
Role of Trientine in Hypertrophic Cardiomyopathy: A Review of Mechanistic Aspects
 , ,
, ,
Abstract
:1. Introduction
2. Role of Trientine in Cardiac Copper Regulation

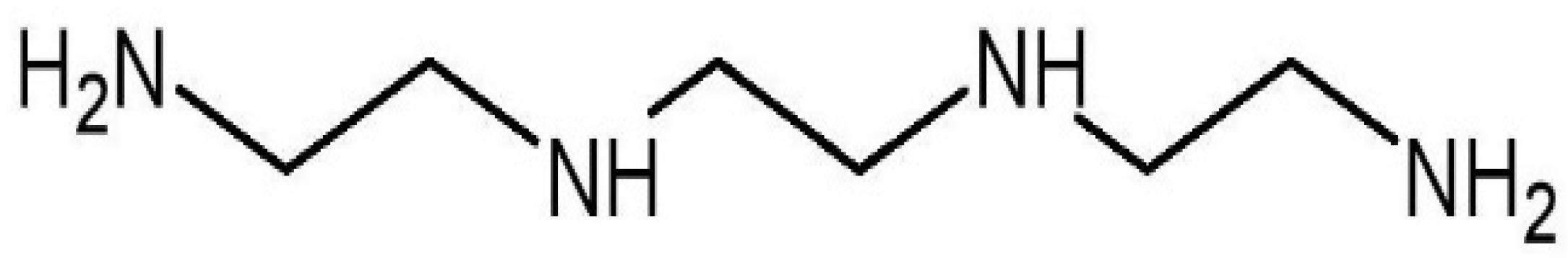{kind=link}
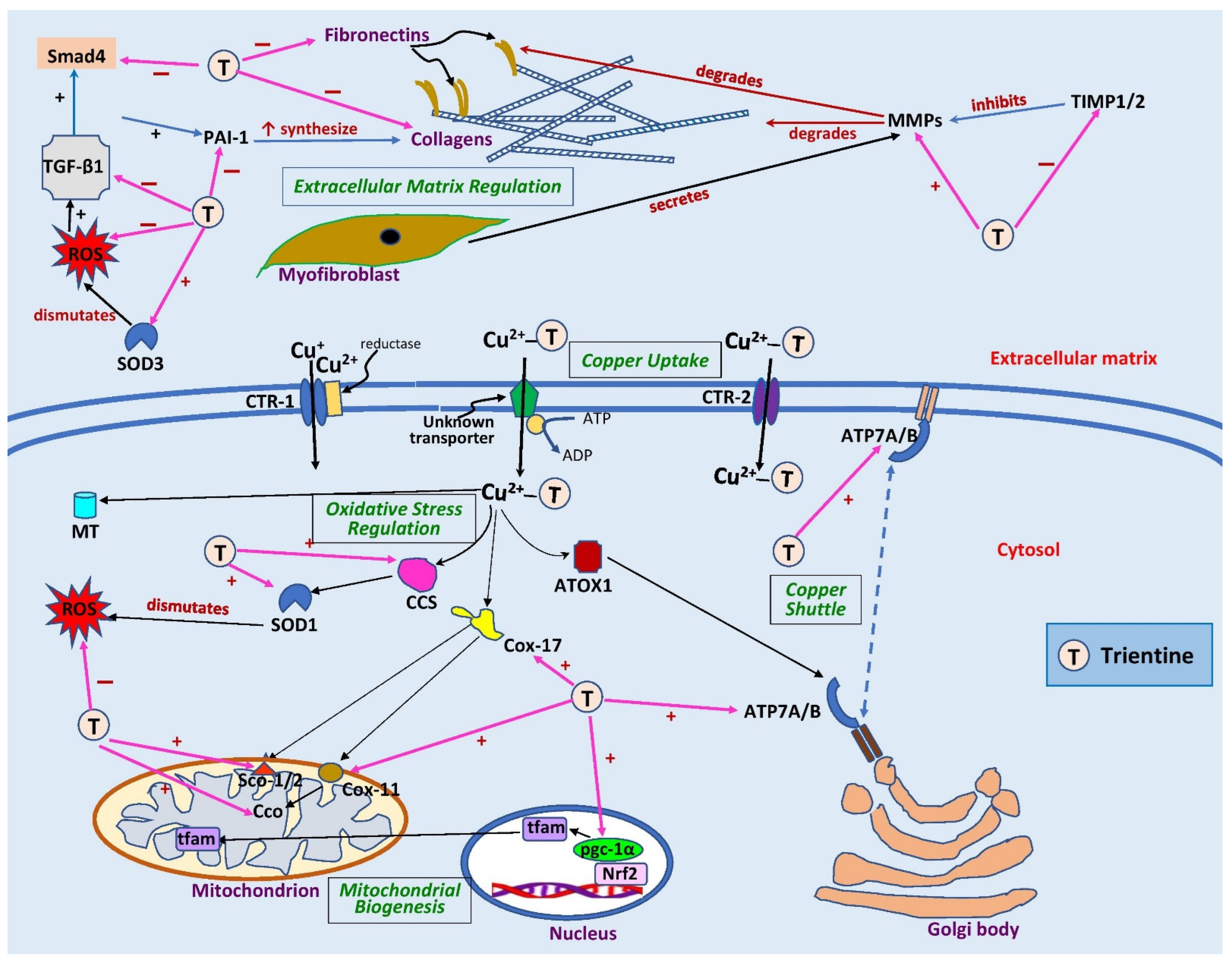{kind=link}
| Study | Type of Model/Subjects | Trientine (TETA) (Dose and Duration) | Findings | Reference |
|---|---|---|---|---|
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ cardiac copper content ↔ CTR-1 mRNA and protein ↑ CTR-2 mRNA and protein | [34] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ cardiac copper content | [38] |
| Animal | Transverse aortic constriction- induced cardiac hypertrophy in rats | 21.9 and 87.6 mg/kg twice daily orally for 6 weeks | TETA (21.9 mg/kg/day): ↑ LV copper content ↑ urinary copper ↓ renal copper content ↔ plasma copper level ↔ CTR-1 and CTR-2 protein TETA (87.6 mg/kg/day): ↓ LV and renal content ↑ urinary copper ↓ renal copper content ↔ plasma copper level ↔ CTR-1 and CTR-2 protein | [14] |
| Animal | Ascending aortic constriction-induced cardiac hypertrophy in rats | 21.9 mg/kg twice daily orally for 6 weeks | ↑ cardiac copper ↔ CTR-1 mRNA and protein ↑ CTR-2 mRNA and protein | [15] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | (a) Intravenous infusion, 60 s once hourly in increasing doses (0.1, 1.0, 10, and 100 mg/kg) (b) 8–11 mg/day in drinking water for 7 weeks (post-treatment) | ↑ Copper urinary excretion ↑ Cardiac total copper | [39] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 30 mg/day in drinking water for 8 weeks (post-treatment) | ↑ LV total copper | [33] |
| Human | Type 2 diabetic patients with LVH (n = 15) | 600 mg twice daily orally for 12 months | ↔ plasma copper ↑ 24 h urinary copper | [40] |
| Human | Patients with hypertrophic cardiomyopathy (n = 20) | 300 mg twice daily orally, increased after 1 week to 600 mg twice daily if tolerated for 6 months | ↔ serum copper | [23] |
3. Effects of Trientine on Mitochondrial Function and Biogenesis
4. Effects of Trientine on Cardiac Function
5. Effects of Trientine on Myocardial Oxidative Stress and Inflammation
6. Effects of Trientine on Extracellular Matrix Regulation and Interstitial Fibrosis
7. Effects of Trientine on Cardiac Structure
8. Conclusions and Directions for Future Studies
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lippi, G.; Sanchis-Gomar, F. Global epidemiology and future trends of heart failure. AME Med. J. 2020, 5, 1–6. [Google Scholar] [CrossRef]
- Savarese, G.; Lund, L.H. Global public health burden of heart failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Mohan, M.; Dihoum, A.; Mordi, I.R.; Choy, A.M.; Rena, G.; Lang, C.C. Left ventricular hypertrophy in diabetic cardiomyopathy: A target for intervention. Front. Cardiovasc. Med. 2021, 8, 746382. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Udelson, J.E.; Maron, M.S. Clinical spectrum and management of heart failure in hypertrophic cardiomyopathy. JACC Heart Fail. 2018, 6, 353–363. [Google Scholar] [CrossRef]
- Chair, S.Y.; Chan, J.Y.W.; Waye, M.M.Y.; Liu, T.; Law, B.M.H.; Chien, W.T. Exploration of potential genetic biomarkers for heart failure: A systematic review. Int. J. Environ. Res. Public Health 2021, 18, 5904. [Google Scholar] [CrossRef]
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar]
- Siti, H.; Jalil, J.; Asmadi, A.Y.; Kamisah, Y. Roles of rutin in cardiac remodeling. J. Funct. Foods 2020, 64, 103606. [Google Scholar] [CrossRef]
- Bomer, N.; Pavez-Giani, M.G.; Grote Beverborg, N.; Cleland, J.G.F.; van Veldhuisen, D.J.; van der Meer, P. Micronutrient deficiencies in heart failure: Mitochondrial dysfunction as a common pathophysiological mechanism? J. Intern. Med. 2022, 291, 713–731. [Google Scholar] [CrossRef]
