
Ciclopirox Olamine Induces Proliferation Inhibition and Protective Autophagy in Hepatocellular Carcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
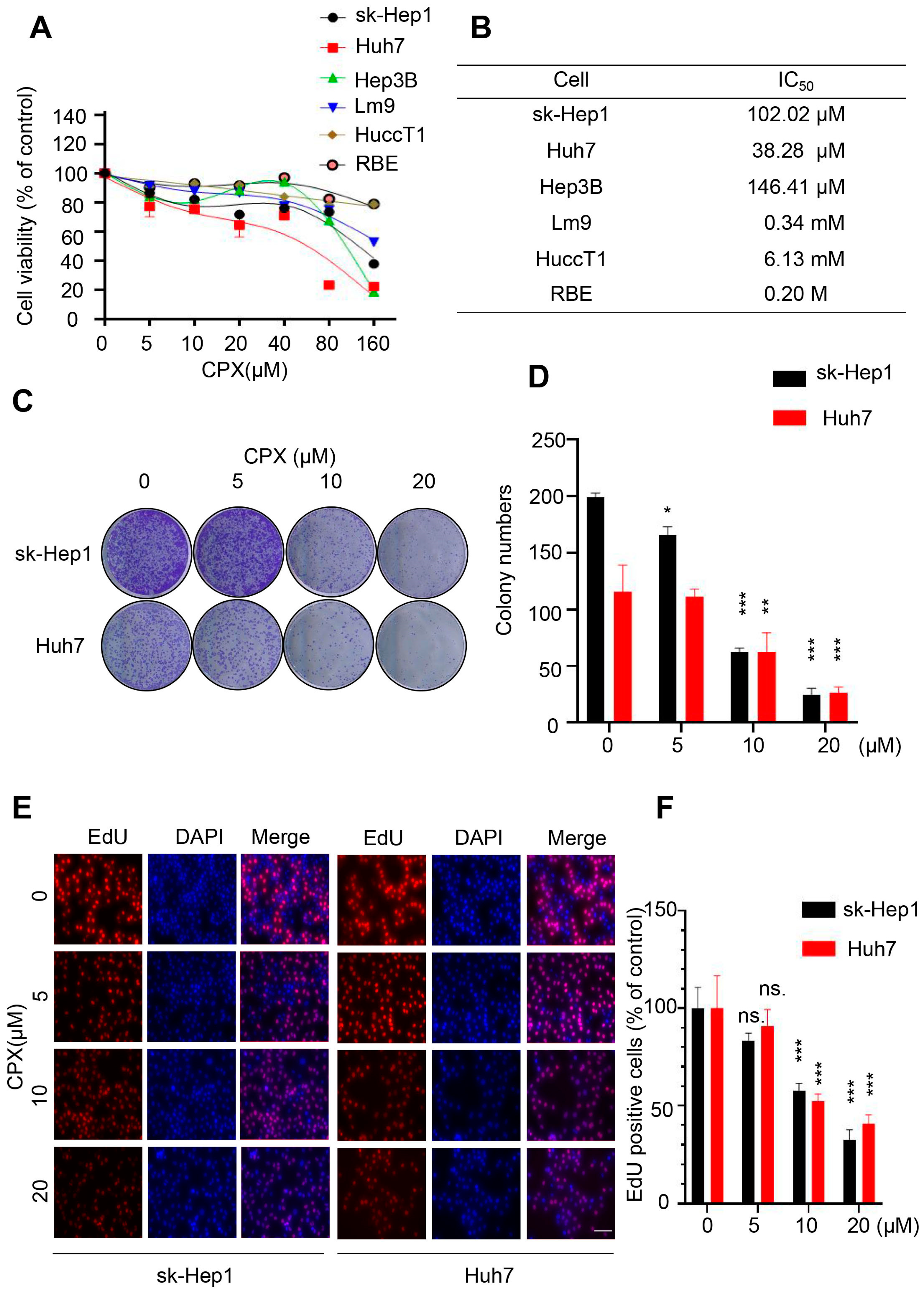
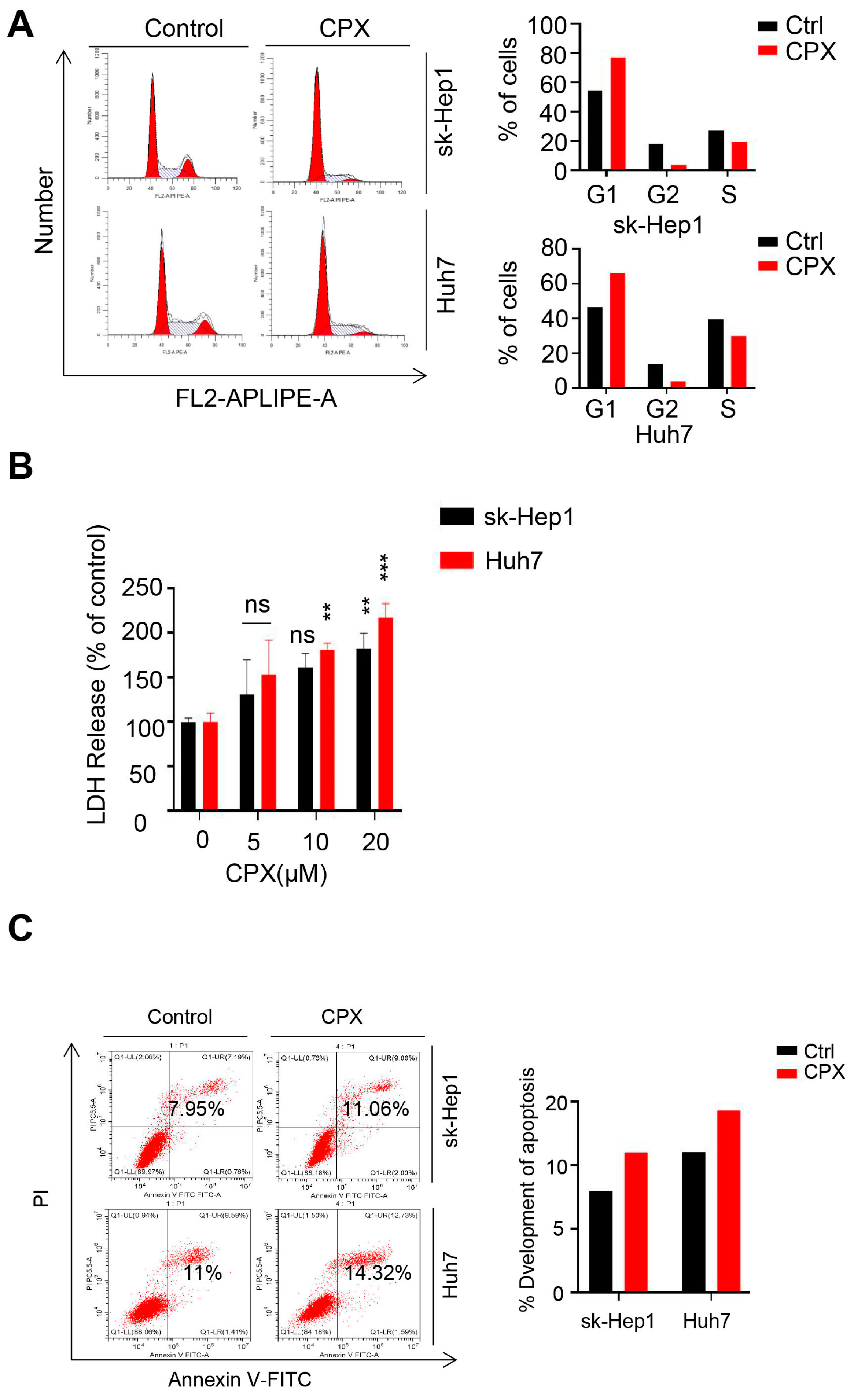
2.1. CPX Inhibits Proliferation in HCC Cells
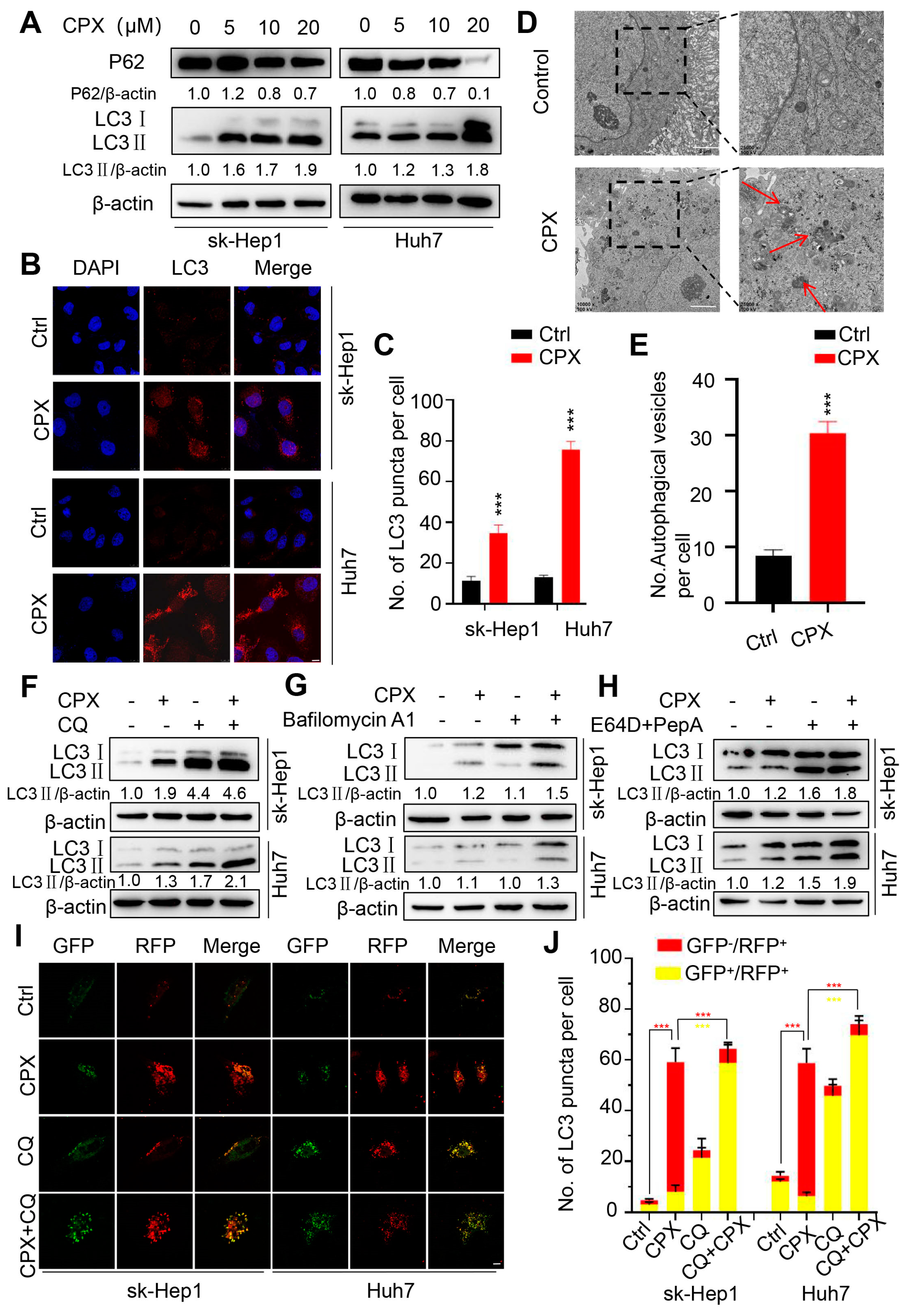
2.2. CPX Induces Autophagy in HCC Cells
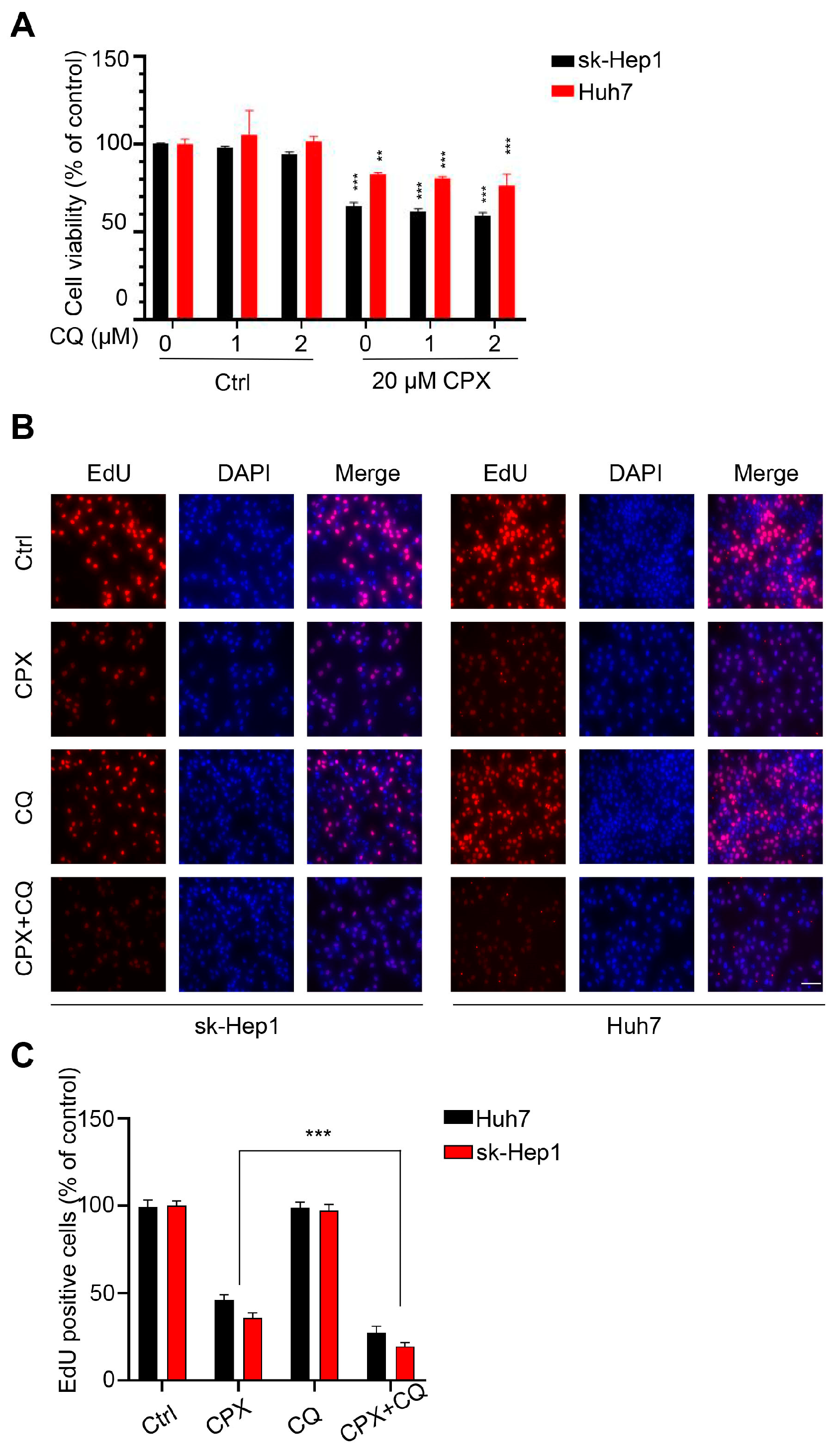
2.3. Autophagy Attenuates CPX-Induced Anti-Cancer Effects
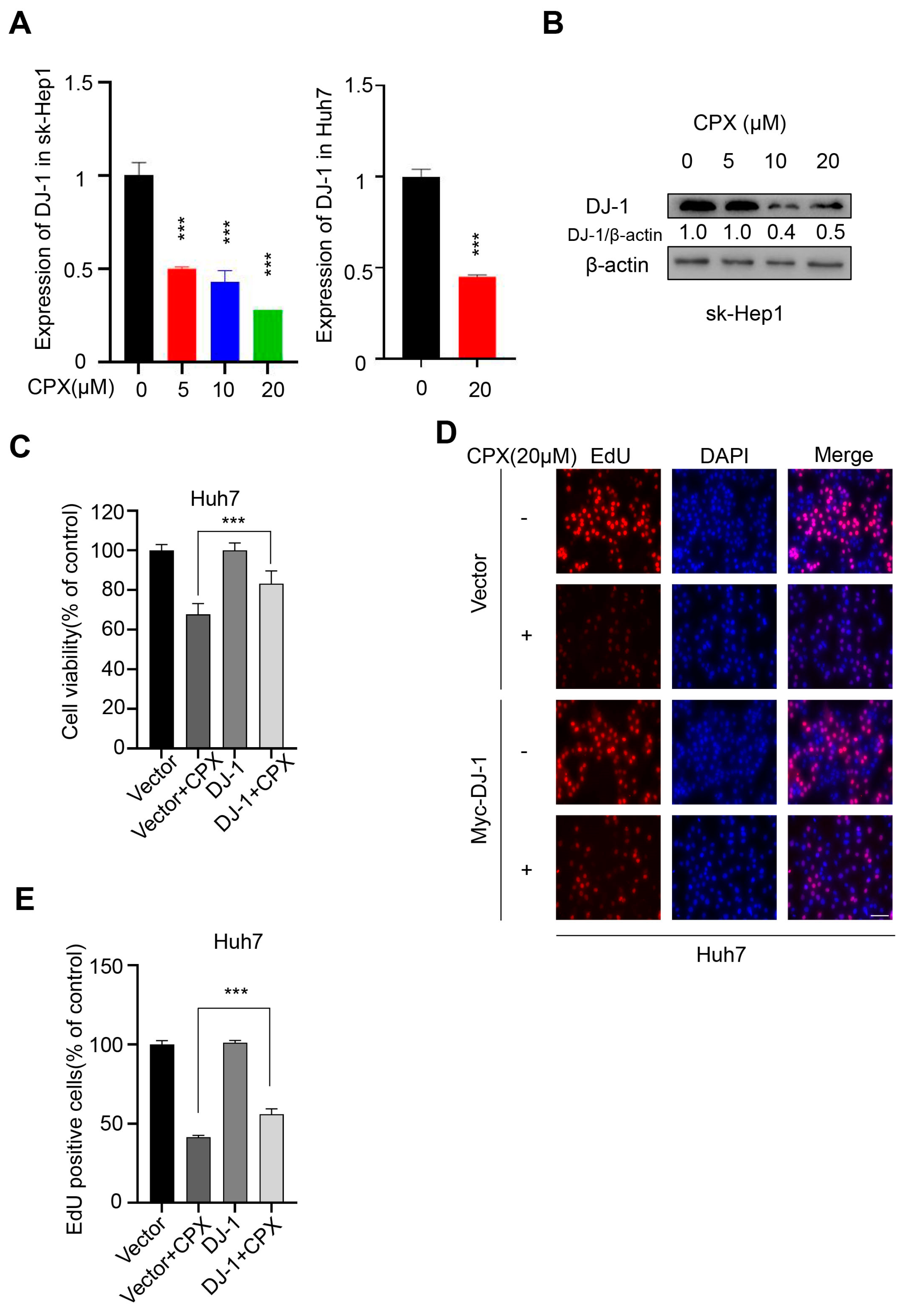
2.4. Downregulation of DJ-1 Is Involved in the Anti-Cancer Effects of CPX
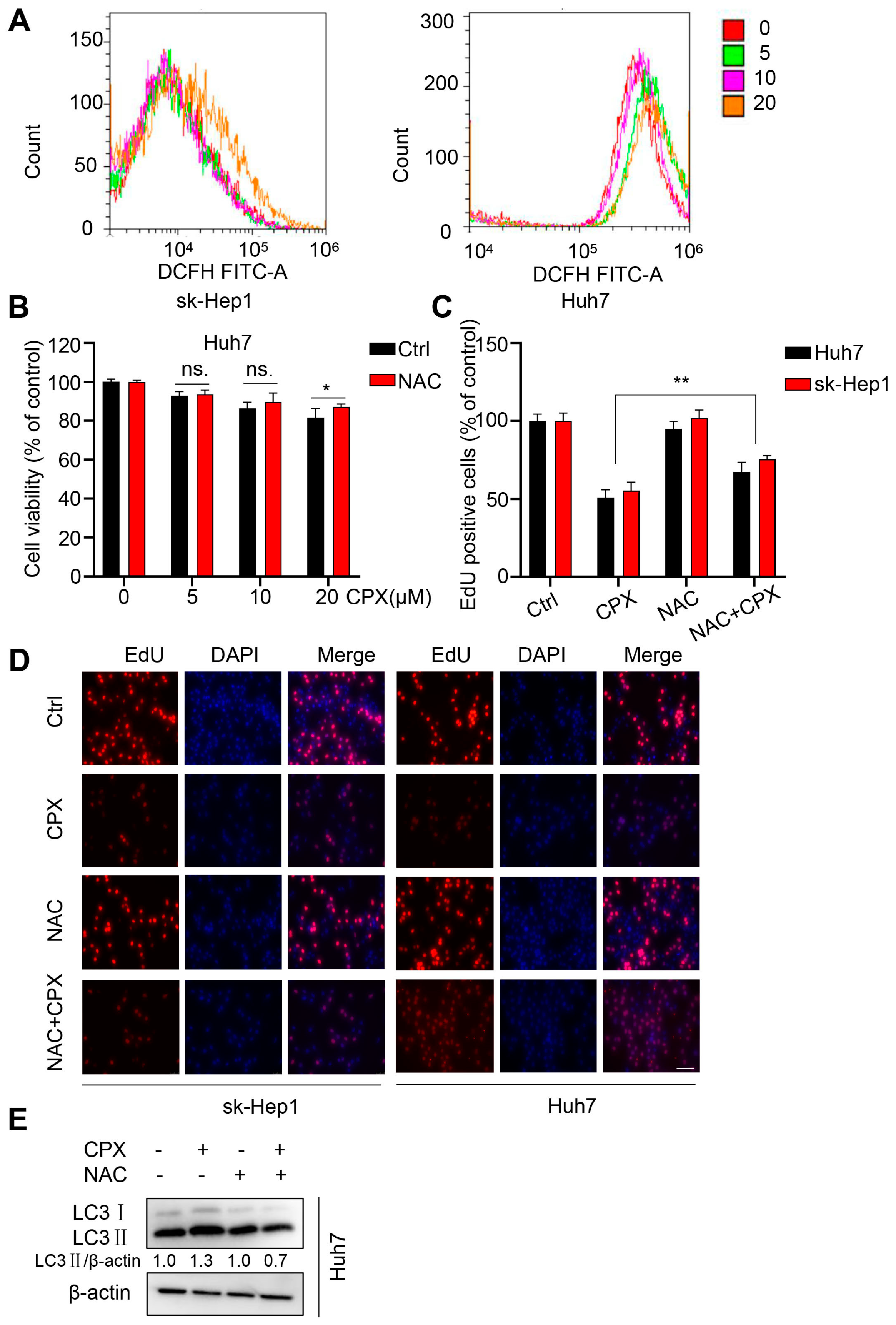
2.5. ROS Are Responsible for the Anti-Cancer Effects of CPX
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies and Reagents
4.3. Cell Viability and Proliferation Assays
4.4. Cell Cycle and Apoptosis Analysis
4.5. Immunoblot
4.6. Transmission Electron Microscopy
4.7. Immunofluorescence
4.8. Transfection
4.9. Quantitative RT-PCR (qRT-PCR)
4.10. Reactive Oxygen Species (ROS) Measurement
4.11. Glycogen Staining
4.12. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CPX | Ciclopirox olamine |
| CQ | chloroquine |
| Baf A1 | Bafilomycin A1 |
| LDH | lactate dehydrogenase |
| NAC | N-acetyl-l-cysteine |
| ROS | reactive oxygen species. |
References
