
A High-Affinity 64Cu-Labeled Ligand for PET Imaging of Hepsin: Design, Synthesis, and Characterization
 , and
, and
Abstract
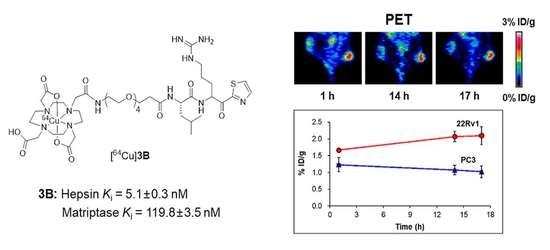
:
1. Introduction
2. Results
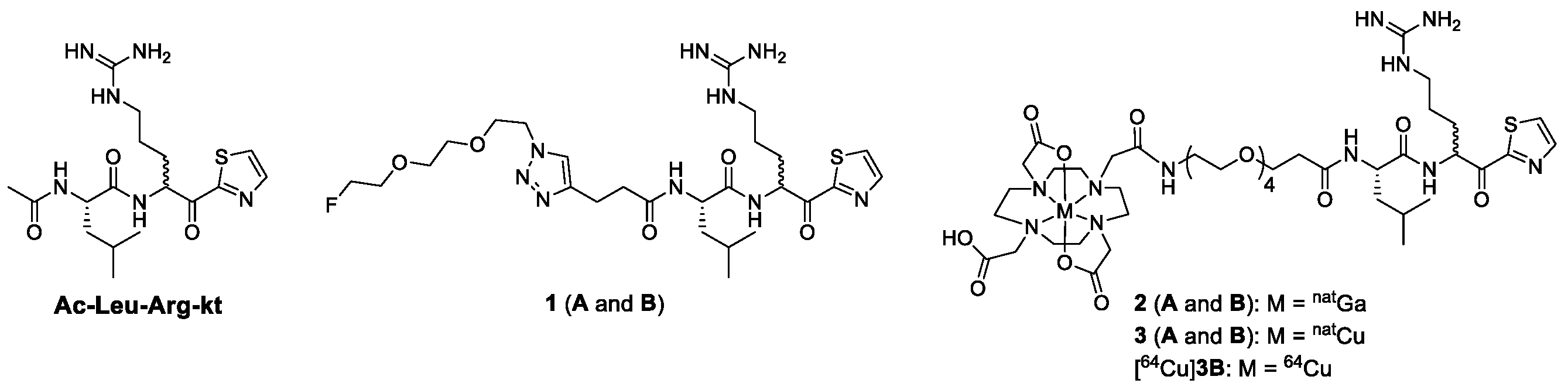
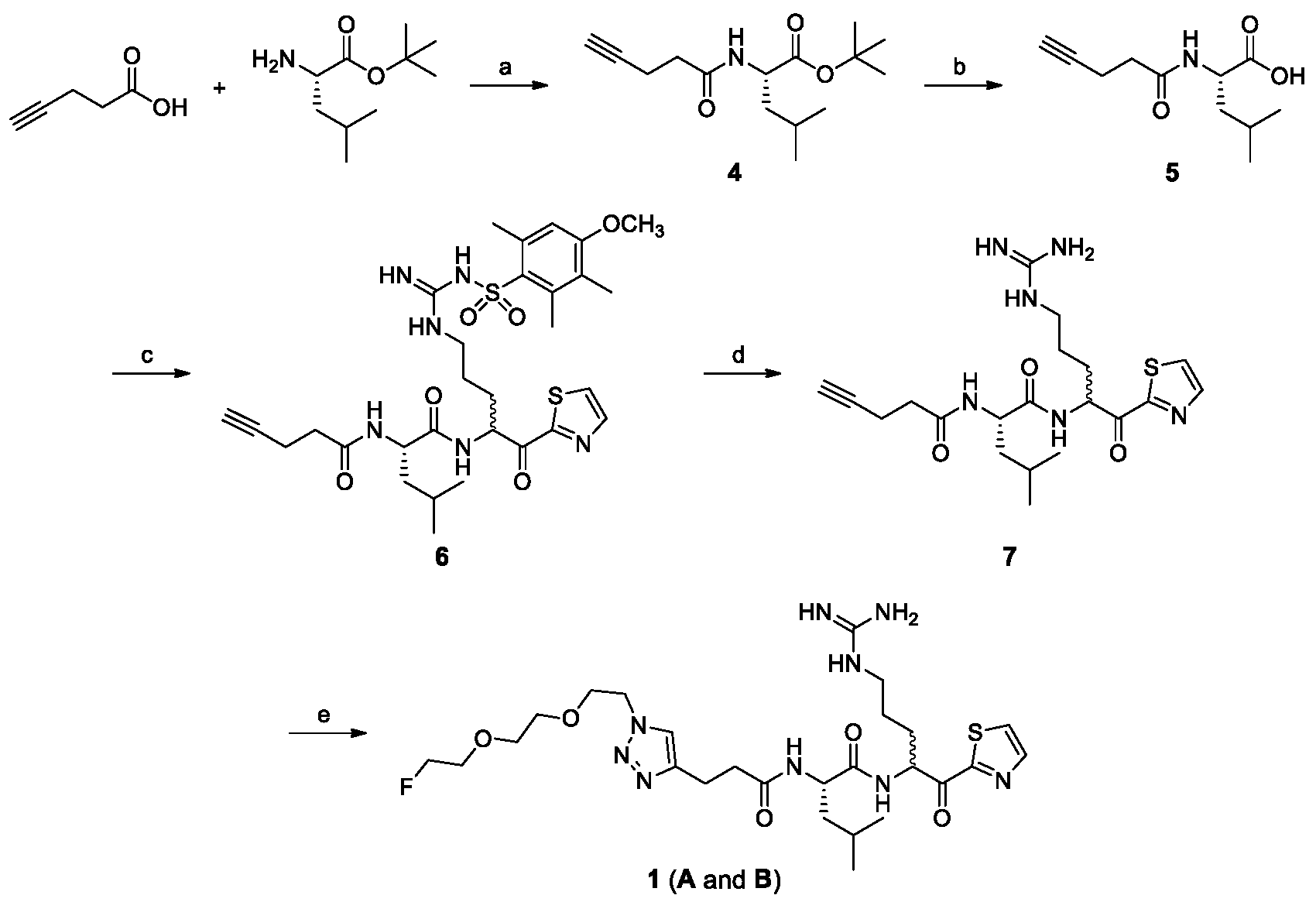
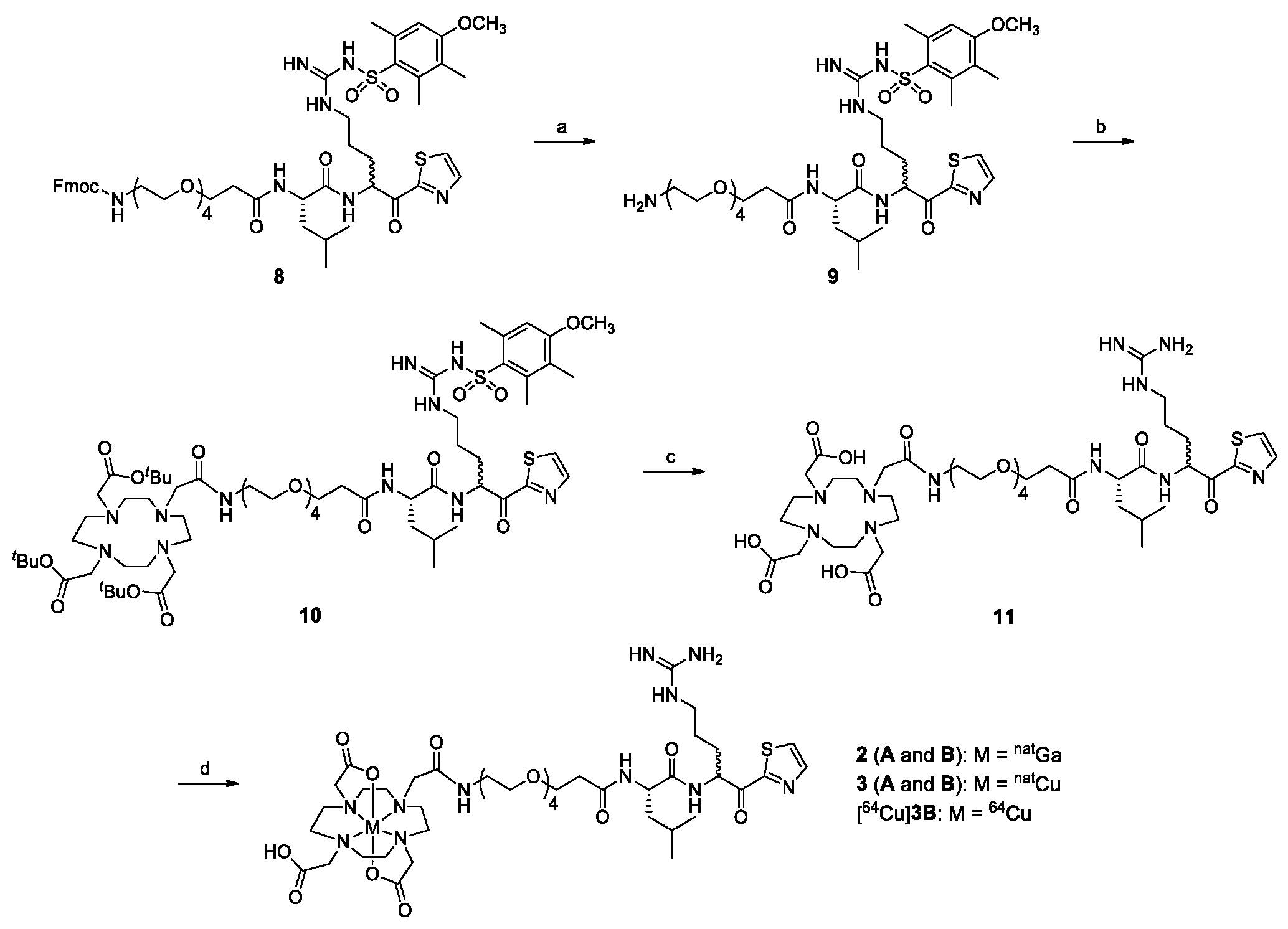
2.1. Chemical Synthesis
2.2. Radiochemical Synthesis
2.3. In Vitro Binding Assay
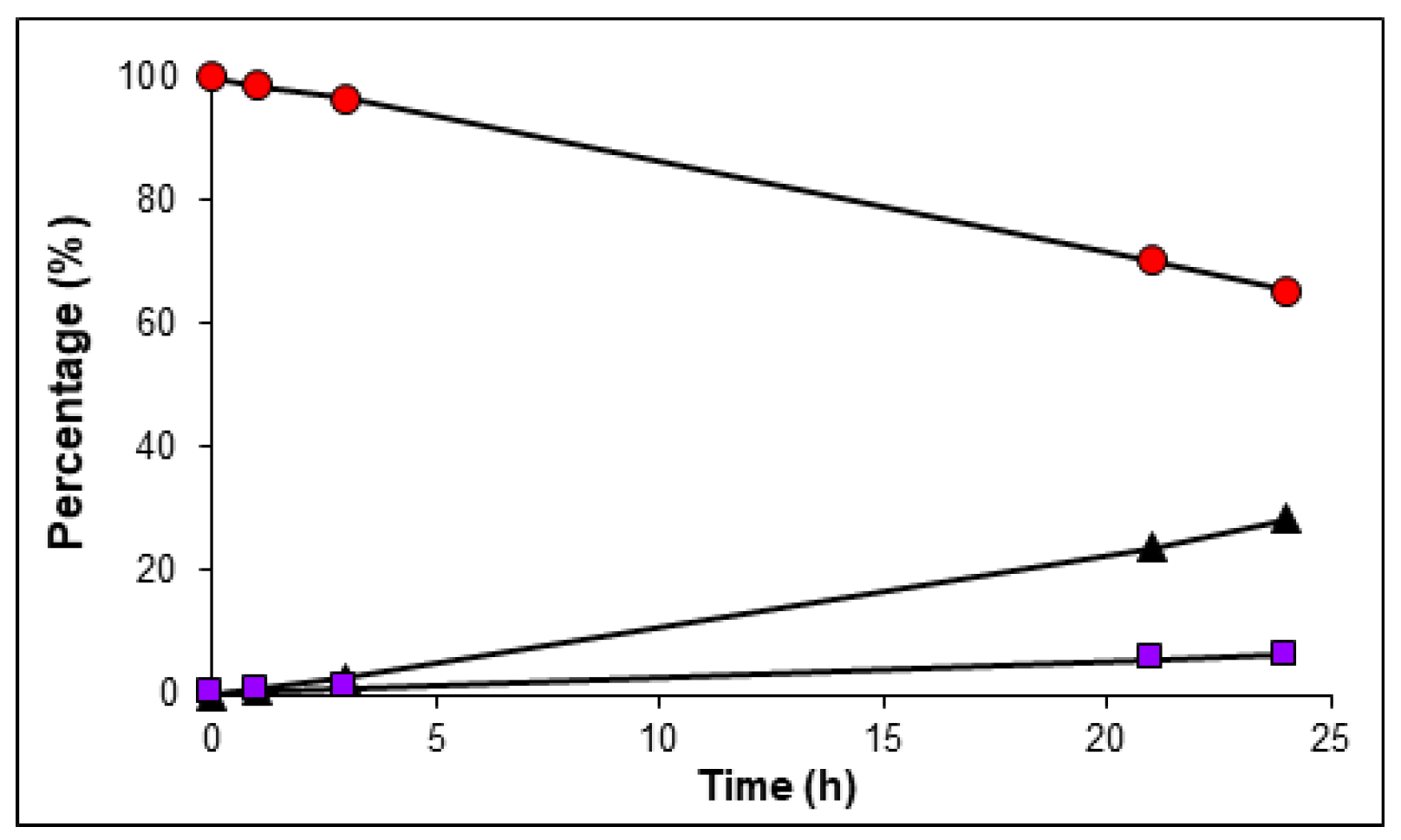
2.4. In Vitro Serum Stability
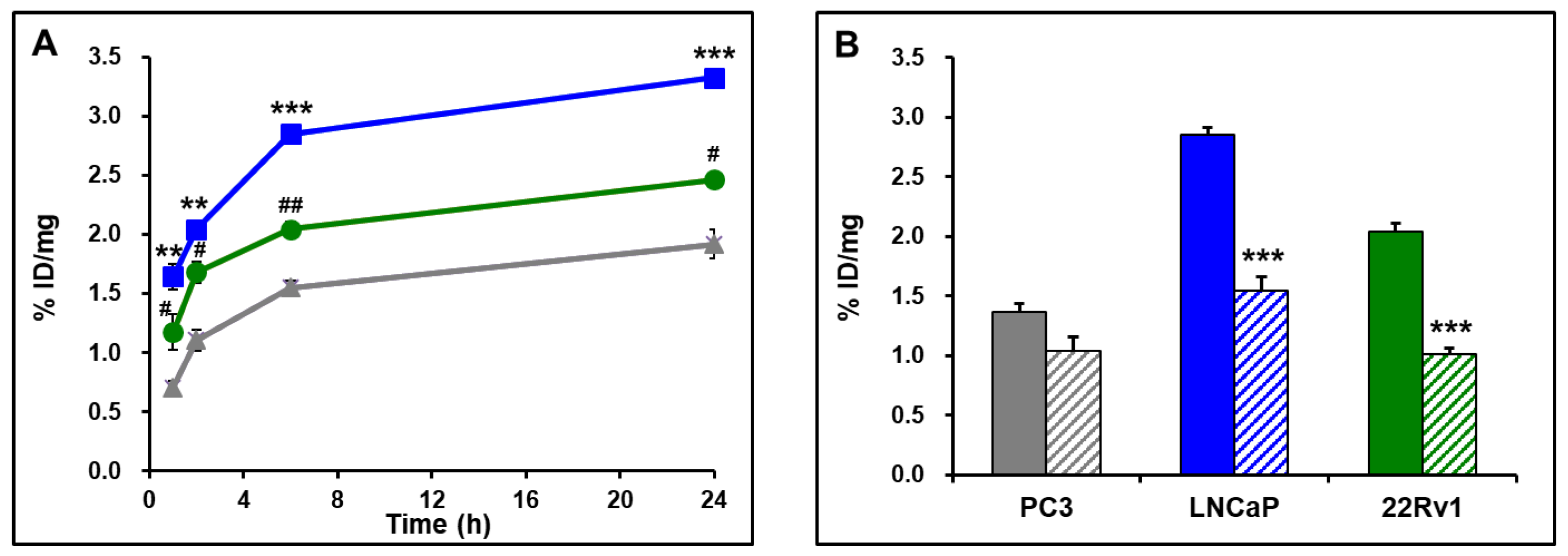
2.5. Cell Binding
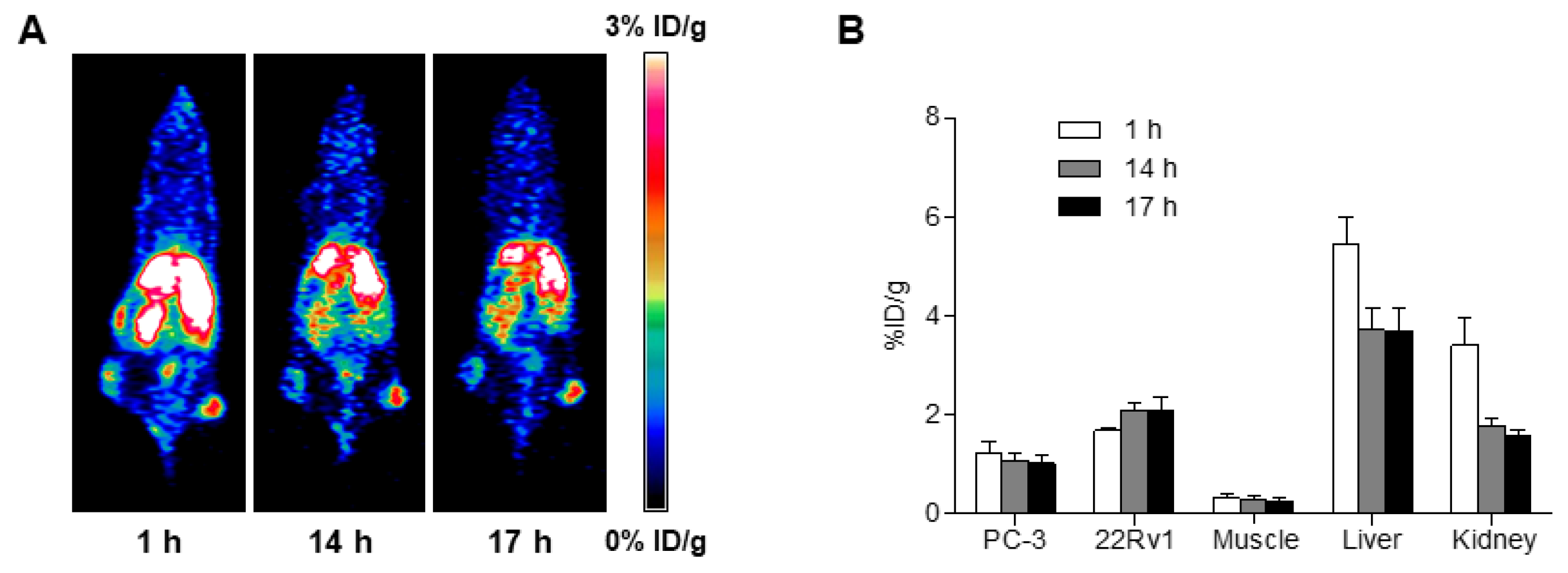
2.6. MicroPET Imaging
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Chemical Synthesis
4.2.1. (S)-tert-Butyl 4-Methyl-2-(pent-4-ynamido)pentanoate (4)
4.2.2. (S)-4-Methyl-2-(pent-4-ynamido)pentanoic acid (5)
4.2.3. N-((2S)-1-(5-(3-((4-Methoxy-2,3,6-trimethylphenyl)sulfonyl)guanidino)-1-oxo-1-(thiazol-2-yl)pentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)pent-4-ynamide (6)
4.2.4. N-((2S)-1-(5-Guanidino-1-oxo-1-(thiazol-2-yl)pentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)pent-4-ynamide (7)
4.2.5. (2S)-2-(3-(1-(2-(2-(2-Fluoroethoxy)ethoxy)ethyl)-1H-1,2,3-triazol-4-yl)propanamido)-N-(5-guanidino-1-oxo-1-(thiazol-2-yl)pentan-2-yl)-4-methylpentanamide (1)
