
Antibiotics and Carbohydrate-Containing Drugs Targeting Bacterial Cell Envelopes: An Overview

 ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
1.1. Bacteria
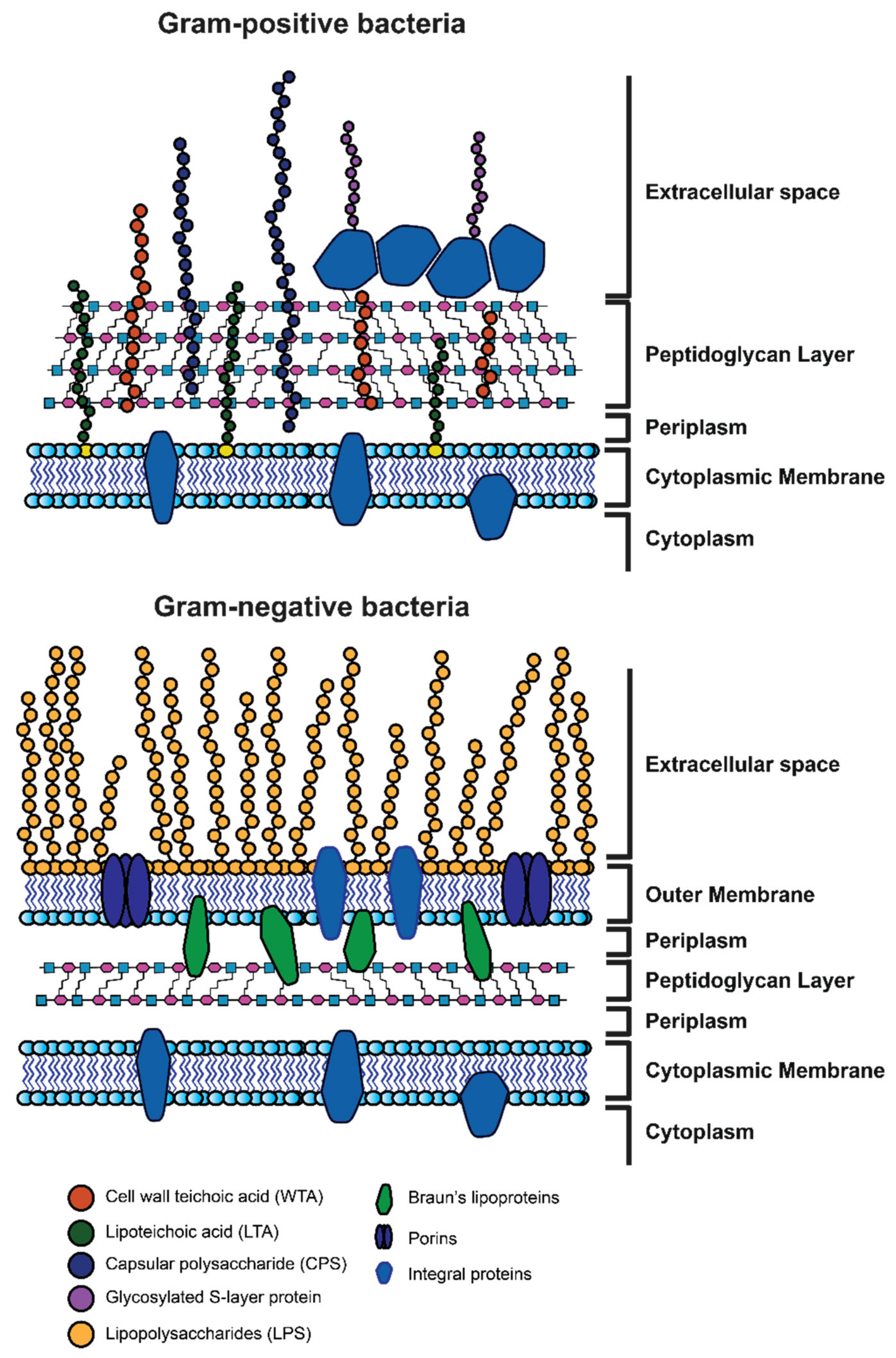
1.1.1. Gram-Positive and Gram-Negative Bacteria: General Remarks
1.1.2. Morphology of Gram-Positive and Gram-Negative Bacteria
1.1.3. The Cytoplasmic Membrane
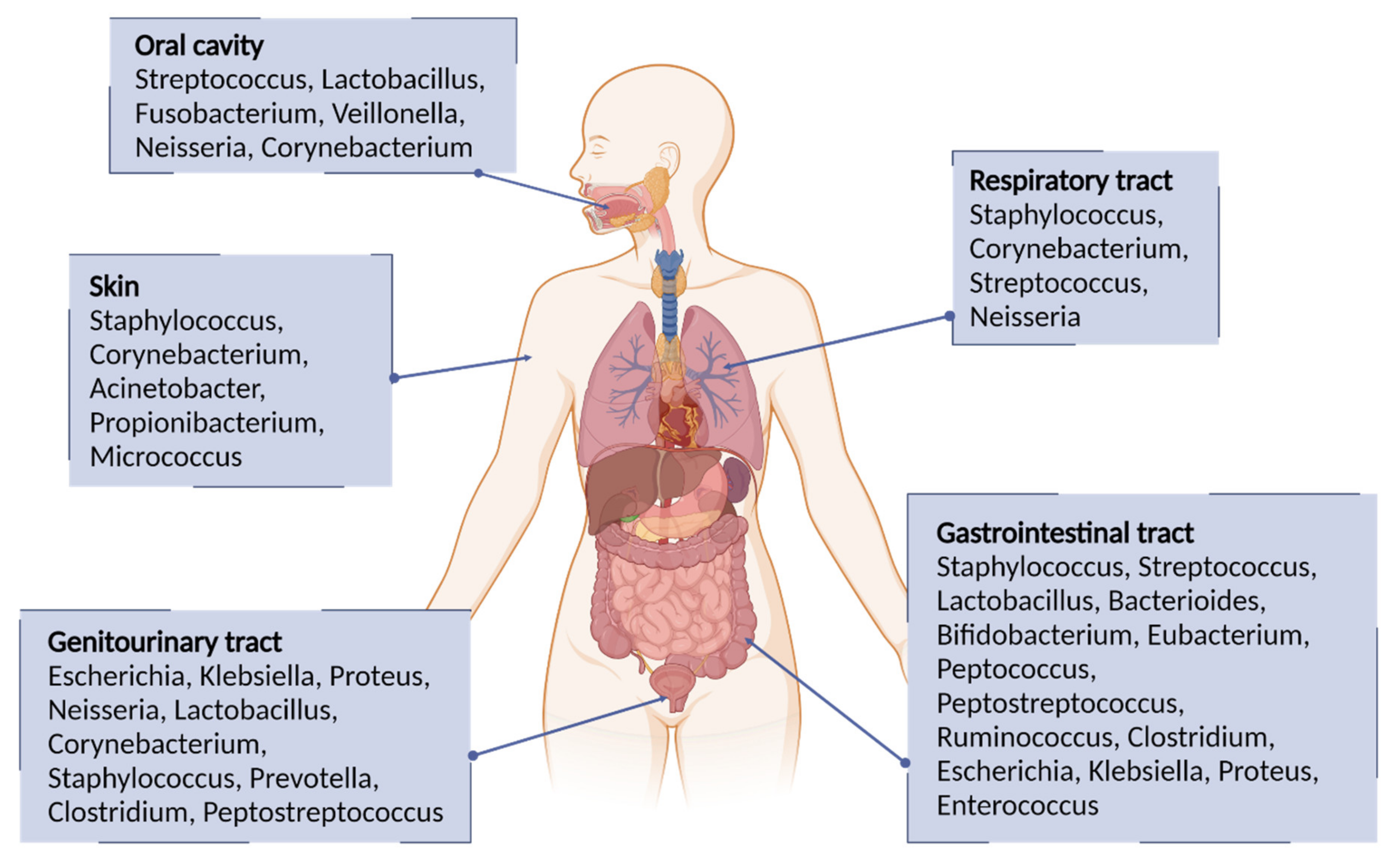
1.1.4. Pathogenic Bacteria
1.2. Bacterial Cell Wall Polysaccharides (CWPs) and Peptidoglycans (PGs): Structure and Functions
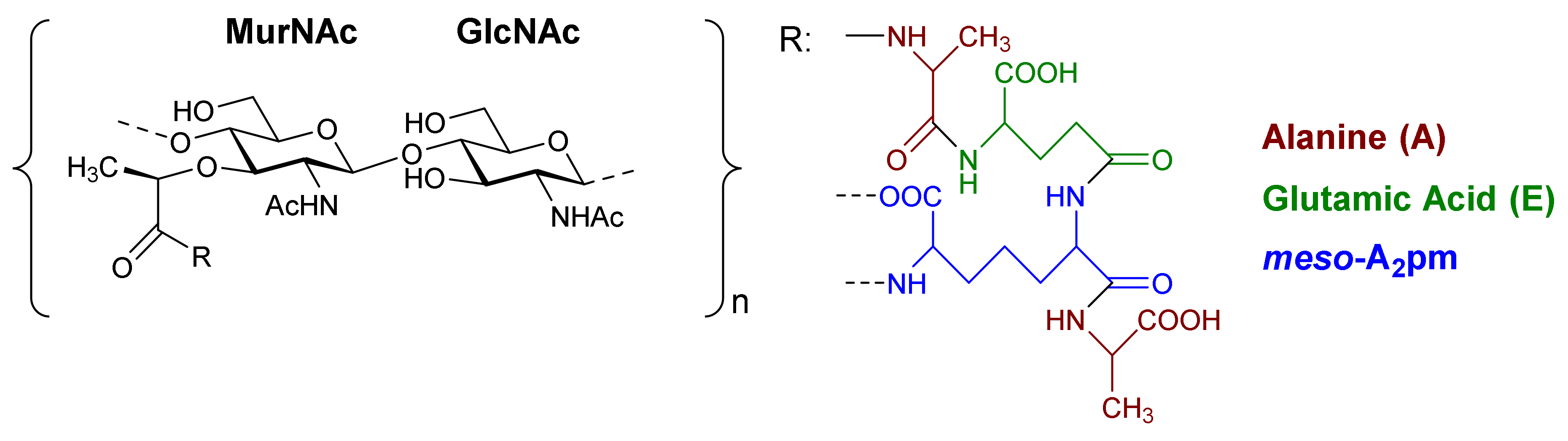
1.2.1. Chemical Structure and Variability of PG and Correlated Functions
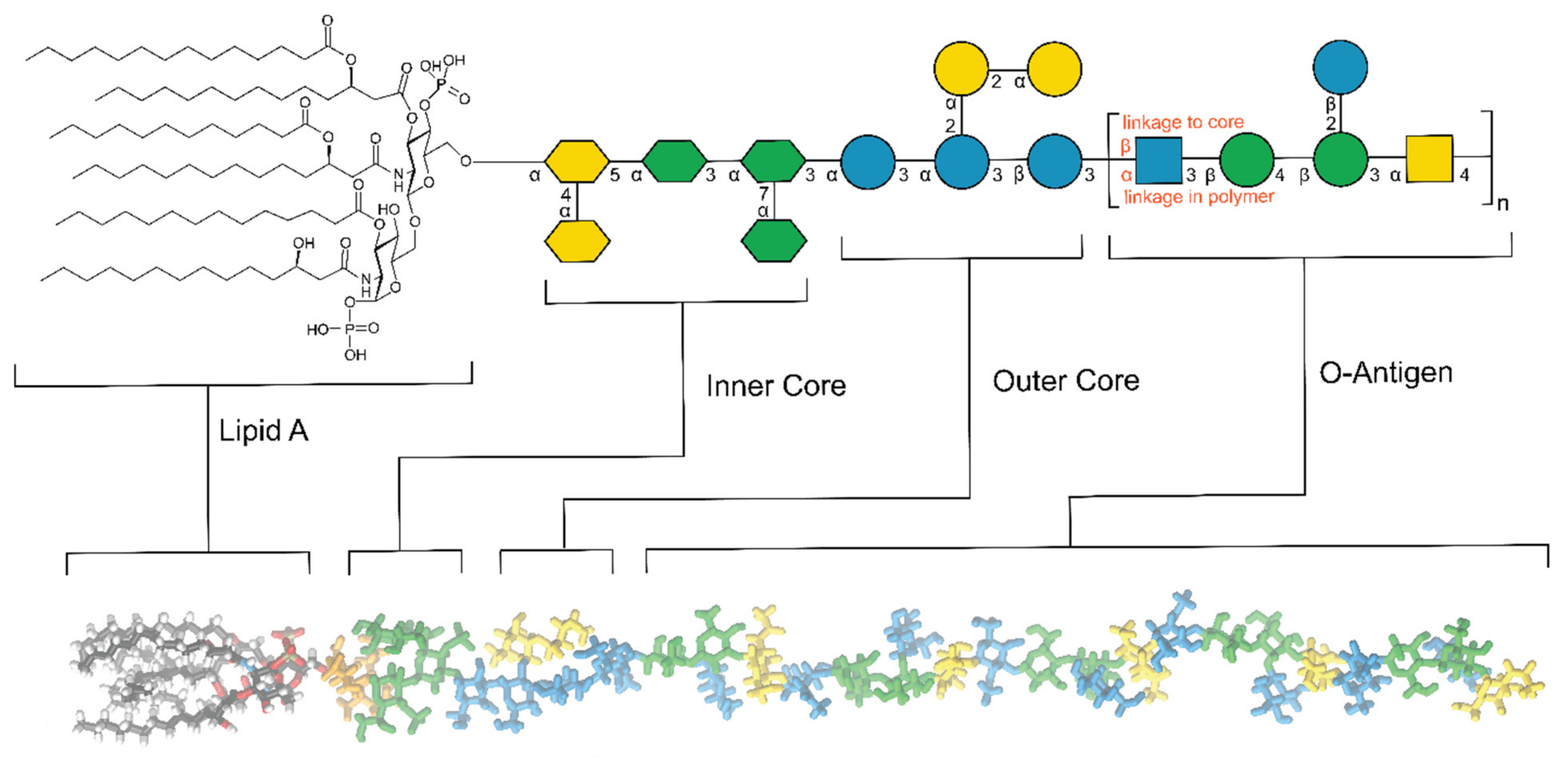
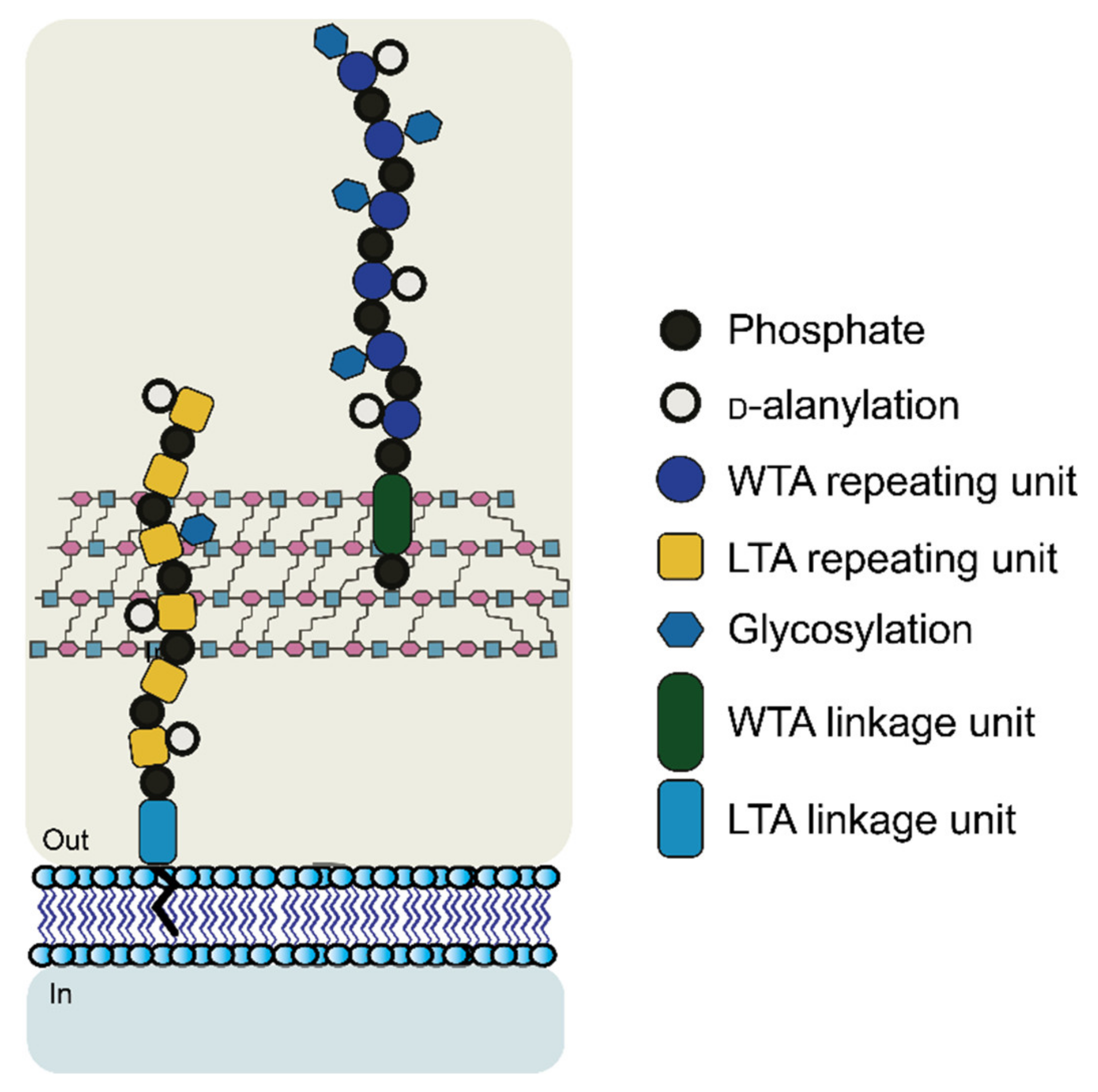
1.2.2. Cell Wall Polysaccharides: Structure and Functions
2. Drugs Targeting CWPs and PGs
2.1. Antibiotics
2.2. Carbohydrate-Based Antibiotics Targeting CWPs and PGs: Glycopeptides and Lipoglycopeptides
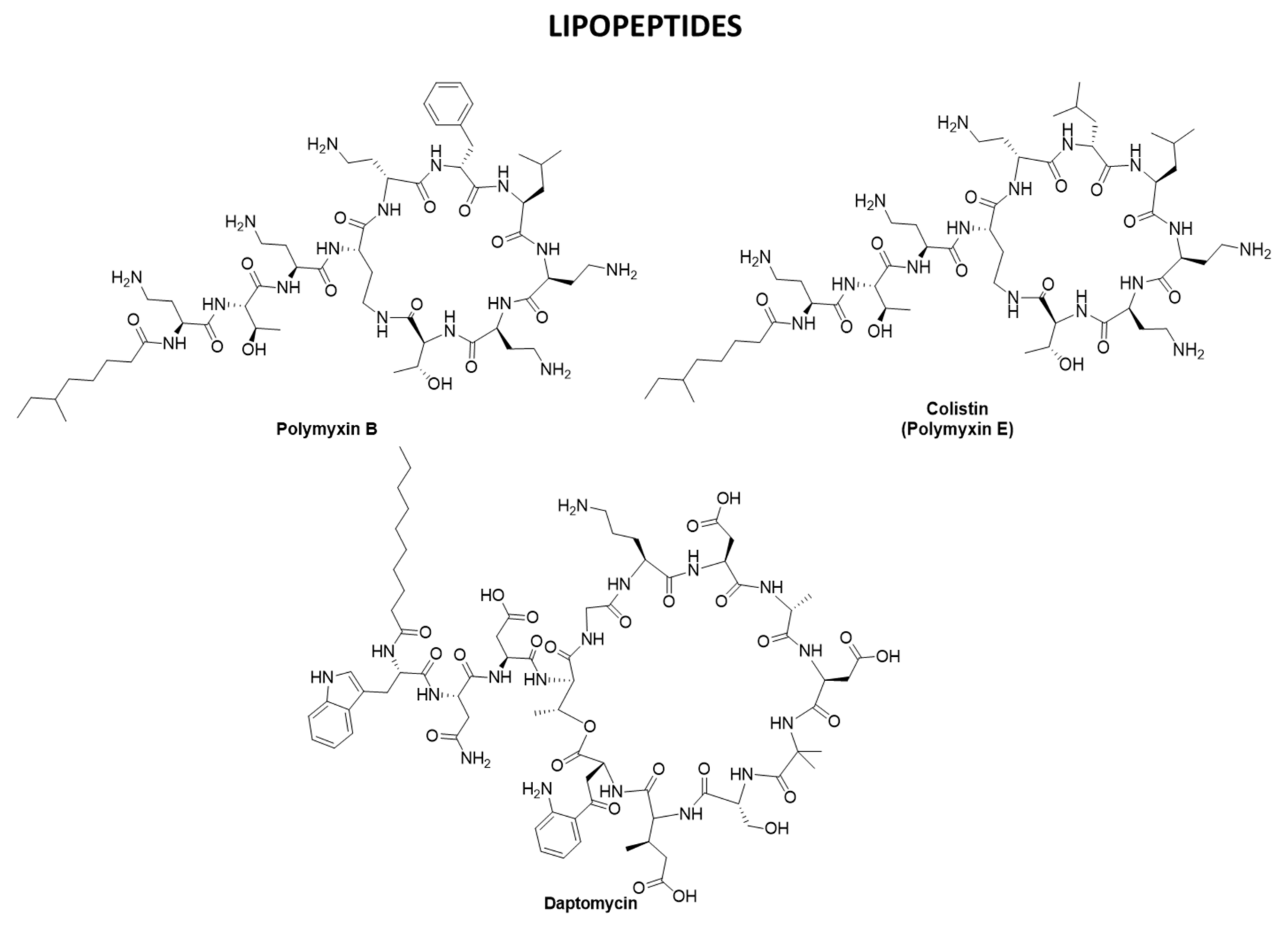
2.3. Antibiotics Targeting CWPs and PGs: β-lactams, Polymyxins, and Daptomycin
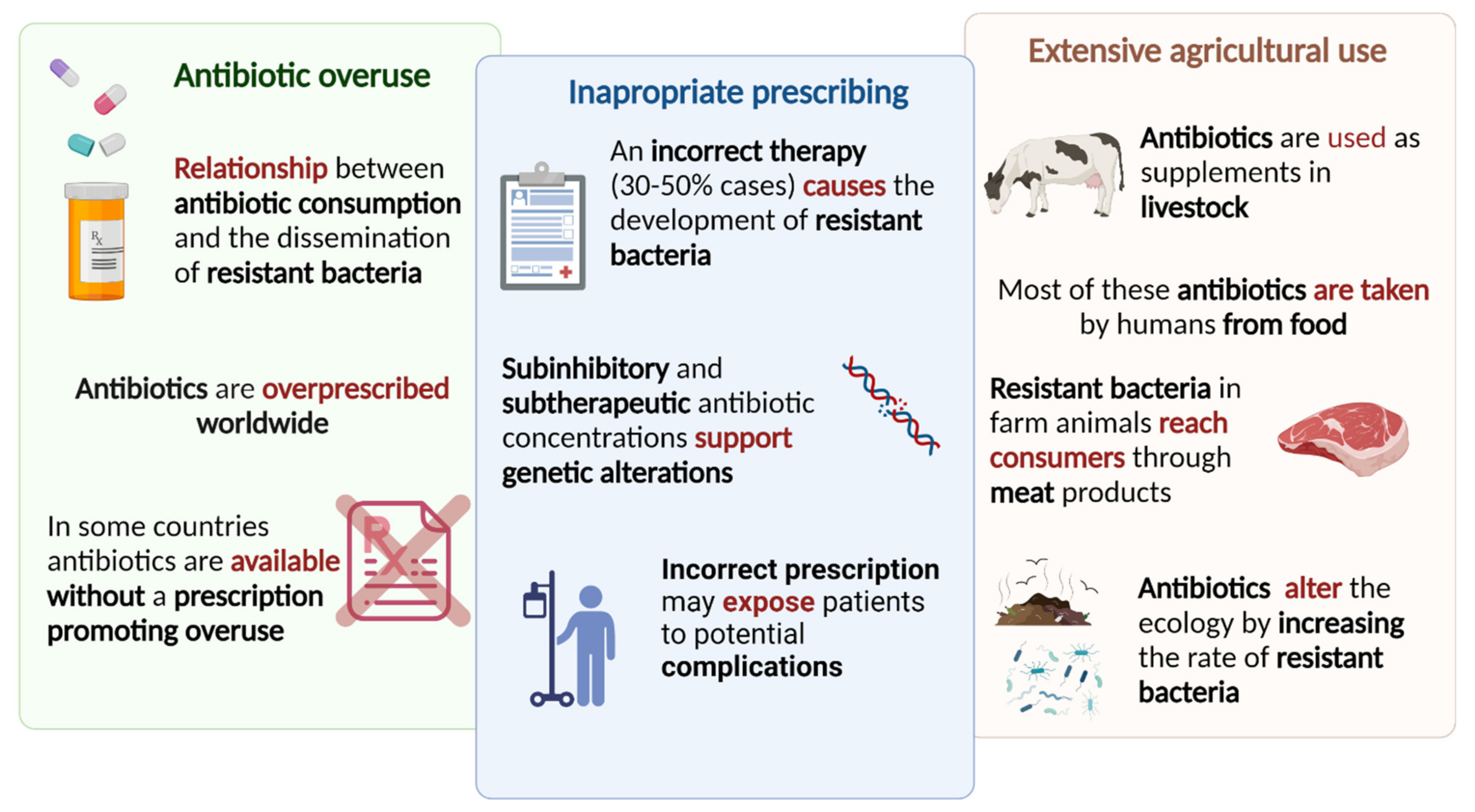
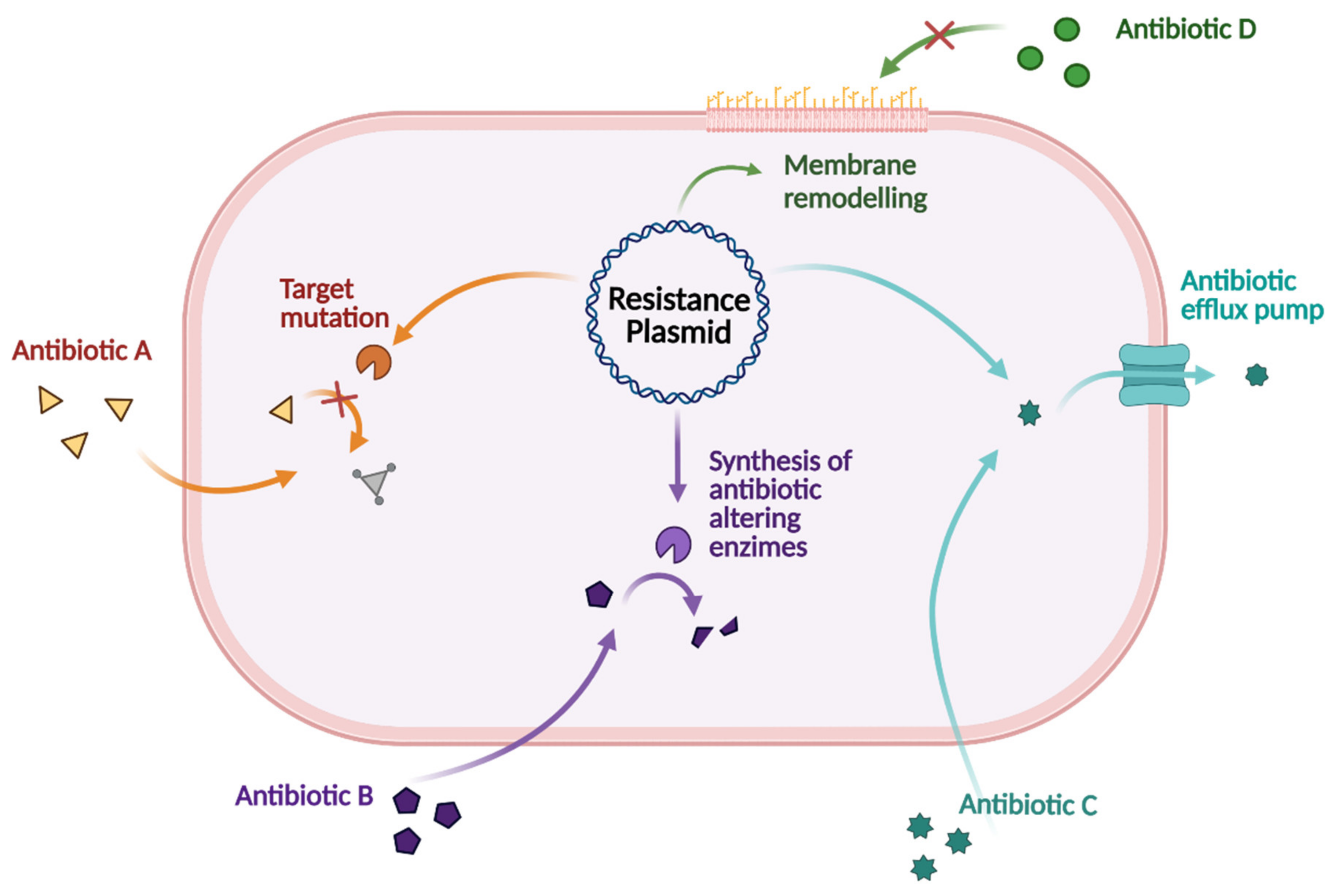
3. Antibiotic Drug Resistance
3.1. Consumption and Therapy
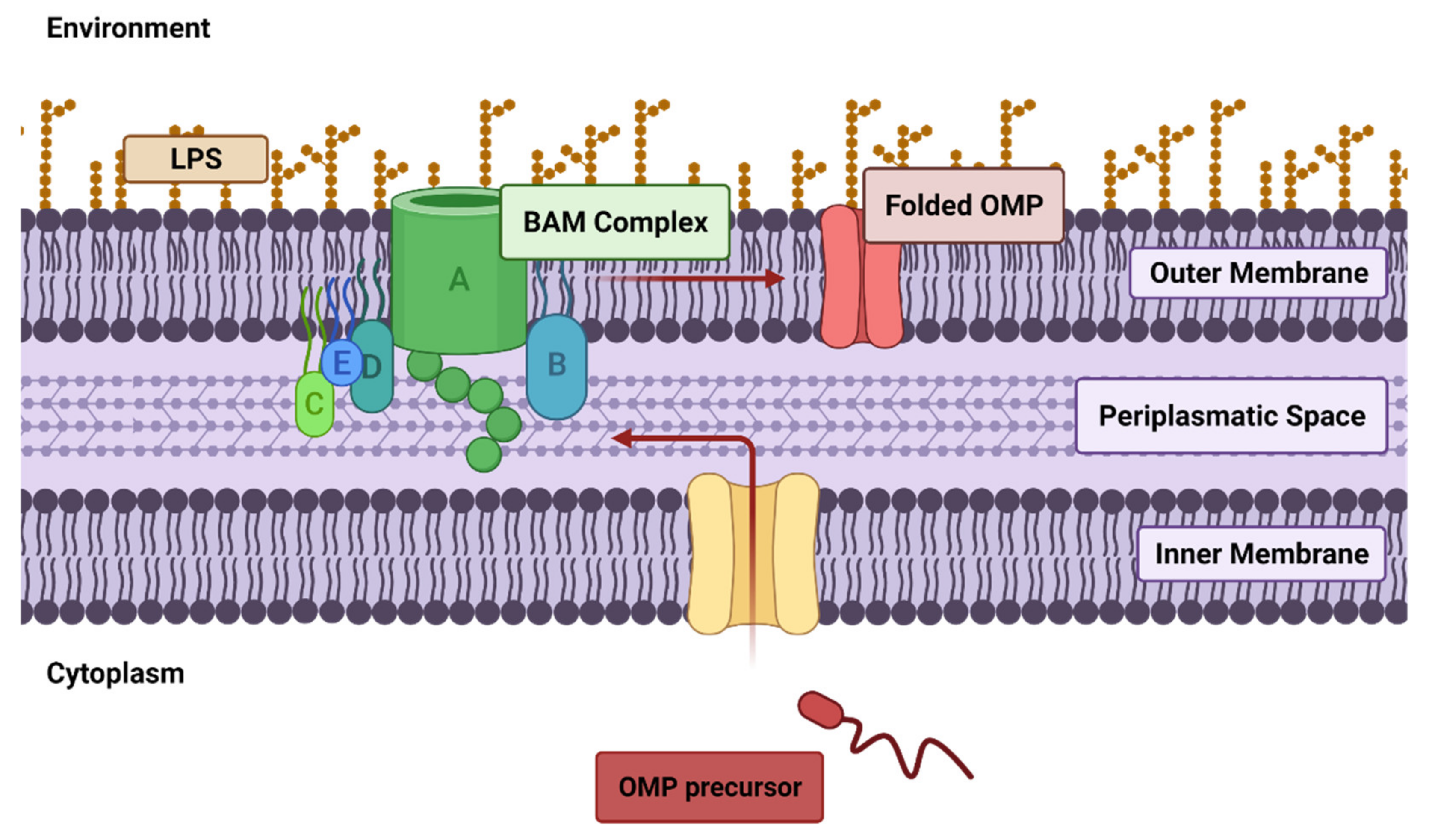
3.2. Drug Resistance Involving the Outer Membrane (OM)
3.3. Carbohydrate-Based Antibacterial Vaccines
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Shi, Y.; Jiang, Y.; Wang, S.; Wang, X.; Zhu, G. Biogeographic Distribution of Comammox Bacteria in Diverse Terrestrial Habitats. Sci. Total Environ. 2020, 717, 137257. [Google Scholar] [CrossRef] [PubMed]
- Mckay, R.M.L.; Prášil, O.; Pechar, L.; Lawrenz, E.; Rozmarynowycz, M.J.; Bullerjahn, G.S. Freshwater Ice as Habitat: Partitioning of Phytoplankton and Bacteria between Ice and Water in Central European Reservoirs. Environ. Microbiol. Rep. 2015, 7, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Sul, W.J.; Oliver, T.A.; Ducklow, H.W.; Amaral-Zettlera, L.A.; Sogin, M.L. Marine Bacteria Exhibit a Bipolar Distribution. Proc. Natl. Acad. Sci. USA 2013, 110, 2342–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louca, S.; Mazel, F.; Doebeli, M.; Parfrey, L.W. A Census-Based Estimate of Earth’s Bacterial and Archaeal Diversity. PLoS Biol. 2019, 17, e3000106. [Google Scholar] [CrossRef] [Green Version]
- Barney, B.M. Aerobic Nitrogen-Fixing Bacteria for Hydrogen and Ammonium Production: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2020, 104, 1383–1399. [Google Scholar] [CrossRef]
- Reis, V.M.; Teixeira, K.R.D.S. Nitrogen Fixing Bacteria in the Family Acetobacteraceae and Their Role in Agriculture. J. Basic Microbiol. 2015, 55, 931–949. [Google Scholar] [CrossRef]
- Kurth, J.M.; den Camp, H.J.M.O.; Welte, C.U. Several Ways One Goal—Methanogenesis from Unconventional Substrates. Appl. Microbiol. Biotechnol. 2020, 104, 6839–6854. [Google Scholar] [CrossRef]
- Tropini, C.; Earle, K.A.; Huang, K.C.; Sonnenburg, J.L. The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host Microbe 2017, 21, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, R.; Tema, G.; Cornu, J.N.; Fusco, F.; McVary, K.; Tubaro, A.; de Nunzio, C. The Urothelium, the Urinary Microbioma and Men LUTS: A Systematic Review. Minerva Urol. E Nefrol. 2020, 72, 712–722. [Google Scholar] [CrossRef]
- Annalisa, N.; Alessio, T.; Claudette, T.D.; Erald, V.; Antonino, D.L.; Nicola, D.D. Gut Microbioma Population: An Indicator Really Sensible to Any Change in Age, Diet, Metabolic Syndrome, and Life-Style. Mediat. Inflamm. 2014, 2014, 901308. [Google Scholar] [CrossRef]
- Requena, T.; Velasco, M. The Human Microbiome in Sickness and in Health. Rev. Clínica Española 2021, 221, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Ladhoff, A.; Pernthaler, A.; Swidsinski, S.; Loening–Baucke, V.; Ortner, M.; Weber, J.; Hoffmann, U.; Schreiber, S.; Dietel, M.; et al. Mucosal Flora in Inflammatory Bowel Disease. Gastroenterology 2002, 122, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, A.L.; Stagg, A.J.; Frame, M.; Graffner, H.; Glise, H.; Falk, P.; Kamm, M.A. The Role of the Gut Flora in Health and Disease, and Its Modification as Therapy. Aliment. Pharmacol. Ther. 2002, 16, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.; Monif, G.R.G. Understanding the Bacterial Flora of the Female Genital Tract. Clin. Infect. Dis. 2001, 32, e69–e77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The Human Skin Microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Kataoka, K. The Intestinal Microbiota and Its Role in Human Health and Disease. J. Med. Investig. 2016, 63, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Kåhrström, C.T.; Pariente, N.; Weiss, U. Intestinal Microbiota in Health and Disease. Nature 2016, 535, 47. [Google Scholar] [CrossRef] [Green Version]
- Patterson, E.E.; Ryan, P.M.; Cryan, J.F.; Dinan, T.G.; Paul Ross, R.; Fitzgerald, G.F.; Stanton, C.E. Gut Microbiota, Obesity and Diabetes. Postgrad. Med. J. 2016, 92, 286–300. [Google Scholar] [CrossRef]
- Dabke, K.; Hendrick, G.; Devkota, S. The Gut Microbiome and Metabolic Syndrome. J. Clin. Investig. 2019, 129, 4050–4057. [Google Scholar] [CrossRef]
- Spielman, L.J.; Gibson, D.L.; Klegeris, A. Unhealthy Gut, Unhealthy Brain: The Role of the Intestinal Microbiota in Neurodegenerative Diseases. Neurochem. Int. 2018, 120, 149–163. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Kesika, P.; Suganthy, N.; Sivamaruthi, B.S.; Chaiyasut, C. Role of Gut-Brain Axis, Gut Microbial Composition, and Probiotic Intervention in Alzheimer’s Disease. Life Sci. 2021, 264, 118627. [Google Scholar] [CrossRef]
- Mirza, A.; Forbes, J.D.; Zhu, F.; Bernstein, C.N.; Van Domselaar, G.; Graham, M.; Waubant, E.; Tremlett, H. The Multiple Sclerosis Gut Microbiota: A Systematic Review. Mult. Scler. Relat. Disord. 2020, 37, 101427. [Google Scholar] [CrossRef]
