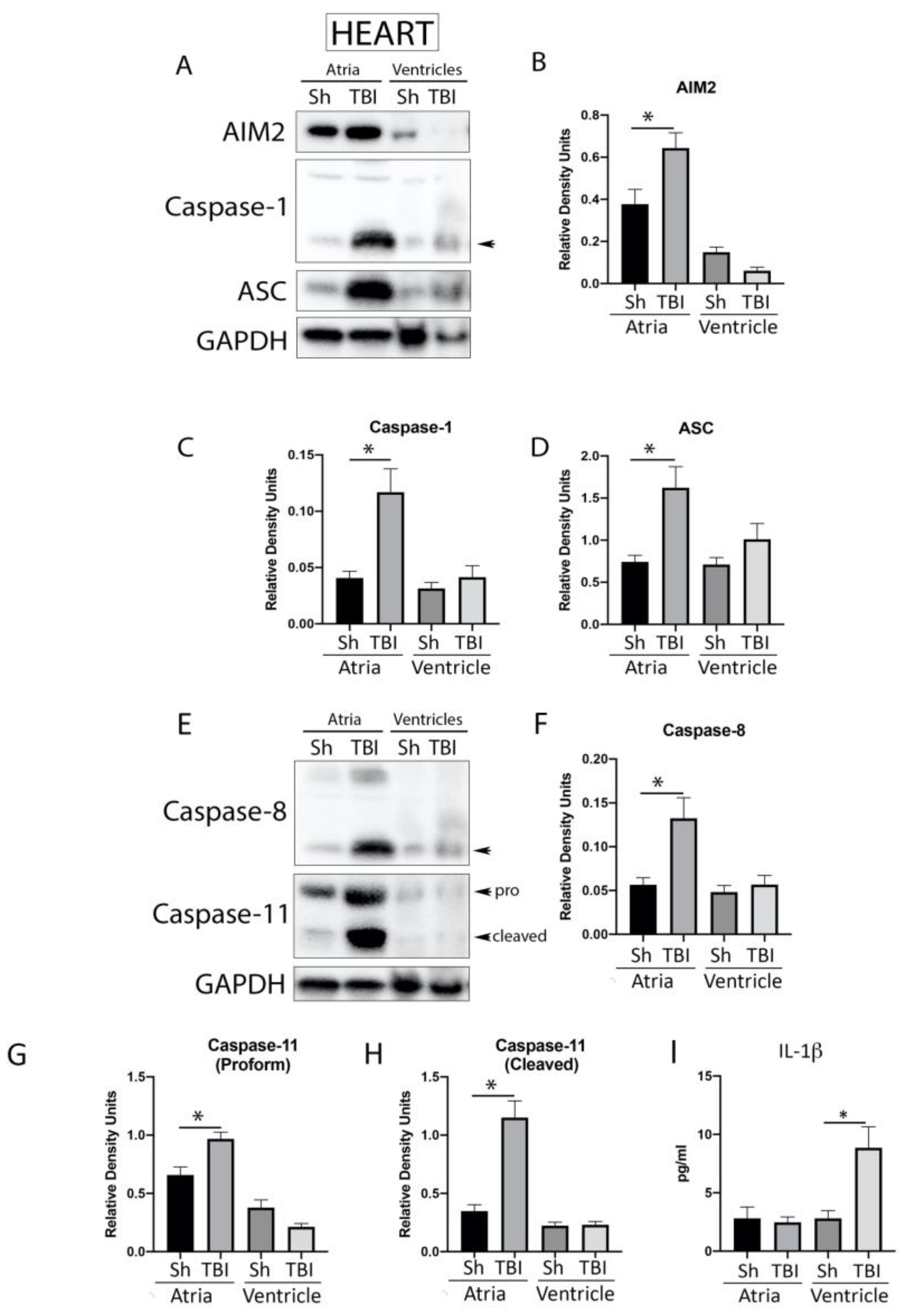
2.4. Inflammasome Signaling Proteins Are Elevated in the Heart after TBI
2.5. TBI Induces Histological Myocardial Abnormalities
2.6. ASC Specks Are Elevated in the Heart of Mice after TBI
2.7. Serum-Derived EV Characterization after TBI
2.8. Serum-Derived EVs Activate the Inflammasome in Cardiovascular Cells
3. Discussion
4. Materials and Methods
4.1. Animals and TBI
4.2. Perfusion/fixation and Immunohistochemistry
4.3. IL-1β Protein Analysis Assay (ECLIA)
4.4. Immunoblotting
4.5. Partial Purification of ASC Specks
4.6. Extracellular Vesicle (EV) Isolation
4.7. Inflammasome Protein Characterization in EVs with the Simple Plex Assay
4.8. Nanoparticle Tracking Analysis (NTA)
4.9. Mass Spectrometry
4.10. Adoptive Transfer of EVs into Cardiovascular Cells
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zygun, D.A.; Kortbeek, J.B.; Fick, G.H.; Laupland, K.B.; Doig, C.J. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit. Care Med. 2005, 33, 654–660. [Google Scholar] [CrossRef]
- McDonald, S.J.; Sharkey, J.M.; Sun, M.; Kaukas, L.M.; Shultz, S.R.; Turner, R.J.; Leonard, A.V.; Brady, R.D.; Corrigan, F. Beyond the Brain: Peripheral Interactions after Traumatic Brain Injury. J. Neurotrauma 2020, 37, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Griesbach, G.S.; Hovda, D.A.; Tio, D.L.; Taylor, A.N. Heightening of the stress response during the first weeks after a mild traumatic brain injury. Neuroscience 2011, 178, 147–158. [Google Scholar] [CrossRef]
- Koiv, L.; Merisalu, E.; Zilmer, K.; Tomberg, T.; Kaasik, A.E. Changes of sympatho-adrenal and hypothalamo-pituitary-adrenocortical system in patients with head injury. Acta Neurol. Scand. 1997, 96, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.J.; Newsome, H.H.; Becker, D.P. Mechanical brain injury: The sympathoadrenal response. J. Neurosurg. 1984, 61, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.N.; Rahman, S.U.; Sanders, N.C.; Tio, D.L.; Prolo, P.; Sutton, R.L. Injury severity differentially affects short- and long-term neuroendocrine outcomes of traumatic brain injury. J. Neurotrauma 2008, 25, 311–323. [Google Scholar] [CrossRef]
- Kerr, N.A.; de Rivero Vaccari, J.P.; Abbassi, S.; Kaur, H.; Zambrano, R.; Wu, S.; Dietrich, W.D.; Keane, R.W. Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. J. Neurotrauma 2018, 35, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Neural-respiratory inflammasome axis in traumatic brain injury. Exp. Neurol. 2020, 323, 113080. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J. Cereb. Blood Flow. Metab. 2009, 29, 1251–1261. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Bastien, D.; Yurcisin, G.; Pineau, I.; Dietrich, W.D.; De Koninck, Y.; Keane, R.W.; Lacroix, S. P2X4 receptors influence inflammasome activation after spinal cord injury. J. Neurosci. 2012, 32, 3058–3066. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Lotocki, G.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. A molecular platform in neurons regulates inflammation after spinal cord injury. J. Neurosci. 2008, 28, 3404–3414. [Google Scholar] [CrossRef] [PubMed]