4.2.6. 1-Amino-N-((2S)-1-((5-(3-((4-methoxy-2,3,6-trimethylphenyl)sulfonyl)guanidino)-1-oxo-1-(thiazol-2-yl)pentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-3,6,9,12-tetraoxapentadecan-15-amide (9)
4.2.7. Tri-tert-butyl 2,2’,2’’-(10-((9S)-1-imino-9-isobutyl-1-(4-methoxy-2,3,6-trimethyl phenylsulfonamido)-8,11,27-trioxo-6-(thiazole-2-carbonyl)-14,17,20,23-tetraoxa-2,7,10,26- tetraazaoctacosan-28-yl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (10)
4.2.8. 2,2′,2″-(10-((9S)-1-Amino-1-imino-9-isobutyl-8,11,27-trioxo-6-(thiazole-2-carbonyl)-14,17,20,23-tetraoxa-2,7,10,26-tetraazaoctacosan-28-yl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid (11)
4.2.9. natGa-DOTA-conjugated 1-amino-N-(1-((5-guanidino-1-oxo-1-(thiazol-2-yl)pentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-3,6,9,12-tetraoxapentadecan-15-amide (2)
4.2.10. natCu-DOTA-conjugated 1-amino-N-(1-((5-guanidino-1-oxo-1-(thiazol-2-yl)pentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-3,6,9,12-tetraoxapentadecan-15-amide (3)
4.3. Radiochemical Synthesis
4.3.1. General Information
4.3.2. Synthesis of Radioligand [64Cu]3B
4.4. In Vitro Binding Assay
4.5. In Vitro Serum Stability
4.6. In Vitro Cell Studies
4.6.1. Cell Lines and Culture
4.6.2. Cell Binding
4.7. In Vivo Studies
4.7.1. Animals
4.7.2. MicroPET Imaging