- Riedel, S.; Morse, S.A.; Mietzner, T.; Miller, S. Jawetz Melnick & Adelbergs Medical Microbiology, 28th ed.; McGraw Hill: Columbus, OH, USA, 2019. [Google Scholar]
- Madigan, M.T.; Martinko, J.M.; Bender, K.S.; Buckley, D.H.; Stahl, D.A. Brock Biology of Microorganisms, 14th ed.; Pearson Education, Inc.: London, UK, 2015. [Google Scholar]
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Sáenz, J.P.; Grosser, D.; Bradley, A.S.; Lagny, T.J.; Lavrynenko, O.; Broda, M.; Simons, K. Hopanoids as Functional Analogues of Cholesterol in Bacterial Membranes. Proc. Natl. Acad. Sci. USA 2015, 112, 11971–11976. [Google Scholar] [CrossRef] [Green Version]
- Mbamala, E.C.; Ben-Shaul, A.; May, S. Domain Formation Induced by the Adsorption of Charged Proteins on Mixed Lipid Membranes. Biophys. J. 2005, 88, 1702–1714. [Google Scholar] [CrossRef] [Green Version]
- Whited, A.M.; Johs, A. The Interactions of Peripheral Membrane Proteins with Biological Membranes. Chem. Phys. Lipids 2015, 192, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Tanghe, A.; van Dijck, P.; Thevelein, J.M. Why Do Microorganisms Have Aquaporins? Trends Microbiol. 2006, 14, 78–85. [Google Scholar] [CrossRef]
- Horne, J.E.; Brockwell, D.J.; Radford, S.E. Role of the Lipid Bilayer in Outer Membrane Protein Folding in Gram-Negative Bacteria. J. Biol. Chem. 2020, 295, 10340–10367. [Google Scholar] [CrossRef]
- Henderson, J.C.; Zimmerman, S.M.; Crofts, A.A.; Boll, J.M.; Kuhns, L.G.; Herrera, C.M.; Trent, M.S. The Power of Asymmetry: Architecture and Assembly of the Gram-Negative Outer Membrane Lipid Bilayer. Annu. Rev. Microbiol. 2016, 70, 255–278. [Google Scholar] [CrossRef]
- Chattaway, F. Microbial Cell Walls and Membranes. Biochem. Educ. 1982, 10, 33. [Google Scholar] [CrossRef]
- Mengin-Lecreulx, D.; Lemaitre, B. Structure and Metabolism of Peptidoglycan and Molecular Requirements Allowing Its Detection by the Drosophila Innate Immune System. J. Endotoxin Res. 2005, 11, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmer, W.; Höltje, J.V. Morphogenesis of Escherichia coli. Curr. Opin. Microbiol. 2001, 4, 625–633. [Google Scholar] [CrossRef]
- Moulder, J.W. Why Is Chlamydia Sensitive to Penicillin in the Absence of Peptidoglycan? Infect. Agents. Dis. 1993, 2, 87–99. [Google Scholar] [PubMed]
- Tamura, A.; Ohashi, N.; Urakami, H.; Miyamura, S. Classification of Rickettsia tsutsugamushi in a New Genus, Orientia Gen. Nov., as Orientia tsutsugamushi Comb. Nov. Int. J. Syst. Bacteriol. 1995, 45, 589–591. [Google Scholar] [CrossRef] [Green Version]
- Schleifer, K.H.; Kandler, O. Peptidoglycan Types of Bacterial Cell Walls and Their Taxonomic Implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Doyle, R.J.; Dziarski, R. The Bacterial Cell: Peptidoglycan. J. Clin. Pathol. 2002, 1, 137–153. [Google Scholar] [CrossRef] [Green Version]
- Dramsi, S.; Magnet, S.; Davison, S.; Arthur, M. Covalent Attachment of Proteins to Peptidoglycan. FEMS Microbiol. Rev. 2008, 32, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, F.C.; Baddiley, J. A Continuum of Anionic Charge: Structures and Functions of d -Alanyl-Teichoic Acids in Gram-Positive Bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 686–723. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial Peptidoglycan (Murein) Hydrolases. FEMS Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef] [Green Version]
- Dziarski, R. Recognition of Bacterial Peptidoglycan by the Innate Immune System. Cell. Mol. Life Sci. 2003, 60, 1793–1804. [Google Scholar] [CrossRef]
- Rosenthal, R.S.; Dziarski, R. Isolation of Peptidoglycan and Soluble Peptidoglycan Fragments. Methods 1994, 235, 253–285. [Google Scholar]
- Park, J.T.; Uehara, T. How Bacteria Consume Their Own Exoskeletons (Turnover and Recycling of Cell Wall Peptidoglycan). Microbiol. Mol. Biol. Rev. 2008, 72, 211–227. [Google Scholar] [CrossRef] [Green Version]
- Reith, J.; Mayer, C. Peptidoglycan Turnover and Recycling in Gram-Positive Bacteria. Appl. Microbiol. Biotechnol. 2011, 92, 1–11. [Google Scholar] [CrossRef]
- Mayer, C.; Kluj, R.M.; Mühleck, M.; Walter, A.; Unsleber, S.; Hottmann, I.; Borisova, M. Bacteria’s Different Ways to Recycle Their Own Cell Wall. Int. J. Med. Microbiol. 2019, 309, 151326. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How Antibiotics Kill Bacteria: From Targets to Networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, W. Structural Variation in the Glycan Strands of Bacterial Peptidoglycan. FEMS Microbiol. Rev. 2008, 32, 287–306. [Google Scholar] [CrossRef]
- Zipperle, G.F.; Ezzell, J.W.; Doyle, R.J. Glucosamine Substitution and Muramidase Susceptibility in Bacillus Anthracis. Can. J. Microbiol. 1984, 30, 553–559. [Google Scholar] [CrossRef]
- Hayashi, H.; Araki, Y.; Ito, E. Occurrence of Glucosamine Residues with Free Amino Groups in Cell Wall Peptidoglycan from Bacilli as a Factor Responsible for Resistance to Lysozyme. J. Bacteriol. 1973, 113, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Atrih, A.; Bacher, G.; Allmaier, G.; Williamson, M.P.; Foster, S.J. Analysis of Peptidoglycan Structure from Vegetative Cells of Bacillus subtilis 168 and Role of PBP 5 in Peptidoglycan Maturation. J. Bacteriol. 1999, 181, 3956–3966. [Google Scholar] [CrossRef] [Green Version]
- Ohno, N.; Yadomae, T.; Miyazaki, T. Identification of 2-Amino-2-Deoxyglucose Residues in the Peptidoglucan of Streptococcus pneumoniae. Carbohydr. Res. 1982, 107, 152–155. [Google Scholar] [CrossRef]