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qin, F.; Meng, Q.; Dong, M. Protein tyrosine phosphatase receptor type D (PTPRD)-mediated signaling pathways for the potential treatment of hepatocellular carcinoma: A narrative review. Ann. Transl. Med. 2020, 8, 1192. [Google Scholar] [CrossRef]
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular Senescence: Immunosurveillance and Future Senescence-Induced Therapy in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 589908. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; González-Lucán, M.; Donapetry-García, C.; Fernández-Fernández, C.; Ameneiros-Rodríguez, E. Glycogen metabolism in humans. BBA Clin. 2016, 5, 85–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.; Wang, M.; Hu, J.; Li, S.; Zhao, S.; Li, H.; Liu, L. Prognostic value of the albumin-bilirubin grade in patients with hepatocellular carcinoma and other liver diseases. Ann. Transl. Med. 2020, 8, 553. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Y. Recent Advances in Liver Cancer Stem Cells: Non-coding RNAs, Oncogenes and Oncoproteins. Front. Cell Dev. Biol. 2020, 8, 548335. [Google Scholar] [CrossRef]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in liver cancer and possible treatment approaches. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188314. [Google Scholar] [CrossRef]
- Al-Zubaydi, F.; Gao, D.; Kakkar, D.; Li, S.; Adler, D.; Holloway, J.; Szekely, Z.; Gu, Z.; Chan, N.; Kumar, S.; et al. Breast intraductal nanoformulations for treating ductal carcinoma in situ I: Exploring metal-ion complexation to slow ciclopirox release, enhance mammary persistence and efficacy. J. Control. Release 2020, 323, 71–82. [Google Scholar] [CrossRef]
- Braun, J.A.; Herrmann, A.L.; Blase, J.I.; Frensemeier, K.; Bulkescher, J.; Scheffner, M.; Galy, B.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Effects of the antifungal agent ciclopirox in HPV-positive cancer cells: Repression of viral E6/E7 oncogene expression and induction of senescence and apoptosis. Int. J. Cancer 2020, 146, 461–474. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; He, Y.; Xiang, J.; Zhou, J.; Wan, X.; You, J.; Du, K.; Li, Y.; Cui, L.; Wang, Y.; et al. ROS generation attenuates the anti-cancer effect of CPX on cervical cancer cells by inducing autophagy and inhibiting glycophagy. Redox Biol. 2022, 53, 102339. [Google Scholar] [CrossRef]
- Londoño, M.C.; Abraldes, J.G.; Altamirano, J.; Decaens, T.; Forns, X. Clinical trial watch: Reports from the AASLD Liver Meeting®, Boston, November 2014. J. Hepatol. 2015, 62, 1196–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhang, L.; Wang, M.; Zhou, L.; Feng, X.; Yu, L.; Lan, J.; Gao, W.; Zhang, C.; Bu, Y.; et al. CPX Targeting DJ-1 Triggers ROS-induced Cell Death and Protective Autophagy in Colorectal Cancer. Theranostics 2019, 9, 5577–5594. [Google Scholar] [CrossRef] [PubMed]