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.B.S.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.J.; Autio, K.A.; Roach, M., 3rd; Scher, H.I. High-risk prostate cancer-classification and therapy. Nat. Rev. Clin. Oncol. 2014, 11, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Dupont, A.; Suburu, R.; Cusan, L.; Tremblay, M.; Gomez, J.L.; Emond, J. Serum prostate specific antigen as pre-screening test for prostate cancer. J. Urol. 1992, 147, 846–851. [Google Scholar] [CrossRef]
- Monda, J.M.; Barry, M.J.; Oesterling, J.E. Prostate specific antigen cannot distinguish stage T1a (A1) prostate cancer from benign prostatic hyperplasia. J. Urol. 1994, 151, 1291–1295. [Google Scholar] [CrossRef]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Delineation of prognostic biomarkers in prostate cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef]
- Luo, J.; Duggan, D.J.; Chen, Y.; Sauvageot, J.; Ewing, C.M.; Bittner, M.L.; Trent, J.M.; Isaacs, W.B. Human prostate cancer and benign prostatic hyperplasia: Molecular dissection by gene expression profiling. Cancer Res. 2001, 61, 4683–4688. [Google Scholar] [PubMed]
- Magee, J.A.; Araki, T.; Patil, S.; Ehrig, T.; True, L.; Humphrey, P.A.; Catalona, W.J.; Watson, M.A.; Milbrandt, J. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 2001, 61, 5692–5696. [Google Scholar]