- Medeiros, D.M. Perspectives on the role and relevance of copper in cardiac disease. Biol. Trace Elem. Res. 2017, 176, 10–19. [Google Scholar] [CrossRef]
- Klevay, L.M. Cardiovascular disease from copper deficiency—A history. J. Nutr. 2000, 130, 489S–492S. [Google Scholar] [CrossRef]
- Fu, C.; Lizhao, J.; Luo, Z.; Wang, T.; Grapperhaus, C.A.; Ding, X.; Kang, Y.J. Active uptake of hydrophilic copper complex Cu(ii)-TETA in primary cultures of neonatal rat cardiomyocytes. Metallomics 2019, 11, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Getz, J.; Lin, D.; Medeiros, D.M. The cardiac copper chaperone proteins Sco1 and CCS are up-regulated, but Cox 1 and Cox4 are down-regulated, by copper deficiency. Biol. Trace Elem. Res. 2011, 143, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Olivares, R.W.I.; Postma, G.C.; Schapira, A.; Iglesias, D.E.; Valdez, L.B.; Breininger, E.; Gazzaneo, P.D.; Minatel, L. Biochemical and morphological alterations in hearts of copper-deficient bovines. Biol. Trace Elem. Res. 2019, 189, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, C.; Liu, Y.; Sun, X.; Ding, X.; Qiu, L.; Han, P.; James Kang, Y. Trientine selectively delivers copper to the heart and suppresses pressure overload-induced cardiac hypertrophy in rats. Exp. Biol. Med. 2018, 243, 1141–1152. [Google Scholar] [CrossRef]
- Liu, Y.; Xiao, Y.; Liu, J.; Feng, L.; Kang, Y.J. Copper-induced reduction in myocardial fibrosis is associated with increased matrix metalloproteins in a rat model of cardiac hypertrophy. Metallomics 2018, 10, 201–208. [Google Scholar] [CrossRef]
- Nath, R. Copper deficiency and heart disease: Molecular basis, recent advances and current concepts. Int. J. Biochem. Cell. Biol. 1997, 29, 1245–1254. [Google Scholar] [CrossRef]
- Cobine, P.A.; Moore, S.A.; Leary, S.C. Getting out what you put in: Copper in mitochondria and its impacts on human disease. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 118867. [Google Scholar] [CrossRef]
- Jiang, Y.; Reynolds, C.; Xiao, C.; Feng, W.; Zhou, Z.; Rodriguez, W.; Tyagi, S.C.; Eaton, J.W.; Saari, J.T.; Kang, Y.J. Dietary copper supplementation reverses hypertrophic cardiomyopathy induced by chronic pressure overload in mice. J. Exp. Med. 2007, 204, 657–666. [Google Scholar] [CrossRef]
- Koracevic, G.; Stojanovic, M.; Lovic, D.; Zdravkovic, M.; Sakac, D. Certain beta blockers (e.g., bisoprolol) may be reevaluated in hypertension guidelines for patients with left ventricular hypertrophy to diminish the ventricular arrhythmic risk. J. Hum. Hypertens. 2021, 35, 564–576. [Google Scholar] [CrossRef]
- Enzan, N.; Matsushima, S.; Ide, T.; Kaku, H.; Tohyama, T.; Funakoshi, K.; Higo, T.; Tsutsui, H. Beta-blocker use is associated with prevention of left ventricular remodeling in recovered dilated cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e019240. [Google Scholar] [CrossRef]
- Alvarez, C.K.; Cronin, E.; Baker, W.L.; Kluger, J. Heart failure as a substrate and trigger for ventricular tachycardia. J. Interv. Card. Electrophysiol. 2019, 56, 229–247. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, N.H.; Jalil, J.; Zainalabidin, S.; Saleh, M.S.M.; Asmadi, A.Y.; Kamisah, Y. Molecular mechanisms of sacubitril/valsartan in cardiac remodeling. Front. Pharmacol. 2022, 13, 892460. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.; Miller, C.; Farrant, J.P.; Polturi, R.; Clark, D.; Ray, S.; Cooper, G.; Schmitt, M. Copper chelation in patients with hypertrophic cardiomyopathy. Open Heart 2022, 9, e001803. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. The protection conferred by chelation therapy in post-MI diabetics might be replicated by high-dose zinc supplementation. Med. Hypotheses 2015, 84, 451–455. [Google Scholar] [CrossRef]