- Linden, T.; Katschinski, D.M.; Eckhardt, K.; Scheid, A.; Pagel, H.; Wenger, R.H. The antimycotic ciclopirox olamine induces HIF-1alpha stability, VEGF expression, and angiogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 761–763. [Google Scholar] [CrossRef]
- Yang, J.; Milasta, S.; Hu, D.; AlTahan, A.M.; Interiano, R.B.; Zhou, J.; Davidson, J.; Low, J.; Lin, W.; Bao, J.; et al. Targeting Histone Demethylases in MYC-Driven Neuroblastomas with Ciclopirox. Cancer Res. 2017, 77, 4626–4638. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Shen, T.; Luo, Y.; Liu, L.; Chen, W.; Xu, B.; Han, X.; Pang, J.; Rivera, C.A.; Huang, S. The antitumor activity of the fungicide ciclopirox. Int. J. Cancer 2010, 127, 2467–2477. [Google Scholar] [CrossRef] [Green Version]
- Minden, M.D.; Hogge, D.E.; Weir, S.J.; Kasper, J.; Webster, D.A.; Patton, L.; Jitkova, Y.; Hurren, R.; Gronda, M.; Goard, C.A.; et al. Oral ciclopirox olamine displays biological activity in a phase I study in patients with advanced hematologic malignancies. Am. J. Hematol. 2014, 89, 363–368. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Katayama, M.; Kawaguchi, T.; Berger, M.S.; Pieper, R.O. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007, 14, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Resaz, R.; Vanni, C.; Segalerba, D.; Sementa, A.R.; Mastracci, L.; Grillo, F.; Murgia, D.; Bosco, M.C.; Chou, J.Y.; Barbieri, O.; et al. Development of hepatocellular adenomas and carcinomas in mice with liver-specific G6Pase-α deficiency. Dis. Model. Mech. 2014, 7, 1083–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, L.H.; Cho, J.-H.; Estrella, A.; Smyth, J.A.; Wu, R.; Chengsupanimit, T.; Brown, L.M.; Weinstein, D.A.; Lee, Y.M. Liver Glycogen Phosphorylase Deficiency Leads to Profibrogenic Phenotype in a Murine Model of Glycogen Storage Disease Type VI. Hepatol. Commun. 2019, 3, 1544–1555. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Li, J.; Zhang, W.; Xiao, C.; Zhang, S.; Nian, C.; Li, J.; Su, D.; Chen, L.; Zhao, Q.; et al. Glycogen accumulation and phase separation drives liver tumor initiation. Cell 2021, 184, 5559–5576. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Bernier, K.M.; Morrison, L.A. Antifungal drug ciclopirox olamine reduces HSV-1 replication and disease in mice. Antivir. Res. 2018, 156, 102–106. [Google Scholar] [CrossRef]
- Mihailidou, C.; Papakotoulas, P.; Papavassiliou, A.G.; Karamouzis, M.V. Superior efficacy of the antifungal agent ciclopirox olamine over gemcitabine in pancreatic cancer models. Oncotarget 2018, 9, 10360–10374. [Google Scholar] [CrossRef] [Green Version]