- Vollmer, W.; Tomasz, A. The PgdA Gene Encodes for a Peptidoglycan N-Acetylglucosamine Deacetylase in Streptococcus pneumoniae. J. Biol. Chem. 2000, 275, 20496–20501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, J.S.; Parajuli, P.; Ruda, A.; Li, J.; Pohane, A.A.; Zamakhaeva, S.; Rahman, M.M.; Chang, J.C.; Gogos, A.; Kenner, C.W.; et al. PplD Is a De-N-Acetylase of the Cell Wall Linkage Unit of Streptococcal Rhamnopolysaccharides. Nat. Commun. 2022, 13, 590. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Sharon, N.; Assaf, Y.; Chipman, D.M. Mechanism of Lysozyme Catalysis: Role of Ground-State Strain in Subsite D in Hen Egg-White and Human Lysozymes. Biochemistry 1977, 16, 423–431. [Google Scholar] [CrossRef]
- Ragland, S.A.; Criss, A.K. From Bacterial Killing to Immune Modulation: Recent Insights into the Functions of Lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef]
- Amano, K.; Hayashi, H.; Araki, Y.; Ito, E. The Action of Lysozyme on Peptidoglycan with N-Unsubstituted Glucosamine Residues. Eur. J. Biochem. 1977, 76, 299–307. [Google Scholar] [CrossRef]
- Amano, K.-I.; Araki, Y.; Ito, E. Effect of N-Acyl Substitution at Glucosamine Residues on Lysozyme-Catalyzed Hydrolysis of Cell-Wall Peptidoglycan and Its Oligosaccharides. Eur. J. Biochem. 1980, 107, 547–553. [Google Scholar] [CrossRef]
- Westmacott, D.; Perkins, H.R. Effects of Lysozyme on Bacillus cereus 569: Rupture of Chains of Bacteria and Enhancement of Sensitivity to Autolysins. J. Gen. Microbiol. 1979, 115, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Adam, A.; Petit, J.F.; Wietzerbin-Falszpan, J.; Sinay, P.; Thomas, D.W.; Lederer, E. L’acide N-Glycolyl-Muramique, Constituant Des Parois de Mycobacterium smegmatis: Identification Par Spectrometrie de Masse. FEBS Lett. 1969, 4, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K.; Kudo, T.; Suzuki, K.I.; Nakase, T. A New Rapid Method of Glycolate Test by Diethyl Ether Extraction, Which Is Applicable to a Small Amount of Bacterial Cells of Less than One Milligram. J. Gen. Appl. Microbiol. 1999, 45, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Sutcliffe, I.C. Cell Envelope Composition and Organisation in the Genus Rhodococcus. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 1998, 74, 49–58. [Google Scholar] [CrossRef]
- Azuma, I.; Thomas, D.; Adam, A.; Ghuysen, J.; Bonaly, R.; Petit, J.; Lederer, E. Occurence of N-Glucosylmuramic Acid in Bacterial Cell Walls. Biochim. Biophys. Acta 1970, 20, 444–451. [Google Scholar] [CrossRef]
- Waksman, S.A.; Schatz, A.; Reynolds, D.M. Production of Antibiotic Substances by Actinomycetes. Ann. N. Y. Acad. Sci. 2010, 1213, 112–124. [Google Scholar] [CrossRef]
- Brennan, P.J.; Nikaido, H. The Envelope of Mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. [Google Scholar] [CrossRef]
- Sekiguchi, J.; Akeo, K.; Yamamoto, H.; Khasanov, F.K.; Alonso, J.C.; Kuroda, A. Nucleotide Sequence and Regulation of a New Putative Cell Wall Hydrolase Gene, CwlD, Which Affects Germination in Bacillus subtilis. J. Bacteriol. 1995, 177, 5582–5589. [Google Scholar] [CrossRef] [Green Version]
- Popham, D.L.; Helin, J.; Costello, C.E.; Setlow, P. Muramic Lactam in Peptidoglycan of Bacillus subtilis Spores Is Required for Spore Outgrowth but Not for Spore Dehydration or Heat Resistance. Proc. Natl. Acad. Sci. USA 1996, 93, 15405–15410. [Google Scholar] [CrossRef] [Green Version]
- Woodward, R.; Yi, W.; Li, L.; Zhao, G.; Eguchi, H.; Sridhar, P.R.; Guo, H.; Song, J.K.; Motari, E.; Cai, L.; et al. In Vitro Bacterial Polysaccharide Biosynthesis: Defining the Functions of Wzy and Wzz. Nat. Chem. Biol. 2010, 6, 418–423. [Google Scholar] [CrossRef]
- Chirakkal, H.; O’Rourke, M.; Atrih, A.; Foster, S.J.; Moir, A. Analysis of Spore Cortex Lytic Enzymes and Related Proteins in Bacillus subtilis Endoscope Germination. Microbiology 2002, 148, 2383–2392. [Google Scholar] [CrossRef] [Green Version]
- Abrams, A. O-Acetyl Groups in the Cell Wall of Streptococcus faecalis. J. Biol. Chem. 1958, 230, 949–959. [Google Scholar] [CrossRef]
- Brumfitt, W.; Wardlaw, A.C.; Park, J.T. Development of Lysozyme-Resistance in Micrococcus lysodiekticus and Its Association with an Increased O-Acetyl Content of the Cell Wall. Nature 1958, 181, 1783–1784. [Google Scholar] [CrossRef]
- Clarke, A.J.; Dupont, C. O-Acetylated Peptidoglycan: Its Occurrence, Pathobiological Significance, and Biosynthesis. Can. J. Microbiol. 1992, 38, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J. Extent of Peptidoglycan O Acetylation in the Tribe Proteeae. J. Bacteriol. 1993, 175, 4550–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weadge, J.T.; Pfeffer, J.M.; Clarke, A.J. Identification of a New Family of Enzymes with Potential O-Acetylpeptidoglycan Esterase Activity in Both Gram-Positive and Gram-Negative Bacteria. BMC Microbiol. 2005, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, C.; Clarke, A.J. Evidence for N → O Acetyl Migration as the Mechanism for O Acetylation of Peptidoglycan in Proteus mirabilis. J. Bacteriol. 1991, 173, 4318–4324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Götz, F. Why Are Pathogenic Staphylococci so Lysozyme Resistant? The Peptidoglycan O-Acetyltransferase OatA Is the Major Determinant for Lysozyme Resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787. [Google Scholar] [CrossRef]
- Dupont, C.; Clarke, A.J. Dependence of Lysozyme-Catalyzed Solubilization of Proteus mirabilis Peptidoglycan on the Extent of O-Acetylation. Eur. J. Biochem. 1991, 195, 763–769. [Google Scholar] [CrossRef]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The Penicillin-Binding Proteins: Structure and Role in Peptidoglycan Biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [Green Version]
- Burghaus, P.; Johannsen, L.; Naumann, D.; Labischinski, H.; Bradaczek, H.; Giesbrecht, P. The Influence of Different Antibiotics on the Degree of O-Acetylation of Staphylococcal Cell Walls. In The Target of Penicillin; De Gruyter: Berlin, Germany, 1983; pp. 317–322. [Google Scholar] [CrossRef]
- Sidow, T.; Johannsen, L.; Labischinski, H. Penicillin-Induced Changes in the Cell Wall Composition of Staphylococcus aureus before the Onset of Bacteriolysis. Arch. Microbiol. 1990, 154, 73–81. [Google Scholar] [CrossRef]