- Lu, J.; Gong, D.; Choong, S.Y.; Xu, H.; Chan, Y.K.; Chen, X.; Fitzpatrick, S.; Glyn-Jones, S.; Zhang, S.; Nakamura, T.; et al. Copper(II)-selective chelation improves function and antioxidant defences in cardiovascular tissues of rats as a model of diabetes: Comparisons between triethylenetetramine and three less copper-selective transition-metal-targeted treatments. Diabetologia 2010, 53, 1217–1226. [Google Scholar] [CrossRef]
- Cooper, G.J. Therapeutic potential of copper chelation with triethylenetetramine in managing diabetes mellitus and Alzheimer’s disease. Drugs 2011, 71, 1281–1320. [Google Scholar] [CrossRef]
- Lynch, E.N.; Campani, C.; Innocenti, T.; Dragoni, G.; Forte, P.; Galli, A. Practical insights into chronic management of hepatic Wilson’s disease. World J. Clin. Cases 2022, 10, 4334–4347. [Google Scholar] [CrossRef]
- Roberts, E.A. Update on the diagnosis and management of Wilson disease. Curr. Gastroenterol. Rep. 2018, 20, 56. [Google Scholar] [CrossRef]
- Mandal, T.; Kar, S.; Maji, S.; Sen, S.; Gupta, A. Structural and functional diversity among the members of ctr, the membrane copper transporter family. J. Membr. Biol. 2020, 253, 459–468. [Google Scholar] [CrossRef]
- McCarty, M.F. Nutraceutical, dietary, and lifestyle options for prevention and treatment of ventricular hypertrophy and heart failure. Int. J. Mol. Sci. 2021, 22, 3321. [Google Scholar] [CrossRef]
- Ford, E.S. Serum copper concentration and coronary heart disease among US adults. Am. J. Epidemiol. 2000, 151, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- He, W.; James Kang, Y. Ischemia-induced copper loss and suppression of angiogenesis in the pathogenesis of myocardial infarction. Cardiovasc. Toxicol. 2013, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ward, M.L.; Phillips, A.R.; Zhang, S.; Kennedy, J.; Barry, B.; Cannell, M.B.; Cooper, G.J. Protection of the heart by treatment with a divalent-copper-selective chelator reveals a novel mechanism underlying cardiomyopathy in diabetic rats. Cardiovasc. Diabetol. 2013, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, H.; Amarsingh, G.V.; Cheung, C.C.; Hogl, S.; Narayanan, U.; Zhang, L.; McHarg, S.; Xu, J.; Gong, D.; et al. Diabetic cardiomyopathy is associated with defective myocellular copper regulation and both defects are rectified by divalent copper chelation. Cardiovasc. Diabetol. 2014, 13, 100. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Crisponi, G.; Crespo-Alonso, M.; Lachowicz, J.I.; Szewczuk, Z.; Cooper, G.J. Complex formation equilibria of Cu(II) and Zn(II) with triethylenetetramine and its mono- and di-acetyl metabolites. Dalton Trans. 2013, 42, 6161–6170. [Google Scholar] [CrossRef]
- Bertinato, J.; Duval, S.; L’Abbé, M.R. Copper transporter 2 content is lower in liver and heart of copper-deficient rats. Int. J. Mol. Sci. 2010, 11, 4741–4749. [Google Scholar] [CrossRef]
- Lutsenko, S. Copper trafficking to the secretory pathway. Metallomics 2016, 8, 840–852. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, H.; Amarsingh, G.V.; Cheung, C.C.H.; Wu, D.; Narayanan, U.; Zhang, L.; Cooper, G.J.S. Restoration of myocellular copper-trafficking proteins and mitochondrial copper enzymes repairs cardiac function in rats with diabetes-evoked heart failure. Metallomics 2020, 12, 259–272. [Google Scholar] [CrossRef]
- Cooper, G.J.; Phillips, A.R.; Choong, S.Y.; Leonard, B.L.; Crossman, D.J.; Brunton, D.H.; Saafi, L.; Dissanayake, A.M.; Cowan, B.R.; Young, A.A.; et al. Regeneration of the heart in diabetes by selective copper chelation. Diabetes 2004, 53, 2501–2508. [Google Scholar] [CrossRef]