- Wu, Q.; Parry, G. Hepsin and prostate cancer. Front. Biosci. 2007, 12, 5052–5059. [Google Scholar] [CrossRef]
- Lu, L.; Cole, A.; Huang, D.; Wang, Q.; Guo, Z.; Yang, W.; Lu, J. Clinical significance of hepsin and underlying signaling pathways in prostate cancer. Biomolecules 2022, 12, 203. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Ho, J.D.; Luong, C.; Ghate, M.; Sprengeler, P.A.; Mortara, K.; Shrader, W.D.; Sperandio, D.; Chan, H.; McGrath, M.E.; et al. The structure of the extracellular region of human hepsin reveals a serine protease domain and a novel scavenger receptor cysteine-rich (SRCR) domain. Structure 2003, 11, 1123–1131. [Google Scholar] [CrossRef]
- Kelly, K.A.; Setlur, S.R.; Ross, R.; Anbazhagan, R.; Waterman, P.; Rubin, M.A.; Weissleder, R. Detection of early prostate cancer using a hepsin-targeted imaging agent. Cancer Res. 2008, 68, 2286–2291. [Google Scholar] [CrossRef] [PubMed]
- Subedi, M.; Minn, I.; Chen, J.; Kim, Y.H.; Ok, K.; Jung, Y.W.; Pomper, M.G.; Byun, Y. Design, synthesis and biological evaluation of PSMA/hepsin-targeted heterobivalent ligands. Eur. J. Med. Chem. 2016, 118, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kwon, H.; Choi, D.; Lim, T.; Minn, I.; Son, S.-H.; Byun, Y. Design and synthesis of dye-conjugated hepsin inhibitors. Bioorg. Chem. 2019, 89, 102990. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Martin, N.E.; Modi, M. Pegylation: A novel process for modifying pharmacokinetics. Clin. Pharmacokinet. 2001, 40, 539–551. [Google Scholar] [CrossRef]