- Abulafia, D.P.; de Rivero Vaccari, J.P.; Lozano, J.D.; Lotocki, G.; Keane, R.W.; Dietrich, W.D. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow. Metab. 2009, 29, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Scott, X.O.; Ferrer Marcelo, Y.; Almeida, V.W.; Blackwelder, P.L.; Yavagal, D.R.; Peterson, E.C.; Starke, R.M.; Dietrich, W.D.; Keane, R.W.; et al. Netosis and Inflammasomes in Large Vessel Occlusion Thrombi. Front. Pharmacol. 2020, 11, 607287. [Google Scholar] [CrossRef]
- Scott, X.O.; Stephens, M.E.; Desir, M.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The Inflammasome Adaptor Protein ASC in Mild Cognitive Impairment and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 4674. [Google Scholar] [CrossRef]
- Desu, H.L.; Plastini, M.; Illiano, P.; Bramlett, H.M.; Dietrich, W.D.; de Rivero Vaccari, J.P.; Brambilla, R.; Keane, R.W. IC100: A novel anti-ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J. Neuroinflammation 2020, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Cabrera Ranaldi, E.; Nuytemans, K.; Martinez, A.; Luca, C.C.; Keane, R.W.; de Rivero Vaccari, J.P. Proof-of-Principle Study of Inflammasome Signaling Proteins as Diagnostic Biomarkers of the Inflammatory Response in Parkinson’s Disease. Pharmaceuticals 2023, 16, 883. [Google Scholar] [CrossRef]
- Govindarajan, V.; de Rivero Vaccari, J.P.; Keane, R.W. Role of inflammasomes in multiple sclerosis and their potential as therapeutic targets. J. Neuroinflammation 2020, 17, 260. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Brand, F., 3rd; Adamczak, S.; Lee, S.W.; Perez-Barcena, J.; Wang, M.Y.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W. Exosome-mediated inflammasome signaling after central nervous system injury. J. Neurochem. 2016, 136 (Suppl. S1), 39–48. [Google Scholar] [CrossRef]
- Kerr, N.; Garcia-Contreras, M.; Abbassi, S.; Mejias, N.H.; Desousa, B.R.; Ricordi, C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. Inflammasome Proteins in Serum and Serum-Derived Extracellular Vesicles as Biomarkers of Stroke. Front. Mol. Neurosci. 2018, 11, 309. [Google Scholar] [CrossRef]
- van der Bilt, I.A.; Hasan, D.; Vandertop, W.P.; Wilde, A.A.; Algra, A.; Visser, F.C.; Rinkel, G.J. Impact of cardiac complications on outcome after aneurysmal subarachnoid hemorrhage: A meta-analysis. Neurology 2009, 72, 635–642. [Google Scholar] [CrossRef]
- Zygun, D. Non-neurological organ dysfunction in neurocritical care: Impact on outcome and etiological considerations. Curr. Opin. Crit. Care 2005, 11, 139–143. [Google Scholar] [CrossRef]