- Cooper, G.J.; Young, A.A.; Gamble, G.D.; Occleshaw, C.J.; Dissanayake, A.M.; Cowan, B.R.; Brunton, D.H.; Baker, J.R.; Phillips, A.R.; Frampton, C.M.; et al. A copper(II)-selective chelator ameliorates left-ventricular hypertrophy in type 2 diabetic patients: A randomised placebo-controlled study. Diabetologia 2009, 52, 715–722. [Google Scholar] [CrossRef]
- Baynes, J.W.; Murray, D.B. The metal chelators, trientine and citrate, inhibit the development of cardiac pathology in the Zucker diabetic rat. Exp. Diabetes Res. 2009, 2009, 696378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, D.; Lu, J.; Chen, X.; Choong, S.Y.; Zhang, S.; Chan, Y.-K.; Glyn-Jones, S.; Gamble, G.D.; Phillips, A.R.; Cooper, G.J. Molecular changes evoked by triethylenetetramine treatment in the extracellular matrix of the heart and aorta in diabetic rats. Mol. Pharmacol. 2006, 70, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M.; Kaplan, J.H. Copper transporters and copper chaperones: Roles in cardiovascular physiology and disease. Am. J. Physiol. Cell. Physiol. 2018, 315, C186–C201. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Y.; Shi, H.; Peng, Y.; Fan, X.; Li, C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflügers Arch. 2020, 472, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Vogt, S.; Ruppert, V.; Pankuweit, S.; Paletta, J.P.J.; Rhiel, A.; Weber, P.; Irqsusi, M.; Cybulski, P.; Ramzan, R. Myocardial insufficiency is related to reduced subunit 4 content of cytochrome c oxidase. J. Cardiothorac. Surg. 2018, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, M. Role of creatine in the heart: Health and disease. Nutrients 2021, 13, 1215. [Google Scholar] [CrossRef]
- Wibowo, P.G.; Charman, S.J.; Okwose, N.C.; Velicki, L.; Popovic, D.; Hollingsworth, K.G.; Macgowan, G.A.; Jakovljevic, D.G. Association between cardiac high-energy phosphate metabolism and whole body metabolism in healthy female adults. Physiol. Res. 2021, 70, 393–399. [Google Scholar] [CrossRef]
- Burrage, M.K.; Hundertmark, M.; Valkovič, L.; Watson, W.D.; Rayner, J.; Sabharwal, N.; Ferreira, V.M.; Neubauer, S.; Miller, J.J.; Rider, O.J.; et al. Energetic basis for exercise-induced pulmonary congestion in heart failure with preserved ejection fraction. Circulation 2021, 144, 1664–1678. [Google Scholar] [CrossRef]
- Abou, R.; van der Bijl, P.; Bax, J.J.; Delgado, V. Global longitudinal strain: Clinical use and prognostic implications in contemporary practice. Heart 2020, 106, 1438–1444. [Google Scholar] [CrossRef]
- Lu, J.; Pontré, B.; Pickup, S.; Choong, S.Y.; Li, M.; Xu, H.; Gamble, G.D.; Phillips, A.R.; Cowan, B.R.; Young, A.A.; et al. Treatment with a copper-selective chelator causes substantive improvement in cardiac function of diabetic rats with left-ventricular impairment. Cardiovasc. Diabetol. 2013, 12, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jüllig, M.; Chen, X.; Hickey, A.J.; Crossman, D.J.; Xu, A.; Wang, Y.; Greenwood, D.R.; Choong, Y.S.; Schönberger, S.J.; Middleditch, M.J.; et al. Reversal of diabetes-evoked changes in mitochondrial protein expression of cardiac left ventricle by treatment with a copper(II)-selective chelator. Proteom. Clin. Appl. 2007, 1, 387–399. [Google Scholar] [CrossRef]
- Siti, H.N.; Jalil, J.; Asmadi, A.Y.; Kamisah, Y. Effects of quercetin on cardiac function in pressure overload and postischemic cardiac injury in rodents: A systematic review and meta-analysis. Cardiovasc. Drugs Ther. 2022, 36, 15–29. [Google Scholar] [CrossRef]