- Varki, A. Nothing in Glycobiology Makes Sense, except in the Light of Evolution. Cell 2006, 126, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Patel, D.S.; Ståhle, J.; Park, S.-J.; Kern, N.R.; Kim, S.; Lee, J.; Cheng, X.; Valvano, M.A.; Holst, O.; et al. CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J. Chem. Theory Comput. 2019, 15, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Furevi, A. Structural and Conformational Analysis of Bacterial Polysaccharides Using NMR Spectroscopy. Ph.D. Thesis, Stockholm University, Stockholm, Sweden, 2022. [Google Scholar]
- Brade, H.; Opal, S.M.; Vogel, S.N.; Morrison, D.C. Endotoxin in Health and Disease; Marcel Dekker: New York, NY, USA, 1999. [Google Scholar]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, T.J.O.; Raetz, C.R.H. The Active Site of Escherichia coli UDP-N-Acetylglucosamine Acyltransferase. J. Biol. Chem. 1999, 274, 27047–27055. [Google Scholar] [CrossRef] [Green Version]
- Rietschel, E.T. Handbook of Endotoxin, Vol.1; Elsevier: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Lugtenberg, B.; Van Alphen, L. Molecular Architecture and Functioning of the Outer Membrane of Escherichia Coli and Other Gram-Negative Bacteria. BBA Rev. Biomembr. 1983, 737, 51–115. [Google Scholar] [CrossRef]
- Weintraub, A.; Zähringer, U.; Wollenweber, H.W.; Seydel, U.; Rietschel, E. Structural Characterization of the Lipid A Component of Bacteroides fragilis Strain NCTC 9343 Lipopolysaccharide. Eur. J. Biochem. 1989, 183, 425–431. [Google Scholar] [CrossRef]
- Wollenweber, H.W.; Rietschel, E.T. Analysis of Lipopolysaccharide (Lipid A) Fatty Acids. J. Microbiol. Methods 1990, 11, 195–211. [Google Scholar] [CrossRef]
- Salimath, P.V.; Weckesser, J.; Strittmatter, W.; Mayer, H. Structural Studies on the Non-Toxic Lipid A from Rhodopseudomonas sphaeroides ATCC 17023. Eur. J. Biochem. 1983, 136, 195–200. [Google Scholar] [CrossRef]
- Weckesser, J.; Mayer, H. Different Lipid A Types in Lipopolysaccharides of Phototrophic and Related Non-Phototrophic Bacteria. FEMS Microbiol. Lett. 1988, 54, 143–153. [Google Scholar] [CrossRef]
- Moran, A.P.; Zähringer, U.; Seydel, U.; Scholz, D.; Stutz, P.; Rietschel, E.T. Structural Analysis of the Lipid A Component of Campylobacter jejuni CCUG 10936 (Serotype O:2) Lipopolysaccharide. Eur. J. Biochem. 1991, 198, 459–469. [Google Scholar] [CrossRef]
- Zhou, X.-Y.; Li, L.-X.; Zhang, Z.; Duan, S.-C.; Huang, Y.-W.; Luo, Y.-Y.; Mu, X.-D.; Chen, Z.-W.; Qin, Y.; Hu, J.; et al. Chemical Synthesis and Antigenic Evaluation of Inner Core Oligosaccharides from Acinetobacter baumannii Lipopolysaccharide. Angew. Chem. Int. Ed. 2022, 61, e202204420. [Google Scholar] [CrossRef]
- Vaara, M. Agents That Increase the Permeability of the Outer Membrane. Microbiol. Rev. 1992, 56, 395–411. [Google Scholar] [CrossRef]
- Liu, B.; Furevi, A.; Perepelov, A.V.; Guo, X.; Cao, H.; Wang, Q.; Reeves, P.R.; Knirel, Y.A.; Wang, L.; Widmalm, G. Structure and Genetics of Escherichia coli O Antigens. FEMS Microbiol. Rev. 2020, 44, 655–683. [Google Scholar] [CrossRef] [Green Version]
- Lundborg, M.; Modhukur, V.; Widmalm, G. Glycosyltransferase Functions of E. coli O-Antigens. Glycobiology 2009, 20, 366–368. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, J.; Wang, X.; Xu, C.; Han, T.; Guo, X. Genetic Characterization of the O-Antigen and Development of a Molecular Serotyping Scheme for Enterobacter cloacae. Front. Microbiol. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Toukach, P.V.; Egorova, K.S. Carbohydrate Structure Database Merged from Bacterial, Archaeal, Plant and Fungal Parts. Nucleic Acids Res. 2016, 44, D1229–D1236. [Google Scholar] [CrossRef]
- Brown, S.; Santa Maria, J.P.; Walker, S. Wall Teichoic Acids of Gram-Positive Bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef] [Green Version]
- Weidenmaier, C.; Peschel, A. Teichoic Acids and Related Cell-Wall Glycopolymers in Gram-Positive Physiology and Host Interactions. Nat. Rev. Microbiol. 2008, 6, 276–287. [Google Scholar] [CrossRef]
- Swoboda, J.G.; Campbell, J.; Meredith, T.C.; Walker, S. Wall Teichoic Acid Function, Biosynthesis, and Inhibition. ChemBioChem 2009, 11, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, K.; Owens, T.W.; Kahne, D.; Walker, S. Substrate Preferences Establish the Order of Cell Wall Assembly in Staphylococcus aureus. J. Am. Chem. Soc. 2018, 140, 2442–2445. [Google Scholar] [CrossRef]
- Araki, Y.; Ito, E. Linkage Units in Cell Walls of Gram-Positive Bacteria. Crit. Rev. Microbiol. 1989, 17, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Kojima, N.; Araki, Y.; Ito, E. Structure of the Linkage Units between Ribitol Teichoic Acids and Peptidoglycan. J. Bacteriol. 1985, 161, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Endl, J.; Seidl, H.P.; Fiedler, F.; Schleider, K.H. Chemical Composition and Structure of Cell Wall Teichoic Acids of Staphylococci. Arch. Microbiol. 1983, 135, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Endl, J.; Seidl, P.H.; Fiedler, F.; Schleifer, K.H. Determination of Cell Wall Teichoic Acid Structure of Staphylococci by Rapid Chemical and Serological Screening Methods. Arch. Microbiol. 1984, 137, 272–280. [Google Scholar] [CrossRef]
- Naumova, I.B.; Shashkov, A.S.; Tul’Skaya, E.M.; Streshinskaya, G.M.; Kozlova, Y.I.; Potekhina, N.V.; Evtushenko, L.I.; Stackebrandt, E. Cell Wall Teichoic Acids: Structural Diversity, Species Specificity in the Genus Nocardiopsis, and Chemotaxonomic Perspective. FEMS Microbiol. Rev. 2001, 25, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Shashkov, A.S.; Potekhina, N.V.; Naumova, I.B.; Evtushenko, L.I.; Widmalm, G. Cell Wall Teichoic Acids of Actinomadura Viridis VKM Ac-1315T. Eur. J. Biochem. 1999, 262, 688–695. [Google Scholar] [CrossRef]