- Biso, S.; Wongrakpanich, S.; Agrawal, A.; Yadlapati, S.; Kishlyansky, M.; Figueredo, V. A Review of Neurogenic Stunned Myocardium. Cardiovasc. Psychiatry Neurol. 2017, 2017, 5842182. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Lackner, I.; Gebhard, F.; Miclau, T.; Kalbitz, M. Trauma, a Matter of the Heart-Molecular Mechanism of Post-Traumatic Cardiac Dysfunction. Int. J. Mol. Sci. 2021, 22, 737. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and regulation of cellular inflammasomes: Gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow. Metab. 2014, 34, 369–375. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Therapeutics targeting the inflammasome after central nervous system injury. Transl. Res. 2016, 167, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Xu, X.; Chen, F.; Kong, X.; Wang, H.; Zhang, J.; Lei, P. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 2018, 1697, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Irrera, N.; Russo, M.; Pallio, G.; Bitto, A.; Mannino, F.; Minutoli, L.; Altavilla, D.; Squadrito, F. The Role of NLRP3 Inflammasome in the Pathogenesis of Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 6204. [Google Scholar] [CrossRef]
- Kerr, N.A.; de Rivero Vaccari, J.P.; Weaver, C.; Dietrich, W.D.; Ahmed, T.; Keane, R.W. Enoxaparin Attenuates Acute Lung Injury and Inflammasome Activation after Traumatic Brain Injury. J. Neurotrauma 2021, 38, 646–654. [Google Scholar] [CrossRef]
- Kerr, N.A.; de Rivero Vaccari, J.P.; Umland, O.; Bullock, M.R.; Conner, G.E.; Dietrich, W.D.; Keane, R.W. Human Lung Cell Pyroptosis Following Traumatic Brain Injury. Cells 2019, 8, 69. [Google Scholar] [CrossRef]
- McLeod, A.A.; Neil-Dwyer, G.; Meyer, C.H.; Richardson, P.L.; Cruickshank, J.; Bartlett, J. Cardiac sequelae of acute head injury. Br. Heart J. 1982, 47, 221–226. [Google Scholar] [CrossRef]
- Cuisinier, A.; Maufrais, C.; Payen, J.F.; Nottin, S.; Walther, G.; Bouzat, P. Myocardial function at the early phase of traumatic brain injury: A prospective controlled study. Scand. J. Trauma. Resusc. Emerg. Med. 2016, 24, 129. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.; Smith, M. Cardiovascular complications of brain injury. Contin. Educ. Anaesth. Crit. Care Pain 2012, 12, 67–71. [Google Scholar] [CrossRef]
- Bass-Stringer, S.; Ooi, J.Y.Y.; McMullen, J.R. Clusterin is regulated by IGF1-PI3K signaling in the heart: Implications for biomarker and drug target discovery, and cardiotoxicity. Arch. Toxicol. 2020, 94, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Brandstetter, C.; Holz, F.G.; Krohne, T.U. Complement Component C5a Primes Retinal Pigment Epithelial Cells for Inflammasome Activation by Lipofuscin-mediated Photooxidative Damage. J. Biol. Chem. 2015, 290, 31189–31198. [Google Scholar] [CrossRef]
- Huet, F.; Dupuy, A.M.; Duflos, C.; Reis, C.A.; Kuster, N.; Aguilhon, S.; Cristol, J.P.; Roubille, F. Soluble urokinase-type plasminogen activator receptor strongly predicts global mortality in acute heart failure patients: Insight from the STADE-HF registry. Future Sci. OA 2021, 7, FSO697. [Google Scholar] [CrossRef]