- Migisha, R.; Agaba, D.C.; Katamba, G.; Miranda, S.L.; Muyingo, A.; Siedner, M.J. High prevalence of prolonged QTc interval among individuals in ambulatory diabetic care in southwestern Uganda. Int. J. Diabetes Dev. Ctries. 2021, 41, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Y.; Zhu, Y.; Wang, Y.; Liu, X.; Nie, X.; Zhao, J.; Wang, W.; Cheng, J. Rhynchophylline regulates calcium homeostasis by antagonizing ryanodine receptor 2 phosphorylation to improve diabetic cardiomyopathy. Front. Pharmacol. 2022, 13, 882198. [Google Scholar] [CrossRef] [PubMed]
- Salim, S.; Yunos, N.; Jauri, M.; Kamisah, Y. Cardiotonic effects of cardiac glycosides from plants of Apocynaceae family. Chula. Med. J. 2020, 64, 449–456. [Google Scholar]
- Tveita, T.; Arteaga, G.M.; Han, Y.S.; Sieck, G.C. Cardiac troponin-I phosphorylation underlies myocardial contractile dysfunction induced by hypothermia rewarming. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H726–H731. [Google Scholar] [CrossRef]
- Cheng, Y.S.; Dai, D.Z.; Ji, H.; Zhang, Q.; Dai, Y. Sildenafil and FDP-Sr attenuate diabetic cardiomyopathy by suppressing abnormal expression of myocardial CASQ2, FKBP12.6, and SERCA2a in rats. Acta Pharmacol. Sin. 2011, 32, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Filomena, M.C.; Yamamoto, D.L.; Carullo, P.; Medvedev, R.; Ghisleni, A.; Piroddi, N.; Scellini, B.; Crispino, R.; D’Autilia, F.; Zhang, J.; et al. Myopalladin knockout mice develop cardiac dilation and show a maladaptive response to mechanical pressure overload. eLife 2021, 10, e58313. [Google Scholar] [CrossRef] [PubMed]
- Kamisah, Y.; Zuhair, J.S.F.; Juliana, A.H.; Jaarin, K. Parkia speciosa empty pod prevents hypertension and cardiac damage in rats given N(G)-nitro-l-arginine methyl ester. Biomed. Pharmacother. 2017, 96, 291–298. [Google Scholar] [CrossRef]
- Ge, Q.; Zhao, L.; Ren, X.M.; Ye, P.; Hu, Z.Y. LCZ696, an angiotensin receptor-neprilysin inhibitor, ameliorates diabetic cardiomyopathy by inhibiting inflammation, oxidative stress and apoptosis. Exp. Biol. Med. 2019, 244, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.; Shi, W.; Ma, H.; Yan, D.; Luo, P.; Guo, J.; Li, C.; Lin, J.; Zhang, C.; Li, S.; et al. Alleviation of inflammation and oxidative stress in pressure overload-induced cardiac remodeling and heart failure via IL-6/STAT3 inhibition by raloxifene. Oxid. Med. Cell. Longev. 2021, 2021, 6699054. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.M.; Sun, L.B.; Zheng, J.S.; Wang, X.X.; Chen, D.X.; Li, N. Copper chelation by trientine dihydrochloride inhibits liver RFA-induced inflammatory responses in vivo. Inflamm. Res. 2016, 65, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Mustafa, N.; Jalil, J.; Jubri, Z.; Kamisah, Y. Modulation of NOX4 and MAPK signaling pathways by Parkia speciosa empty pods in H9c2 cardiomyocytes exposed to H2O2. Indian J. Pharm. Sci. 2019, 81, 1029–1035. [Google Scholar] [CrossRef]
- Robinett, N.G.; Peterson, R.L.; Culotta, V.C. Eukaryotic copper-only superoxide dismutases (SODs): A new class of SOD enzymes and SOD-like protein domains. J. Biol. Chem. 2018, 293, 4636–4643. [Google Scholar] [CrossRef]
- Lewandowski, Ł.; Urbanowicz, I.; Kepinska, M.; Milnerowicz, H. Concentration/activity of superoxide dismutase isozymes and the pro-/antioxidative status, in context of type 2 diabetes and selected single nucleotide polymorphisms (genes: INS, SOD1, SOD2, SOD3)—Preliminary findings. Biomed. Pharmacother. 2021, 137, 111396. [Google Scholar] [CrossRef]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef]
- Syamsunarno, M.R.A.; Safitri, R.; Kamisah, Y. Protective effects of Caesalpinia sappan Linn. and its bioactive compounds on cardiovascular organs. Front. Pharmacol. 2021, 12, 725745. [Google Scholar] [CrossRef]