- Kristian, S.A.; Datta, V.; Weidenmaier, C.; Kansal, R.; Fedtke, I.; Peschel, A.; Gallo, R.L.; Nizet, V. D-Alanylation of Teichoic Acids Promotes Group A Streptococcus Antimicrobial Peptide Resistance, Neutrophil Survival, and Epithelial Cell Invasion. J. Bacteriol. 2005, 187, 6719–6725. [Google Scholar] [CrossRef] [Green Version]
- Kristian, S.A.; Lauth, X.; Nizet, V.; Goetz, F.; Neumeister, B.; Peschel, A.; Landmann, R. Alanylation of Teichoic Acids Protects Staphylococcus aureus against Toll-like Receptor 2-Dependent Host Defense in a Mouse Tissue Cage Infection Model. J. Infect. Dis. 2003, 188, 414–423. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.V.; Kristian, S.A.; Weidenmaier, C.; Faigle, M.; Van Kessel, K.P.M.; Van Strijp, J.A.G.; Goütz, F.; Neumeister, B.; Peschel, A. Staphylococcus aureus Strains Lacking D-Alanine Modifications of Teichoic Acids Are Highly Susceptible to Human Neutrophil Killing and Are Virulence Attenuated in Mice. J. Infect. Dis. 2002, 186, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Götz, F. Inactivation of the Dlt Operon in Staphylococcus aureus Confers Sensitivity to Defensins, Protegrins, and Other Antimicrobial Peptides. J. Biol. Chem. 1999, 274, 8405–8410. [Google Scholar] [CrossRef] [Green Version]
- Peschel, A.; Vuong, C.; Otto, M.; Gotz, F. The D-Alanine Residues of Staphylococcus aureus Teichoic Acids Alter the Susceptibility to Vancomycin and the Activity of Autolytic Enzymes. Antimicrob. Agents Chemother. 2000, 44, 2845–2847. [Google Scholar] [CrossRef] [Green Version]
- Vadyvaloo, V.; Arous, S.; Gravesen, A.; Héchard, Y.; Chauhan-Haubrock, R.; Hastings, J.W.; Rautenbach, M. Cell-Surface Alterations in Class IIa Bacteriocin-Resistant Listeria Monocytogenes Strains. Microbiology 2004, 150, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.; Loach, D.M.; Alqumber, M.; Rockel, C.; Hermann, C.; Pfitzenmaier, M.; Tannock, G.W. D-Alanyl Ester Depletion of Teichoic Acids in Lactobacillus reuteri 100-23 Results in Impaired Colonization of the Mouse Gastrointestinal Tract. Environ. Microbiol. 2007, 9, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Xia, G.; Luhachack, L.G.; Campbell, J.; Meredith, T.C.; Chen, C.; Winstel, V.; Gekeler, C.; Irazoqui, J.E.; Peschel, A.; et al. Methicillin Resistance in Staphylococcus aureus Requires Glycosylated Wall Teichoic Acids. Proc. Natl. Acad. Sci. USA 2012, 109, 18909–18914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, W. Glycolipids, Phosphoglycolipids, and Sulfoglycolipids. In Handbook of Lipid Research, Vol.6; Morris, K., Ed.; Prenum Press: New York, NY, USA, 1990; pp. 123–234. [Google Scholar]
- Gründling, A.; Schneewind, O. Synthesis of Glycerol Phosphate Lipoteichoic Acid in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2007, 104, 8478–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, A.J.; Karatsa-Dodgson, M.; Gründling, A. Two-Enzyme Systems for Glycolipid and Polyglycerolphosphate Lipoteichoic Acid Synthesis in Listeria Monocytogenes. Mol. Microbiol. 2009, 74, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Mäki, M.; Renkonen, R. Biosynthesis of 6-Deoxyhexose Glycans in Bacteria. Glycobiology 2004, 14, 1R–15R. [Google Scholar] [CrossRef] [Green Version]
- Facklam, R. What Happened to the Streptococci: Overview of Taxonomic and Nomenclature Changes. Clin. Microbiol. Rev. 2002, 15, 613–630. [Google Scholar] [CrossRef] [Green Version]
- Köhler, W. The Present State of Species within the Genera Streptococcus and Enterococcus. Int. J. Med. Microbiol. 2007, 297, 133–150. [Google Scholar] [CrossRef]
- Neelamegham, S.; Aoki-Kinoshita, K.; Bolton, E.; Frank, M.; Lisacek, F.; Lütteke, T.; O’Boyle, N.; Packer, N.H.; Stanley, P.; Toukach, P.; et al. Updates to the Symbol Nomenclature for Glycans Guidelines. Glycobiology 2019, 29, 620–624. [Google Scholar] [CrossRef]
- McMillan, D.J.; Vu, T.; Bramhachari, P.V.; Kaul, S.Y.; Bouvet, A.; Shaila, M.S.; Karmarkar, M.G.; Sriprakash, K.S. Molecular Markers for Discriminating Streptococcus pyogenes and S. dysgalactiae Subspecies equisimilis. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 585–589. [Google Scholar] [CrossRef]
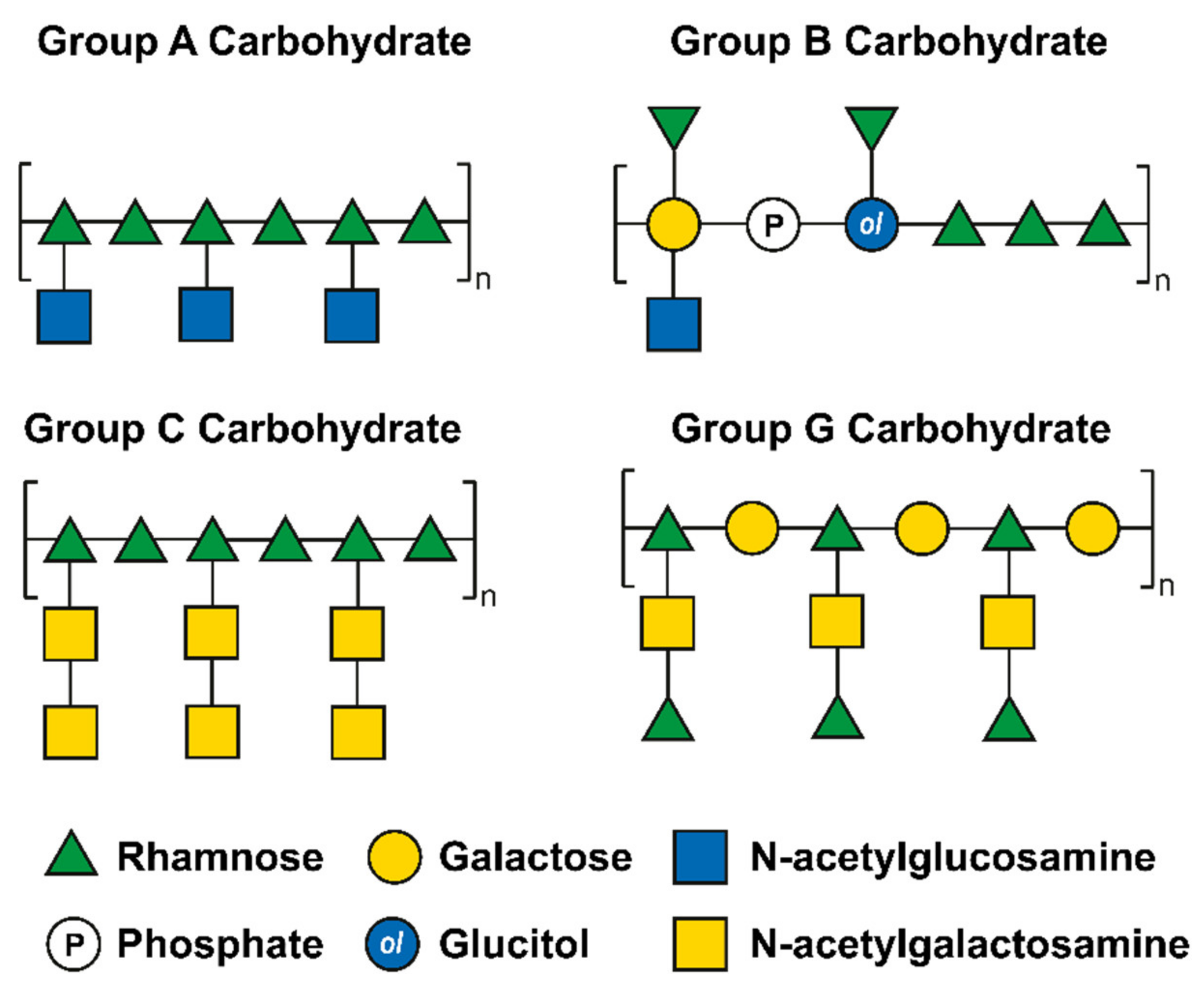
- Swanson, J.; Gotschlich, E.C. Electron Microscopic Studies on Streptococci II. Group A Carbohydrate. J. Exp. Med. 1973, 138, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Wagner, B.; Ryc, M. An Electron Microscopic Study of the Location of Peptidoglycan in Group A and C Streptococcal Cell Walls. J. Gen. Microbiol. 1978, 108, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.; Wagner, M.; Kubin, V.; Ryc, M. Immunoelectron Microscopic Study of the Location of Group-Specific and Protein Type-Specific Antigens of Group B Streptococci. J. Gen. Microbiol. 1980, 118, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutcliffe, I.C.; Black, G.W.; Harrington, D.J. Bioinformatic Insights into the Biosynthesis of the Group B Carbohydrate in Streptococcus agalactiae. Microbiology 2008, 154, 1354–1363. [Google Scholar] [CrossRef] [Green Version]
- Chapot-Chartier, M.P. Interactions of the Cell-Wall Glycopolymers of Lactic Acid Bacteria with Their Bacteriophages. Front. Microbiol. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, L.E.; Gilmore, M.S. The Capsular Polysaccharide of Enterococcus faecalis and Its Relationship to Other Polysaccharides in the Cell Wall. Proc. Natl. Acad. Sci. USA 2002, 99, 1574–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, F.; Singh, K.V.; Bourgogne, A.; Zeng, J.; Murray, B.E. Further Characterization of the Epa Gene Cluster and Epa Polysaccharides of Enterococcus faecalis. Infect. Immun. 2009, 77, 3759–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurlow, L.R.; Thomas, V.C.; Hancock, L.E. Capsular Polysaccharide Production in Enterococcus faecalis and Contribution of CpsF to Capsule Serospecificity. J. Bacteriol. 2009, 191, 6203–6210. [Google Scholar] [CrossRef] [Green Version]