- Clement, P.M.; Hanauske-Abel, H.M.; Wolff, E.C.; Kleinman, H.K.; Park, M.H. The antifungal drug ciclopirox inhibits deoxyhypusine and proline hydroxylation, endothelial cell growth and angiogenesis in vitro. Int. J. Cancer 2002, 100, 491–498. [Google Scholar] [CrossRef]
- Shen, T.; Shang, C.; Zhou, H.; Luo, Y.; Barzegar, M.; Odaka, Y.; Wu, Y.; Huang, S. Ciclopirox inhibits cancer cell proliferation by suppression of Cdc25A. Genes Cancer 2017, 8, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, H.; Zhang, G.; Gu, L.; Zhang, Y.; Gao, J.; Wei, J.; Ma, Z. Antileukemia Effect of Ciclopirox Olamine Is Mediated by Downregulation of Intracellular Ferritin and Inhibition β-Catenin-c-Myc Signaling Pathway in Glucocorticoid Resistant T-ALL Cell Lines. PLoS ONE 2016, 11, e0161509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhard, Y.; McDermott, S.P.; Wang, X.; Gronda, M.; Venugopal, A.; Wood, T.E.; Hurren, R.; Datti, A.; Batey, R.A.; Wrana, J.; et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood 2009, 114, 3064–3073. [Google Scholar] [CrossRef]