- Lu, D.Y.; Lin, C.P.; Wu, C.H.; Cheng, T.M.; Pan, J.P. Plasma haptoglobin level can augment NT-proBNP to predict poor outcome in patients with severe acute decompensated heart failure. J. Investig. Med. 2019, 67, 20–27. [Google Scholar] [CrossRef]
- Sawicki, K.T.; Chang, H.C.; Ardehali, H. Role of heme in cardiovascular physiology and disease. J. Am. Heart Assoc. 2015, 4, e001138. [Google Scholar] [CrossRef]
- Yayama, K.; Hiyoshi, H.; Okamoto, H. Expressions of bradykinin B2-receptor, kallikrein and kininogen mRNAs in the heart are altered in pressure-overload cardiac hypertrophy in mice. Biol. Pharm. Bull. 2001, 24, 34–38. [Google Scholar] [CrossRef]
- Rizoli, S.B.; Jaja, B.N.; Di Battista, A.P.; Rhind, S.G.; Neto, A.C.; da Costa, L.; Inaba, K.; da Luz, L.T.; Nascimento, B.; Perez, A.; et al. Catecholamines as outcome markers in isolated traumatic brain injury: The COMA-TBI study. Crit. Care 2017, 21, 37. [Google Scholar] [CrossRef]
- Lu, J.; Goh, S.J.; Tng, P.Y.; Deng, Y.Y.; Ling, E.A.; Moochhala, S. Systemic inflammatory response following acute traumatic brain injury. Front Biosci. 2009, 14, 3795–3813. [Google Scholar] [CrossRef]
- Kalbitz, M.; Schwarz, S.; Weber, B.; Bosch, B.; Pressmar, J.; Hoenes, F.M.; Braun, C.K.; Horst, K.; Simon, T.P.; Pfeifer, R.; et al. Cardiac Depression in Pigs after Multiple Trauma–Characterization of Posttraumatic Structural and Functional Alterations. Sci. Rep. 2017, 7, 17861. [Google Scholar] [CrossRef]
- Dumas, A.; Amiable, N.; de Rivero Vaccari, J.P.; Chae, J.J.; Keane, R.W.; Lacroix, S.; Vallieres, L. The inflammasome pyrin contributes to pertussis toxin-induced IL-1beta synthesis, neutrophil intravascular crawling and autoimmune encephalomyelitis. PLoS Pathog. 2014, 10, e1004150. [Google Scholar] [CrossRef]
- International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell 1997, 90, 797–807. [Google Scholar] [CrossRef]
- Korkmaz, C.; Cansu, D.U.; Cansu, G.B. Familial Mediterranean fever: The molecular pathways from stress exposure to attacks. Rheumatology 2020, 59, 3611–3621. [Google Scholar] [CrossRef] [PubMed]
- Horstmann, J.P.; Marzi, I.; Relja, B. Adrenergic stimulation alters the expression of inflammasome components and interleukins in primary human monocytes. Exp. Ther. Med. 2016, 11, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.; Lee, S.W.; Perez-Barcena, J.; Crespi, C.; Ibanez, J.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. Inflammasome proteins as biomarkers of traumatic brain injury. PLoS ONE 2018, 13, e0210128. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.P.; Mim, C.; Hadad, R.; Cyr, B.; Stefansdottir, T.A.; Keane, R.W. Mechanism of action of IC 100, a humanized IgG4 monoclonal antibody targeting apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). Transl. Res. 2023, 251, 27–40. [Google Scholar] [CrossRef]