- Nikolov, A.; Popovski, N. Extracellular matrix in heart disease: Focus on circulating collagen type I and III derived peptides as biomarkers of myocardial fibrosis and their potential in the prognosis of heart failure: A concise review. Metabolites 2022, 12, 297. [Google Scholar] [CrossRef]
- Raman, B.; Ariga, R.; Spartera, M.; Sivalokanathan, S.; Chan, K.; Dass, S.; Petersen, S.E.; Daniels, M.J.; Francis, J.; Smillie, R.; et al. Progression of myocardial fibrosis in hypertrophic cardiomyopathy: Mechanisms and clinical implications. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 157–167. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qiu, L.; Lin, C.; Yang, H.; Fu, H.; Li, R.; Kang, Y.J. Copper-dependent and -independent hypoxia-inducible factor-1 regulation of gene expression. Metallomics 2014, 6, 1889–1893. [Google Scholar] [CrossRef] [PubMed]
- Elsherif, L.; Wang, L.; Saari, J.T.; Kang, Y.J. Regression of dietary copper restriction-induced cardiomyopathy by copper repletion in mice. J. Nutr. 2004, 134, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Hughes, W.M., Jr.; Rodriguez, W.E.; Rosenberger, D.; Chen, J.; Sen, U.; Tyagi, N.; Moshal, K.S.; Vacek, T.; Kang, Y.J.; Tyagi, S.C. Role of copper and homocysteine in pressure overload heart failure. Cardiovasc. Toxicol. 2008, 8, 137–144. [Google Scholar] [CrossRef]
- Mongeon, F.P.; Jerosch-Herold, M.; Coelho-Filho, O.R.; Blankstein, R.; Falk, R.H.; Kwong, R.Y. Quantification of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovasc. Imaging 2012, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Meng, K.; Pu, Y.; Zhang, X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2017, 133, 124–130. [Google Scholar] [CrossRef]
- Umbarkar, P.; Singh, A.P.; Gupte, M.; Verma, V.K.; Galindo, C.L.; Guo, Y.; Zhang, Q.; McNamara, J.W.; Force, T.; Lal, H. Cardiomyocyte SMAD4-dependent TGF-β signaling is essential to maintain adult heart homeostasis. JACC Basic Transl. Sci. 2019, 4, 41–53. [Google Scholar] [CrossRef]
- Rabieian, R.; Boshtam, M.; Zareei, M.; Kouhpayeh, S.; Masoudifar, A.; Mirzaei, H. Plasminogen activator inhibitor type-1 as a regulator of fibrosis. J. Cell. Biochem. 2018, 119, 17–27. [Google Scholar] [CrossRef]
- Liu, Y.; Miao, J. An emerging role of defective copper metabolism in heart disease. Nutrients 2022, 14, 700. [Google Scholar] [CrossRef]
- Rezaei, A.; Heidarian, E. Co-administration of trientine and flaxseed oil on oxidative stress, serum lipids and heart structure in diabetic rats. Indian J. Exp. Biol. 2013, 51, 646–652. [Google Scholar]
- Tsang, K.M.; Knutsen, R.H.; Billington, C.J., Jr.; Lindberg, E.; Steenbock, H.; Fu, Y.P.; Wardlaw-Pickett, A.; Liu, D.; Malide, D.; Yu, Z.X.; et al. Copper-binding domain variation in a novel murine lysyl oxidase model produces structurally inferior aortic elastic fibers whose failure is modified by age, sex, and blood pressure. Int. J. Mol. Sci. 2022, 23, 6749. [Google Scholar] [CrossRef] [PubMed]
- Postma, G.C.; Nicastro, C.N.; Valdez, L.B.; Rukavina Mikusic, I.A.; Grecco, A.; Minatel, L. Decrease lysyl oxidase activity in hearts of copper-deficient bovines. J. Trace Elem. Med. Biol. 2021, 65, 126715. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, E.C.; El Hajj, M.C.; Ninh, V.K.; Bradley, J.M.; Claudino, M.A.; Gardner, J.D. Detrimental role of lysyl oxidase in cardiac remodeling. J. Mol. Cell Cardiol. 2017, 109, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Hadebe, N.; Cour, M.; Lecour, S. The SAFE pathway for cardioprotection: Is this a promising target? Basic Res. Cardiol. 2018, 113, 9. [Google Scholar] [CrossRef]