- Zhang, W.; Oya, S.; Kung, M.P.; Hou, C.; Maier, D.L.; Kung, H.F. F-18 Polyethyleneglycol stilbenes as PET imaging agents targeting Aβ aggregates in the brain. Nucl. Med. Biol. 2005, 32, 799–809. [Google Scholar] [CrossRef]
- Zhang, H.; Schuhmacher, J.; Waser, B.; Wild, D.; Eisenhut, M.; Reubi, J.C.; Maecke, H.R. DOTA-PESIN, a DOTA-conjugated bombesin derivative designed for the imaging and targeted radionuclide treatment of bombesin receptor-positive tumours. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1198–1208. [Google Scholar] [CrossRef]
- Wu, Z.; Li, Z.-B.; Chen, K.; Cai, W.; He, L.; Chin, F.T.; Li, F.; Chen, X. microPET of tumor integrin αvβ3 expression using 18F-labeled PEGylated tetrameric RGD peptide (18F-FPRGD4). J. Nucl. Med. 2007, 48, 1536–1544. [Google Scholar] [CrossRef]
- Beaino, W.; Anderson, C.J. PET imaging of very late antigen-4 in melanoma: Comparison of 68Ga- and 64Cu-labeled NODAGA and CB-TE1A1P-LLP2A conjugates. J. Nucl. Med. 2014, 55, 1856–1863. [Google Scholar] [CrossRef]