- Huang, Z.; Huang, S. Reposition of the Fungicide Ciclopirox for Cancer Treatment. Recent Pat. Anti Cancer Drug Discov. 2021, 16, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Berdun, F.; Bartoli, C.; Steelheart, C.; Alegre, M.; Bayir, H.; Tyurina, Y.Y.; Kagan, V.E.; Salerno, G.; Pagnussat, G.; et al. C-ferroptosis is an iron-dependent form of regulated cell death in cyanobacteria. J. Cell Biol. 2022, 221, e201911005. [Google Scholar] [CrossRef]
- White, E.; Lattime, E.C.; Guo, J.Y. Autophagy Regulates Stress Responses, Metabolism, and Anticancer Immunity. Trends Cancer 2021, 7, 778–789. [Google Scholar] [CrossRef]
- Moosavi, M.A.; Haghi, A.; Rahmati, M.; Taniguchi, H.; Mocan, A.; Echeverría, J.; Gupta, V.K.; Tzvetkov, N.T.; Atanasov, A.G. Phytochemicals as potent modulators of autophagy for cancer therapy. Cancer Lett. 2018, 424, 46–69. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Dou, Q.; Chen, H.-N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N.; et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Liu, R.; Li, J.; Mao, J.; Lei, Y.; Wu, J.; Zeng, J.; Zhang, T.; Wu, H.; Chen, L.; et al. Quercetin induces protective autophagy in gastric cancer cells: Involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling. Autophagy 2011, 7, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Ying, M.; Xie, N.; Lin, G.; Dong, R.; Zhang, J.; Yan, H.; Yang, X.; He, Q.; Yang, B. The oxidation states of DJ-1 dictate the cell fate in response to oxidative stress triggered by 4-hpr: Autophagy or apoptosis? Antioxid. Redox Signal. 2014, 21, 1443–1459. [Google Scholar] [CrossRef] [Green Version]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef]