- Wang, J.; Fan, S.; Xiong, Q.; Niu, Y.; Zhang, X.; Qin, J.; Shi, Y.; Zhang, L. Glucagon-like peptide-1 attenuates cardiac hypertrophy via the AngII/AT1R/ACE2 and AMPK/mTOR/p70S6K pathways. Acta Biochim. Biophys. Sin. 2021, 53, 1189–1197. [Google Scholar] [CrossRef]


| Study | Type of Model/Subjects | Trientine (Dose and Duration) | Findings | Reference |
|---|---|---|---|---|
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ ATP7A ↑ Atp7a mRNA | [34] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ mt and cyto cox17 mRNA and protein ↑ mt and cyto cox11 protein ↑ mt Sco1 protein in LV ↑ mitochondrial Cco activity ↑ mt-coI mRNA ↑ mt-coII mRNA ↔ mt-coIII mRNA ↔ mt-coI protein ↔ mt-coII protein ↔ mt-coIII protein ↔ mt-DNA content ↔ mt-tfam mRNA ↔ mt-ssbp mRNA ↑ pgc-1α mRNA | [38] |
| Human | Patients with hypertrophic cardiomyopathy (n = 20) | 300 mg twice daily orally, increased after 1 week to 600 mg twice daily if tolerated for 6 months | ↔ PCr/ATP ratio | [23] |
| Study | Type of Model/Subjects | Trientine (TETA) (Dose and Duration) | Findings | Reference |
|---|---|---|---|---|
| Human | Type 2 diabetic patients with LVH (n = 15) | 600 mg twice daily orally for 12 months | ↔ LVEDV ↔ LVESV ↔ LVEF ↔ E/E′ | [40] |
| Human | Patients with hypertrophic cardiomyopathy (n = 20) | 300 mg twice daily orally, increased after 1 week to 600 mg daily if tolerated for 6 months | ↓ GLS ↔ LVEDV ↔ LVESV ↔ SV ↔ LVEF ↔ E/A ↔ HR ↔ Native septal T1 ↓ LAESV ↔ LAEDV ↔ LAEF ↓ Total atrial strain ↑ Mean S′ velocity | [23] |
| Animal | Transverse aortic constriction- induced cardiac hypertrophy in rats | 21.9 and 87.6 mg/kg twice daily orally for 6 weeks | Both doses: ↔ LVESV ↔ LVEF | [14] |
| Animal | Ascending aortic constriction-induced cardiac hypertrophy in rats | 21.9 mg/kg twice daily orally for 6 weeks | ↑ LVEF ↑ LVFS | [15] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 8–11 mg/day in drinking water for 7 weeks (post-treatment) | ↑ CO ↑ LV +dp/dtmax ↑ LV −dp/dtmax | [39] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↑ LV −dp/dtmax | [42] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↑ CO | [25] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↑ HR ↑ CO ↑ LVEF ↔ LVEDV ↓ LVESV ↑ SV | [51] |
| Anima | STZ-induced diabetic cardiomyopathy in rats | 30 mg/day in drinking water for 7 weeks (preventive) | Preventive: ↔ HR ↓ QTc interval Post-treatment: ↑ LV +dp/dtmax ↑ peak stress | [33] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↑ LV +dp/dtmax ↑ LV +dp/dtmax ↑ CO | [34] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/d) | ↑ CO ↑ LV +dp/dtmax ↑ LV −dp/dtmax | [39] |
| Animal | Zucker diabetic cardiomyopathy in rats | 1 g/L in drinking water for 22 weeks (post-treatment) | ↔ SV ↓ LVEDP ↓ LVEDV ↓ LVESV ↓ LV volume at +dp/dtmax ↓ LV volume at −dp/dtmax | [41] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 10 mg/day in the drinkingwater for 6 weeks (post-treatment) | ↑ CO | [52] |
| Type of Model/Subjects | Trientine (Dose and Duration) | Findings | Reference |
|---|---|---|---|
| STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ mtSod1 activity ↑ mtSod1 protein ↑ mtCcs protein | [38] |
| STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ LV SOD3 mRNA | [42] |
| STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↑ Total antioxidant potential ↔ TNF-α | [25] |
| STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↑ SOD1 activity ↔ SOD1 protein ↑ Ccs protein ↑CCS ↔ ATOX1 ↑ ATP7A ↔ ATP7B ↔ Mt1 mRNA ↔ Mt2 mRNA ↓ MT (70 kD) protein ↑ MT (45 kD) protein ↑ MT (30 kD) protein | [34] |
| Study | Type of Model/Subjects | Trientine (TETA) (Dose and Duration) | Findings | Reference |
|---|---|---|---|---|