- Lin, J.; Deng, H.; Jin, L.; Pandey, P.; Quinn, J.; Cantin, S.; Rynkiewicz, M.J.; Gorga, J.C.; Bibbins, F.; Celatka, C.A.; et al. Design, synthesis, and biological evaluation of peptidomimetic inhibitors of factor XIa as novel anticoagulants. J. Med. Chem. 2006, 49, 7781–7791. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Desilets, A.; Duchene, D.; Chagnon, F.; Najmanovich, R.; Leduc, R.; Marsault, E. Design and synthesis of potent, selective inhibitors of matriptase. ACS Med. Chem. Lett. 2012, 3, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Kim, Y.; Park, K.; Choi, S.A.; Son, S.-H.; Byun, Y. Structure-based design, synthesis, and biological evaluation of Leu-Arg dipeptide analogs as novel hepsin inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Srikantan, V.; Valladares, M.; Rhim, J.S.; Moul, J.W.; Srivastava, S. Hepsin inhibits cell growth/invasion in prostate cancer cells. Cancer Res. 2002, 62, 6812–6816. [Google Scholar]
- Han, Z.; Harris, P.K.; Jones, D.E.; Chugani, R.; Kim, T.; Agarwal, M.; Shen, W.; Wildman, S.A.; Janetka, J.W. Inhibitors of HGFA, matriptase, and hepsin serine proteases: A nonkinase strategy to block cell signaling in cancer. ACS Med. Chem. Lett. 2014, 11, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.M.; Jones, D.E.; Harris, P.K.; Han, Z.; Wildman, S.A.; Jarvis, C.M.; Janetka, J.W. Structure-based discovery of small molecule hepsin and HGFA protease inhibitors: Evaluation of potency and selectivity derived from distinct binding pockets. Bioorg. Med. Chem. 2015, 23, 2328–2343. [Google Scholar] [CrossRef]