- Theilacker, C.; Holst, O.; Lindner, B.; Huebner, J.; Kaczyński, Z. The Structure of the Wall Teichoic Acid Isolated from Enterococcus faecalis Strain 12030. Carbohydr. Res. 2012, 354, 106–109. [Google Scholar] [CrossRef]
- Pritchard, D.G.; Coligan, J.E.; Speed, S.E.; Gray, B.M. Carbohydrate Fingerprints of Streptococcal Cells. J. Clin. Microbiol. 1981, 13, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Mistou, M.Y.; Sutcliffe, I.C.; Van Sorge, N.M. Bacterial Glycobiology: Rhamnose-Containing Cell Wall Polysaccharides in Gram-Positive Bacteria. FEMS Microbiol. Rev. 2016, 40, 464–479. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M. The Lysis of Group A Hemolytic Streptococci by Extracellular Enzymes of Streptomyces albus. J. Exp. Med. 1952, 96, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Tsukioka, Y.; Yamashita, Y.; Nakano, Y.; Oho, T.; Koga, T. Identification of a Fourth Gene Involved in DTDP-Rhamnose Synthesis in Streptococcus mutans. J. Bacteriol. 1997, 179, 4411–4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliot, É.; Dramsi, S.; Chapot-Chartier, M.P.; Courtin, P.; Kulakauskas, S.; Péchoux, C.; Trieu-Cuot, P.; Mistou, M.Y. Role of the Group B Antigen of Streptococcus agalactiae: A Peptidoglycan-Anchored Polysaccharide Involved in Cell Wall Biogenesis. PLoS Pathog. 2012, 8, e1002756. [Google Scholar] [CrossRef] [PubMed]
- Van der Beek, S.L.; Le Breton, Y.; Ferenbach, A.T.; Chapman, R.N.; van Aalten, D.M.F.; Navratilova, I.; Boons, G.J.; Mciver, K.S.; van Sorge, N.M.; Dorfmueller, H.C. GacA Is Essential for Group A Streptococcus and Defines a New Class of Monomeric DTDP-4-Dehydrorhamnose Reductases (RmlD). Mol. Microbiol. 2015, 98, 946–962. [Google Scholar] [CrossRef] [Green Version]
- Van Sorge, N.M.; Cole, J.N.; Kuipers, K.; Henningham, A.; Aziz, R.K.; Kasirer-Friede, A.; Lin, L.; Berends, E.T.M.; Davies, M.R.; Dougan, G.; et al. The Classical Lancefield Antigen of Group A Streptococcus Is a Virulence Determinant with Implications for Vaccine Design. Cell Host Microbe 2014, 15, 729–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, R.M. Studies on Bacteriophages of Hemolytic Streptococci. I. Factors Influencing the Interaction of Phage and Susceptible Host Cell. J. Exp. Med. 1957, 106, 365–384. [Google Scholar] [CrossRef] [Green Version]
- Fischetti, V.A.; Zabriskie, J.B. Studies on Streptococcal Bacteriophages II. Adsorpion Studies on Group A and Group C Streptococcal Bacteriophages. J. Exp. Med. 1967, 127, 489–505. [Google Scholar] [CrossRef]
- Shibata, Y.; Yamashita, Y.; Van Der Ploeg, J.R. The Serotype-Specific Glucose Side Chain of Rhamnose-Glucose Polysaccharides Is Essential for Adsorption of Bacteriophage M102 to Streptococcus mutans. FEMS Microbiol. Lett. 2009, 294, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Singh, K.V.; Qin, X.; Murray, B.E.; Weinstock, G.M. Analysis of a Gene Cluster of Enterococcus faecalis Involved in Polysaccharide Biosynthesis. Infect. Immun. 2000, 68, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Alphey, M.S.; Pirrie, L.; Torrie, L.S.; Boulkeroua, W.A.; Gardiner, M.; Sarkar, A.; Maringer, M.; Oehlmann, W.; Brenk, R.; Scherman, M.S.; et al. Allosteric Competitive Inhibitors of the Glucose-1-Phosphate Thymidylyltransferase (RmlA) from Pseudomonas aeruginosa. ACS Chem. Biol. 2013, 8, 387–396. [Google Scholar] [CrossRef]
- Sewell, E.W.C.; Brown, E.D. Taking Aim at Wall Teichoic Acid Synthesis: New Biology and New Leads for Antibiotics. J. Antibiot. 2014, 67, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.; Singh, A.K.; Santa Maria, J.P.; Kim, Y.; Brown, S.; Swoboda, J.G.; Mylonakis, E.; Wilkinson, B.J.; Walker, S. Synthetic Lethal Compound Combinations Reveal a Fundamental Connection between Wall Teichoic Acid and Peptidoglycan Biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 2011, 6, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Nicolau, D.P. Carbapenems: A Potent Class of Antibiotics. Expert Opin. Pharmacother. 2008, 9, 23–37. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Wiebe, R.; Dilay, L.; Thomson, K.; Rubinstein, E.; Hoban, D.J.; Noreddin, A.M.; Karlowsky, J.A. Comparative Review of the Carbapenems. Drugs 2007, 67, 1027–1052. [Google Scholar] [CrossRef]
- Tenover, F.C. Mechanisms of Antimicrobial Resistance in Bacteria. Am. J. Med. 2006, 119, S3–S10. [Google Scholar] [CrossRef]
- Watkins, R.R. A Primer on Antimicrobials; Elsevier Inc.: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Belley, A.; McKay, G.A.; Arhin, F.F.; Sarmiento, I.; Beaulieu, S.; Fadhil, I.; Parr, T.R.; Moeck, G. Oritavancin Disrupts Membrane Integrity of Staphylococcus aureus and Vancomycin-Resistant Enterococci to Effect Rapid Bacterial Killing. Antimicrob. Agents Chemother. 2010, 54, 5369–5371. [Google Scholar] [CrossRef] [Green Version]
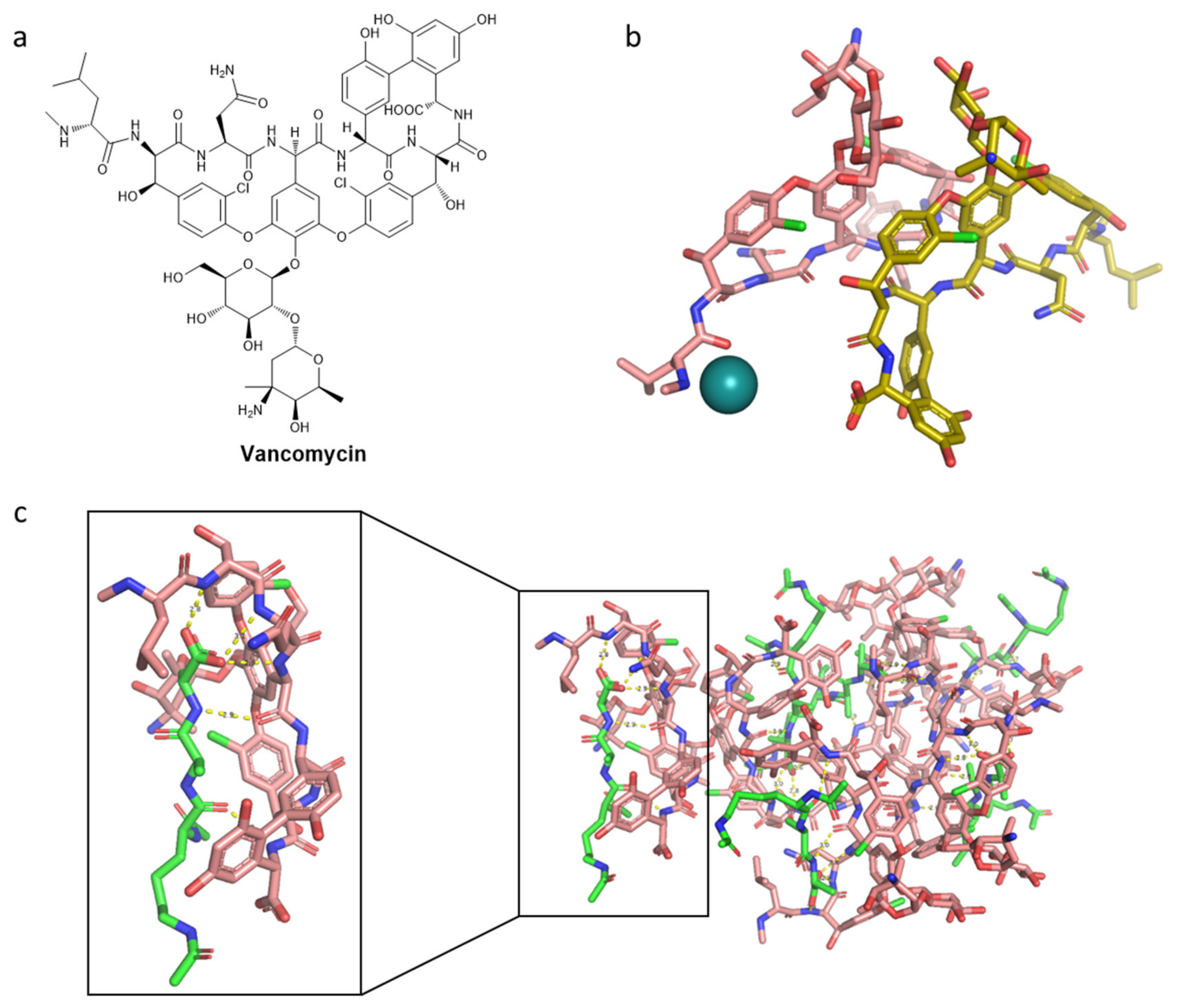
- Griffith, R.S. Vancomycin Use—An Historical Review. J. Antimicrob. Chemother. 1984, 14, 1–5. [Google Scholar] [CrossRef]
- Wilhelm, M.P.; Estes, L. Symposium on Antimicrobial Agents—Part XII. Vancomycin. Mayo Clin. Proc. 1999, 74, 928–935. [Google Scholar] [CrossRef]
- Jacobs, M.R. Emergence of Multiple Resistant Pneumococci. J. Occup. Environ. Med. 1980, 22, 559. [Google Scholar] [CrossRef]