- Cao, J.; Lou, S.; Ying, M.; Yang, B. DJ-1 as a human oncogene and potential therapeutic target. Biochem. Pharmacol. 2015, 93, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Novel Insights into PARK7 (DJ-1), a Potential Anti-Cancer Therapeutic Target, and Implications for Cancer Progression. J. Clin. Med. 2020, 9, 1256. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, X.; Xiang, J.; Fan, H.; Jiang, Y.; Lu, Y.; Zhang, C.; Zhang, Y.; Chen, Q.; Lei, Y. Ciclopirox Olamine Induces Proliferation Inhibition and Protective Autophagy in Hepatocellular Carcinoma. Pharmaceuticals 2023, 16, 113. https://doi.org/10.3390/ph16010113
Wan X, Xiang J, Fan H, Jiang Y, Lu Y, Zhang C, Zhang Y, Chen Q, Lei Y. Ciclopirox Olamine Induces Proliferation Inhibition and Protective Autophagy in Hepatocellular Carcinoma. Pharmaceuticals. 2023; 16(1):113. https://doi.org/10.3390/ph16010113
Chicago/Turabian StyleWan, Xinyan, Junqi Xiang, Hui Fan, Ying Jiang, Yiting Lu, Chundong Zhang, Ying Zhang, Quanmei Chen, and Yunlong Lei. 2023. "Ciclopirox Olamine Induces Proliferation Inhibition and Protective Autophagy in Hepatocellular Carcinoma" Pharmaceuticals 16, no. 1: 113. https://doi.org/10.3390/ph16010113