- Goswami, R.; Wohlfahrt, G.; Tormakangas, O.; Moilanen, A.; Lakshminarasimhan, A.; Nagaraj, J.; Arumugam, K.N.; Mukherjee, S.; Chacko, A.R.; Krishnamurthy, N.R.; et al. Structure-guided discovery of 2-aryl/pyridin-2-yl-1H-indole derivatives as potent and selective hepsin inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 5309–5314. [Google Scholar] [CrossRef]
- Venukadasula, P.K.; Owusu, B.Y.; Bansal, N.; Ross, L.J.; Hobrath, J.V.; Bao, D.; Truss, J.W.; Stackhouse, M.; Messick, T.E.; Klampfer, L.; et al. Design and synthesis of nonpeptide inhibitors of hepatocyte growth factor activation. ACS Med. Chem. Lett. 2016, 7, 177–181. [Google Scholar] [CrossRef]
- Damalanka, V.C.; Han, Z.; Karmakar, P.; O’Donoghue, A.J.; Greca, F.L.; Kim, T.; Pant, S.M.; Helander, J.; Klefstrom, J.; Craik, C.S.; et al. Discovery of selective matriptase and hepsin serine potease inhibitors: Useful chemical tools for cancer cell biology. J. Med. Chem. 2019, 62, 480–490. [Google Scholar] [CrossRef]
- Kwon, H.; Ha, H.; Jeon, H.; Jang, J.; Son, S.-H.; Lee, K.; Park, S.-K.; Byun, Y. Structure-activity relationship studies of dipeptide-based hepsin inhibitors with Arg bioisosteres. Bioorg. Chem. 2021, 107, 104521. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Ki (nM) | Matriptase/ Hepsin | |