- Aber, R.C.; Wennersten, C.; Moellering, R.C. Antimicrobial Susceptibility of Flavobacteria. Antimicrob. Agents Chemother. 1978, 14, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.S.; Matsuhashi, M.; Haskin, M.A.; Strominger, J.L. Lipid-Phosphoacetylmuramyl-Pentapeptide and Lipid-Phosphodisaccharide-Pentapeptide: Presumed Membrane Transport Intermediates in Cell Wall Synthesis. Proc. Natl. Acad. Sci. USA 1965, 53, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Perkins, H.R. Specificity of Combination between Mucopeptide Precursors and Vancomycin or Ristocetin. Biochem. J. 1969, 111, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.; Fitz-James, P.C. Some Differences in the Action of Penicillin, Bacitracin, and Vancomycin on Bacillus megaterium. J. Bacteriol. 1964, 87, 1044–1050. [Google Scholar] [CrossRef] [Green Version]
- Jordan, D.C.; Mallory, H.D. Site of Action of Action of Vancomycin on Staphylococcus aureus. Antimicrob. Agents Chemother. 1964, 10, 489–494. [Google Scholar]
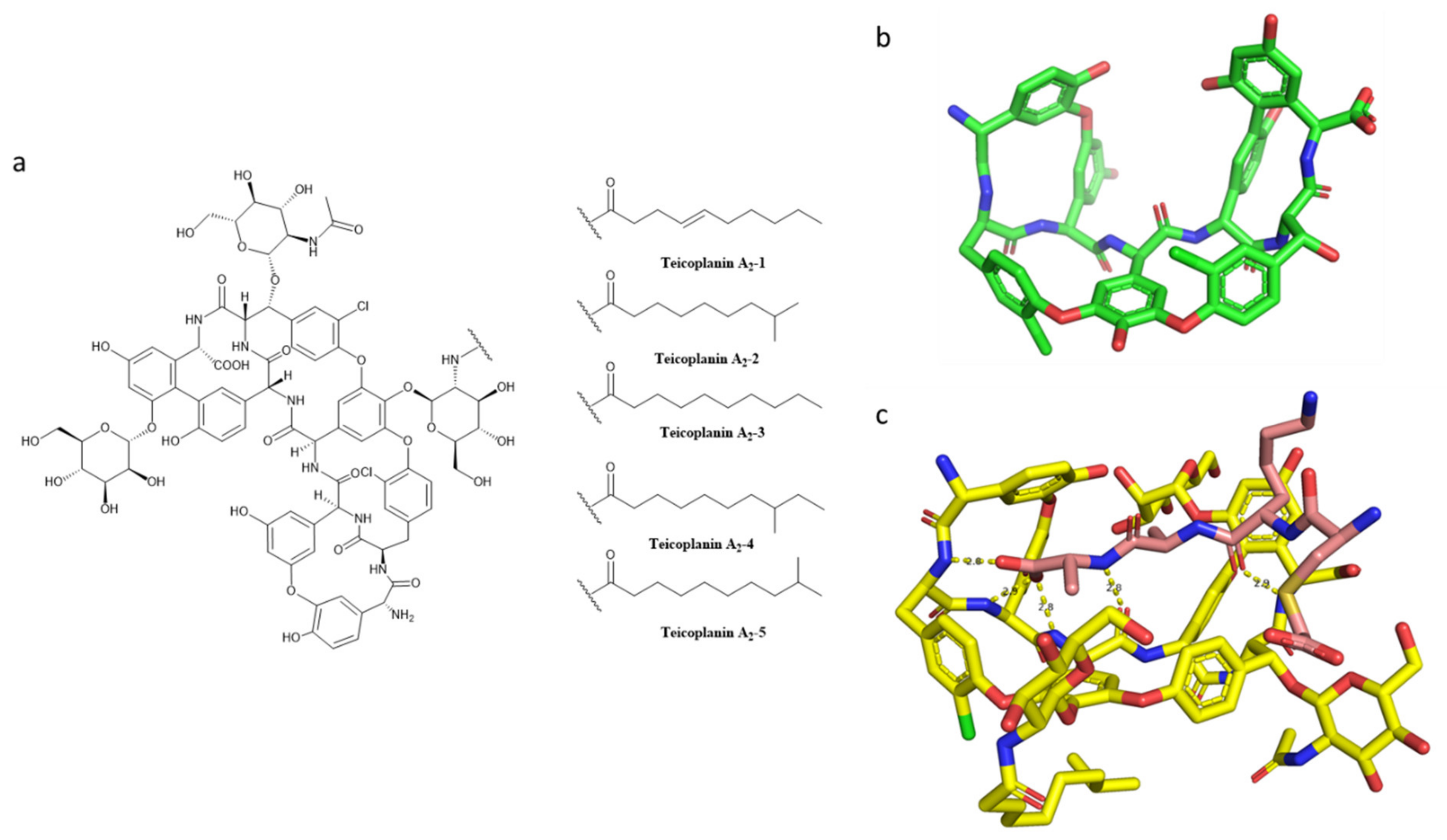
- Campoli-Richards, D.M.; Brogden, R.N.; Faulds, D. Teicoplanin. A Review of Its Antibacterial Activity, Pharmacokinetic Properties and Therapeutic Potential. Drugs 1990, 40, 449–486. [Google Scholar] [CrossRef]
- Parenti, F.; Beretta, G.; Berti, M.; Arioli, V. Teichomycins, New Antibiotics from Actinoplanes Teichomyceticus Nov. Sp. I. Description of the Producer Strain, Fermentation Studies and Biological Properties. J. Antibiot. 1978, 31, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janknegt, R. Teicoplanin in Perspective A Critical Comparison with Vancomycin. Pharm. Weekbl. Sci. 1991, 13, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Parenti, F. Structure and Mechanism of Action of Teicoplanin. J. Hosp. Infect. 1986, 7, 79–83. [Google Scholar] [CrossRef]
- Somma, S.; Gastaldo, L.; Corti, A. Teicoplanin, a New Antibiotic from Actinoplanes teichomyceticus Nov. Sp. Antimicrob. Agents Chemother. 1984, 26, 917–923. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, P.E.; Somner, E.A. Comparison of the Target Sites and Mechanisms of Action of Glycopeptide and Lipoglycodepsipeptide Antibiotics. Drugs Exp. Clin. Res. 1990, 16, 385–389. [Google Scholar]
- Barna, J.C.J.; Williams, D.H.; Stone, D.J.M.; Leung, T.W.C.; Doddrell, D.M. Structure Elucidation of the Teicoplanin Antibiotics. J. Am. Chem Soc. 1984, 106, 4895–4902. [Google Scholar] [CrossRef]
- Parenti, F. Glycopeptide Antibiotics. J. Clin. Pharmacol. 1988, 28, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Bolognino, I.; Carrieri, A.; Purgatorio, R.; Catto, M.; Caliandro, R.; Carrozzini, B.; Belviso, B.D.; Majellaro, M.; Sotelo, E.; Cellamare, S.; et al. Enantiomeric Separation and Molecular Modelling of Bioactive 4-Aryl-3,4-Dihydropyrimidin-2(1H)-One Ester Derivatives on Teicoplanin-Based Chiral Stationary Phase. Separations 2022, 9, 7. [Google Scholar] [CrossRef]
- Kirst, H.A.; Thompson, D.G.; Nicas, T.I. Historical Yearly Usage of Vancomycin. Antimicrob. Agents Chemother 1998, 42, 1303–1304. [Google Scholar] [CrossRef] [Green Version]
- Pakyz, A.L.; Macdougall, C.; Oinonen, M.; Polk, R.E. Trends in Antibacterial Use in US Academic Health Centers. Arch. Intern. Med. 2008, 168, 2254–2260. [Google Scholar] [CrossRef] [Green Version]
- Steinkraus, G.; White, R.; Friedrich, L. Vancomycin MIC Creep in Non-Vancomycin-Intermediate Staphylococcus aureus (VISA), Vancomycin-Susceptible Clinical Methicillin-Resistant S. Aureus (MRSA) Blood Isolates from 2001 05. J. Antimicrob. Chemother. 2007, 60, 788–794. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Phillips, I.; Kaniga, K. Comparative in Vitro Activity of Telavancin (TD-6424), a Rapidly Bactericidal, Concentration-Dependent Anti-Infective with Multiple Mechanisms of Action against Gram-Positive Bacteria. J. Antimicrob. Chemother. 2004, 53, 797–803. [Google Scholar] [CrossRef] [Green Version]
- Saravolatz, L.D.; Stein, G.E.; Johnson, L.B. Telavancin: A Novel Lipoglycopeptide. Clin. Infect. Dis. 2009, 49, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Leadbetter, M.R.; Adams, S.M.; Bazzini Kevin Krause, B.M.; T Lam, B.M.; Linsell, M.S.; Kelly Quast, M.; Shaw, J.; Soriano, E.; Terry Wu, S.X.; Christensen, B.G.; et al. Hydrophobic Vancomycin Derivatives with Improved ADME Properties: Discovery of Telavancin (TD-6424). J. Antibiot. 2004, 57, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.L.; Chang, R.; Debabov, D.V.; Leung, J.; Wu, T.; Krause, K.M.; Sandvik, E.; Hubbard, J.M.; Kaniga, K.; Schmidt, D.E.; et al. Telavancin, a Multifunctional Lipoglycopeptide, Disrupts Both Cell Wall Synthesis and Cell Membrane Integrity in Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2005, 49, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Stryjewski, M.E.; Lentnek, A.; O’Riordan, W.; Pullman, J.; Tambyah, P.A.; Miró, J.M.; Fowler, V.G.; Barriere, S.L.; Kitt, M.M.; Corey, G.R. A Randomized Phase 2 Trial of Telavancin versus Standard Therapy in Patients with Uncomplicated Staphylococcus aureus Bacteremia: The ASSURE Study. BMC Infect. Dis. 2014, 14, 289. [Google Scholar] [CrossRef] [Green Version]
- Holland, T.L.; Chambers, H.F.; Boucher, H.W.; Corey, G.R.; Coleman, R.; Castaneda-Ruiz, B.; Fowler, V.G. Considerations for Clinical Trials of Staphylococcus aureus Bloodstream Infection in Adults. Clin. Infect. Dis. 2019, 68, 865–872. [Google Scholar] [CrossRef] [PubMed]
- A Phase 3 Telavancin Staphylococcus aureus (S. aureus) Bacteremia Trial. 2020. Available online: https://Clinicaltrials.Gov/Ct2/Show/Results/NCT02208063 (accessed on 15 February 2022).
- Burgin, D.J.; Liu, R.; Hsieh, R.C.; Heinzinger, L.R.; Otto, M. Investigational Agents for the Treatment of Methicillin-Resistant Staphylococcus aureus (MRSA) Bacteremia: Progress in Clinical Trials. Expert Opin. Investig. Drugs 2022, 31, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Draghi, D.C.; Benton, B.M.; Krause, K.M.; Thornsberry, C.; Pillar, C.; Sahm, D.F. Comparative Surveillance Study of Telavancin Activity against Recently Collected Gram-Positive Clinical Isolates from across the United States. Antimicrob. Agents Chemother. 2008, 52, 2383–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravolatz, L.D.; Pawlak, J.; Johnson, L.B. Comparative Activity of Telavancin against Isolates of Community-Associated Methicillin-Resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2007, 60, 406–409. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, E.J.C.; Citron, D.M.; Merriam, C.V.; Warren, Y.A.; Tyrrell, K.L.; Fernandez, H.T. In Vitro Activities of the New Semisynthetic Glycopeptide Telavancin (TD-6424), Vancomycin, Daptomycin, Linezolid, and Four Comparator Agents against Anaerobic Gram-Positive Species and Corynebacterium Spp. Antimicrob. Agents Chemother. 2004, 48, 2149–2152. [Google Scholar] [CrossRef] [Green Version]