- Hoang, K.V.; Rajaram, M.V.S.; Curry, H.M.; Gavrilin, M.A.; Wewers, M.D.; Schlesinger, L.S. Complement Receptor 3-Mediated Inhibition of Inflammasome Priming by Ras GTPase-Activating Protein During Francisella tularensis Phagocytosis by Human Mononuclear Phagocytes. Front. Immunol. 2018, 9, 561. [Google Scholar] [CrossRef]
- Dinet, V.; Petry, K.G.; Badaut, J. Brain-Immune Interactions and Neuroinflammation after Traumatic Brain Injury. Front. Neurosci. 2019, 13, 1178. [Google Scholar] [CrossRef]
- Bao, W.; He, F.; Yu, L.; Gao, J.; Meng, F.; Ding, Y.; Zou, H.; Luo, B. Complement cascade on severe traumatic brain injury patients at the chronic unconscious stage: Implication for pathogenesis. Expert. Rev. Mol. Diagn. 2018, 18, 761–766. [Google Scholar] [CrossRef]
- Thelin, E.P.; Just, D.; Frostell, A.; Haggmark-Manberg, A.; Risling, M.; Svensson, M.; Nilsson, P.; Bellander, B.M. Protein profiling in serum after traumatic brain injury in rats reveals potential injury markers. Behav. Brain Res. 2018, 340, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Arbore, G.; Kemper, C. A novel “complement-metabolism-inflammasome axis” as a key regulator of immune cell effector function. Eur. J. Immunol. 2016, 46, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, M.; Hughes, T.R.; Morgan, B.P.; Triantafilou, K. Complementing the inflammasome. Immunology 2016, 147, 152–164. [Google Scholar] [CrossRef]
- Tung, P.; Kopelnik, A.; Banki, N.; Ong, K.; Ko, N.; Lawton, M.T.; Gress, D.; Drew, B.; Foster, E.; Parmley, W.; et al. Predictors of neurocardiogenic injury after subarachnoid hemorrhage. Stroke 2004, 35, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Hilz, M.J.; Wang, R.; Markus, J.; Ammon, F.; Hösl, K.M.; Flanagan, S.R.; Winder, K.; Koehn, J. Severity of traumatic brain injury correlates with long-term cardiovascular autonomic dysfunction. J. Neurol. 2017, 264, 1956–1967. [Google Scholar] [CrossRef]
- Jang, S.H.; Kwon, Y.H.; Lee, S.J. Tachycardia in a patient with mild traumatic brain injury. Clin. Auton. Res. 2020, 30, 87–89. [Google Scholar] [CrossRef]
- Adamczak, S.; Dale, G.; de Rivero Vaccari, J.P.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: Clinical article. J. Neurosurg. 2012, 117, 1119–1125. [Google Scholar] [CrossRef]
- Perez-Barcena, J.; Crespi, C.; Frontera, G.; Llompart-Pou, J.A.; Salazar, O.; Goliney, V.; Ibanez, J.; Bullock, M.R.; de Rivero Vaccari, J.P. Levels of caspase-1 in cerebrospinal fluid of patients with traumatic brain injury: Correlation with intracranial pressure and outcome. J. Neurosurg. 2020, 134, 1644–1649. [Google Scholar] [CrossRef]
- Perez-Barcena, J.; Rodriguez Pilar, J.; Salazar, O.; Crespi, C.; Frontera, G.; Novo, M.A.; Guardiola, M.B.; Llompart-Pou, J.A.; Ibanez, J.; de Rivero Vaccari, J.P. Serum Caspase-1 as an Independent Prognostic Factor in Traumatic Brain Injured Patients. Neurocrit. Care 2021, 36, 527–535. [Google Scholar] [CrossRef]