| Animal | STZ-induced diabetic cardiomyopathy in rats | 8–11 mg/day in drinking water for 7 weeks (post-treatment) | ↓ LV collagen I ↓ LV collagen III | [39] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↓ LV collagen I, III, and IV mRNA ↓ LV fibronectin-I mRNA ↓ LV PAI-1 mRNA ↓ LV TGF-β1 mRNA ↓ LV Smad4 mRNA | [42] |
| Animal | Transverse aortic constriction- induced cardiac hypertrophy in rats | 21.9 and 87.6 mg/kg twice daily orally for 6 weeks | TETA (21.9 mg/kg/day): ↓ cardiac collagen I TETA (87.6 mg/kg/day): ↔ cardiac collagen I | [14] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 30 mg/day in drinking water for 8 weeks (post-treatment) | ↓ LV collagen I ↔ LV collagen III | [33] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↓ TGF-β1 | [25] |
| Animal | Ascending aortic constriction-induced cardiac hypertrophy in rats | 21.9 mg/kg twice daily orally for 6 weeks | ↓ cardiac collagen volume fraction ↓ cardiac hydroxyproline ↔ cardiac collagen I ↓ cardiac collagen III ↔ cardiac active MMP-9 ↓ cardiac MMP-9 mRNA ↑ cardiac active MMP-2 ↔ cardiac MMP-2 mRNA ↓ cardiac TIMP-1 mRNA ↓ cardiac TIMP-2 mRNA | [15] |
| Human | Patients with hypertrophic cardiomyopathy (n = 20) | 300 mg twice daily orally, increased after 1 week to 600 mg twice daily if tolerated for 6 months | ↔ ECV fraction ↓ ECM volume | [23] |
| Study | Type of Model/Subjects | Trientine (TETA) (Dose and Duration) | Findings | Reference |
|---|---|---|---|---|
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↓ HW/BW | [38] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 8–11 mg/day in drinking water for 7 weeks (post-treatment) | ↓ HW/BW | [39] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↓ HW/BW | [42] |
| Animal | Ascending aortic constriction-induced cardiac hypertrophy in rats | 21.9 mg/kg twice daily orally for 6 weeks | ↓ cardiomyocyte size ↔ cardiac vimentin | [15] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↔ HW/BW | [25] |
| Animal | Transverse aortic constriction- induced cardiac hypertrophy in rats | 21.9 and 87.6 mg/kg twice daily orally for 6 weeks | TETA (21.9 mg/kg/day): ↓ LVAWd ↓ LVPWd ↓ HW/TL ↓ cardiomyocyte size TETA (87.6 mg/kg/day): ↔ LVAWd ↔ LVPWd ↔ HW/BW ↔ cardiomyocyte size | [14] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↓ LV mass/BW | [51] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 8–11 mg/day orally for 6 weeks | Improved myocardial structure | [80] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 30 mg/day in drinking water for 7 weeks (preventive) | ↔ HW/BW | [33] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) | ↓ HW/BW | [34] |
| Human | Type 2 diabetic patients with LVH (n = 15) | 600 mg twice daily orally for 12 months | ↓ LVMbsa | [40] |
| Human | Patients with hypertrophic cardiomyopathy (n = 20) | 300 mg twice daily orally, increased after 1 week to 600 mg daily if tolerated for 6 months | ↔ LVMbsa | [23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramli, F.F.; Hashim, S.A.S.; Raman, B.; Mahmod, M.; Kamisah, Y. Role of Trientine in Hypertrophic Cardiomyopathy: A Review of Mechanistic Aspects. Pharmaceuticals 2022, 15, 1145. https://doi.org/10.3390/ph15091145
Ramli FF, Hashim SAS, Raman B, Mahmod M, Kamisah Y. Role of Trientine in Hypertrophic Cardiomyopathy: A Review of Mechanistic Aspects. Pharmaceuticals. 2022; 15(9):1145. https://doi.org/10.3390/ph15091145
Chicago/Turabian StyleRamli, Fitri Fareez, Syed Alhafiz Syed Hashim, Betty Raman, Masliza Mahmod, and Yusof Kamisah. 2022. "Role of Trientine in Hypertrophic Cardiomyopathy: A Review of Mechanistic Aspects" Pharmaceuticals 15, no. 9: 1145. https://doi.org/10.3390/ph15091145