|---|---|---|---|
| Hepsin | Matriptase | ||
| Ac–Leu–Arg–kt | 7.8 ± 2.8 | 56.5 ± 2.7 | 7.2 |
| 1A | 0.9 ± 0.1 | 54.5 ± 4.9 | 60.6 |
| 1B | 0.5 ± 0.1 | 20.0 ± 0.6 | 40.0 |
| 2A | 14.3 ± 1.2 | 166.5 ± 8.2 | 11.6 |
| 2B | 5.7 ± 0.2 | 68.2 ± 2.4 | 12.0 |
| 3A | 15.0 ± 0.6 | 248.9 ± 12.3 | 16.6 |
| 3B | 5.1 ± 0.3 | 119.8 ± 3.5 | 23.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-H.; Zhang, X.; Ha, H.; Kim, J.Y.; Choi, J.Y.; Lee, K.-H.; Byun, Y.; Choe, Y.S. A High-Affinity 64Cu-Labeled Ligand for PET Imaging of Hepsin: Design, Synthesis, and Characterization. Pharmaceuticals 2022, 15, 1109. https://doi.org/10.3390/ph15091109
Park J-H, Zhang X, Ha H, Kim JY, Choi JY, Lee K-H, Byun Y, Choe YS. A High-Affinity 64Cu-Labeled Ligand for PET Imaging of Hepsin: Design, Synthesis, and Characterization. Pharmaceuticals. 2022; 15(9):1109. https://doi.org/10.3390/ph15091109
Chicago/Turabian StylePark, Ji-Hun, Xuran Zhang, Hyunsoo Ha, Jung Young Kim, Joon Young Choi, Kyung-Han Lee, Youngjoo Byun, and Yearn Seong Choe. 2022. "A High-Affinity 64Cu-Labeled Ligand for PET Imaging of Hepsin: Design, Synthesis, and Characterization" Pharmaceuticals 15, no. 9: 1109. https://doi.org/10.3390/ph15091109