- Wu, J.; Dong, E.; Zhang, Y.; Xiao, H. The Role of the Inflammasome in Heart Failure. Front. Physiol. 2021, 12, 709703. [Google Scholar] [CrossRef]
- Higashikuni, Y.; Liu, W.; Numata, G.; Tanaka, K.; Fukuda, D.; Tanaka, Y.; Hirata, Y.; Imamura, T.; Takimoto, E.; Komuro, I.; et al. NLRP3 Inflammasome Activation Through Heart-Brain Interaction Initiates Cardiac Inflammation and Hypertrophy During Pressure Overload. Circulation 2023, 147, 338–355. [Google Scholar] [CrossRef]
- Cyr, B.; de Rivero Vaccari, J.P. Methods to Study Inflammasome Activation in the Central Nervous System: Immunoblotting and Immunohistochemistry. Methods Mol. Biol. 2023, 2696, 223–238. [Google Scholar] [CrossRef]
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; de Rivero Vaccari, J.P. Contribution of the inflammasome to inflammaging. J. Inflamm. 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.P.; Martinez, C.C.; Mejias, N.H.; de Rivero Vaccari, J.P. Sexual dimorphism in inflammasome-containing extracellular vesicles and the regulation of innate immunity in the brain of reproductive senescent females. Neurochem. Int. 2019, 127, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Cyr, B.; Hadad, R.; Keane, R.W.; de Rivero Vaccari, J.P. The Role of Non-canonical and Canonical Inflammasomes in Inflammaging. Front. Mol. Neurosci. 2022, 15, 774014. [Google Scholar] [CrossRef]
- Johnson, N.H.; Hadad, R.; Taylor, R.R.; Rodriguez Pilar, J.; Salazar, O.; Llompart-Pou, J.A.; Dietrich, W.D.; Keane, R.W.; Perez-Barcena, J.; de Rivero Vaccari, J.P. Inflammatory Biomarkers of Traumatic Brain Injury. Pharmaceuticals 2022, 15, 15–660. [Google Scholar] [CrossRef]
- Hadad, R.; Keane, R.W.; de Rivero Vaccari, J.P. Inflammasome signaling proteins as biomarkers of COVID-19. Front. Immunol. 2023, 14, 1014665. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Nishi, M.; Yamaoka, Y.; Miyakawa, K.; Jeremiah, S.S.; Osada, M.; Kimura, Y.; Ryo, A. Phosphopeptide enrichment using Phos-tag technology reveals functional phosphorylation of the nucleocapsid protein of SARS-CoV-2. J. Proteom. 2022, 255, 104501. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keane, R.W.; Hadad, R.; Scott, X.O.; Cabrera Ranaldi, E.d.l.R.M.; Pérez-Bárcena, J.; de Rivero Vaccari, J.P. Neural–Cardiac Inflammasome Axis after Traumatic Brain Injury. Pharmaceuticals 2023, 16, 1382. https://doi.org/10.3390/ph16101382
Keane RW, Hadad R, Scott XO, Cabrera Ranaldi EdlRM, Pérez-Bárcena J, de Rivero Vaccari JP. Neural–Cardiac Inflammasome Axis after Traumatic Brain Injury. Pharmaceuticals. 2023; 16(10):1382. https://doi.org/10.3390/ph16101382
Chicago/Turabian StyleKeane, Robert W., Roey Hadad, Xavier O. Scott, Erika d. l. R. M. Cabrera Ranaldi, Jon Pérez-Bárcena, and Juan Pablo de Rivero Vaccari. 2023. "Neural–Cardiac Inflammasome Axis after Traumatic Brain Injury" Pharmaceuticals 16, no. 10: 1382. https://doi.org/10.3390/ph16101382