- Leuthner, K.D.; Cheung, C.M.; Rybak, M.J. Comparative Activity of the New Lipoglycopeptide Telavancin in the Presence and Absence of Serum against 50 Glycopeptide Non-Susceptible Staphylococci and Three Vancomycin-Resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2006, 58, 338–343. [Google Scholar] [CrossRef]
- Leonard, S.N.; Vidaillac, C.; Rybak, M.J. Activity of Telavancin against Staphylococcus aureus Strains with Various Vancomycin Susceptibilities in an In Vitro Pharmacokinetic/Pharmacodynamic Model with Simulated Endocardial Vegetations. Antimicrob. Agents Chemother. 2009, 53, 2928–2933. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.A. Oritavancin (KIMYRSA™) in Acute Bacterial Skin and Skin Structure Infections: A Profile of Its Use in the USA. Drugs Ther. Perspect. 2022, 38, 57–63. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Schweizer, F.; Karlowsky, J.A. Oritavancin: Mechanism of Action. Clin. Infect. Dis. 2012, 54, S214–S219. [Google Scholar] [CrossRef] [Green Version]
- Guskey, M.T.; Tsuji, B.T. A Comparative Review of the Lipoglycopeptides: Oritavancin, Dalbavancin, and Telavancin. Pharmacotherapy 2010, 30, 80–94. [Google Scholar] [CrossRef]
- Mendes, R.E.; Farrell, D.J.; Sader, H.S.; Jones, R.N. Oritavancin Microbiologic Features and Activity Results from the Surveillance Program in the United States. Clin. Infect. Dis. 2012, 54, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Billeter, M.; Zervos, M.J.; Chen, A.Y.; Dalovisio, J.R.; Kurukularatne, C. Dalbavancin: A Novel Once-Weekly Lipoglycopeptide Antibiotic. Clin. Infect. Dis. 2008, 46, 577–583. [Google Scholar] [CrossRef]
- Mendes, R.E.; Farrell, D.J.; Sader, H.S.; Flamm, R.K.; Jones, R.N. Baseline Activity of Telavancin against Gram-Positive Clinical Isolates Responsible for Documented Infections in U.S. Hospitals (2011–2012) as Determined by the Revised Susceptibility Testing Method. Antimicrob. Agents Chemother. 2015, 59, 702–706. [Google Scholar] [CrossRef] [Green Version]
- Saravolatz, L.D.; Pawlak, J.; Johnson, L.B. In Vitro Susceptibilities and Molecular Analysis of Vancomycin-Intermediate and Vancomycin-Resistant Staphylococcus aureus Isolates. Clin. Infect. Dis. 2012, 55, 582–586. [Google Scholar] [CrossRef] [Green Version]
- Belley, A.; Neesham-Grenon, E.; Arhin, F.F.; McKay, G.A.; Parr, T.R.; Moeck, G. Assessment by Time-Kill Methodology of the Synergistic Effects of Oritavancin in Combination with Other Antimicrobial Agents against Staphylococcus aureus. Antimicrob. Agents Chemother. 2008, 52, 3820–3822. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Raad, I.; Darouiche, R.; Vazquez, J.; Lentnek, A.; Hachem, R.; Hanna, H.; Goldstein, B.; Henkel, T.; Seltzer, E. Efficacy and Safety of Weekly Dalbavancin Therapy for Catheter-Related Bloodstream Infection Caused by Gram-Positive Pathogens. Clin. Infect. Dis. 2005, 40, 374–380. [Google Scholar] [CrossRef]
- Ito, S.; Senoo, A.; Nagatoishi, S.; Ohue, M.; Yamamoto, M.; Tsumoto, K.; Wakui, N. Structural Basis for the Binding Mechanism of Human Serum Albumin Complexed with Cyclic Peptide Dalbavancin. J. Med. Chem. 2020, 63, 14045–14053. [Google Scholar] [CrossRef]
- Xu, X.; Xu, L.; Yuan, G.; Wang, Y.; Qu, Y.; Zhou, M. Synergistic Combination of Two Antimicrobial Agents Closing Each Other’s Mutant Selection Windows to Prevent Antimicrobial Resistance OPEN. Sci. Rep. 2018, 8, 7237. [Google Scholar] [CrossRef]
- Henderson, J.W. The Yellow Brick Road to Penicillin: A Story of Serendipity. Mayo Clin. Proc. 1997, 72, 683–687. [Google Scholar] [CrossRef]
- Terrak, M. Peptidoglycan Glycosyltransferase Inhibition: New Perspectives for an Old Target. Anti-Infect. Agents Med. Chem. 2008, 7, 180–192. [Google Scholar] [CrossRef]
- Miller, E.L. The Penicillins: A Review and Update. J. Midwifery Women’s Health 2002, 47, 426–434. [Google Scholar] [CrossRef]
- Watkins, R.R. Chapter 5—A Primer on Antimicrobials; Elsevier Inc.: Amsterdam, The Netherlands, 1941. [Google Scholar]
- Kahan, F.M.; Kropp, H.; Sundelof, J.G.; Birnbaum, J. Thienamycin: Development of Imipenem-Cilastatin. J. Antimicrob. Chemother. 1983, 12, 1–35. [Google Scholar] [CrossRef]
- Geddes, A.M.; Stille, W. Imipenem: The First Thienamycin Antibiotic. Rev. Infect. Dis. 1985, 7, S353–S356. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [Green Version]
- Chambers, H.F. Other Beta-Lactam Antibiotics. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 5th ed.; Mandell, G.L., Bennett, J.E., Dolin, R., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2000; Volume 1, pp. 291–293. [Google Scholar]
- Neu, H.C.; Novelli, A.; Chin, N.X. In Vitro Activity and Beta-Lactamase Stability of a New Carbapenem, SM-7338. Antimicrob. Agents Chemother. 1989, 33, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, Y.; Yoshida, Y.; Saitoh, K.; Nemoto, M.; Yamaguchi, A.; Sawai, T. Characteristics of Aztreonam as a Substrate, Inhibitor and Inducer for Beta-Lactamases. J. Antibiot. 1990, 43, 403–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykes, R.B.; Bonner, D.P. Discovery and Development of the Monobactams. Rev. Infect. Dis. 1985, 7, S579–S593. [Google Scholar] [CrossRef]
- Gutmann, L.; Vincent, S.; Billot-Klein, D.; Acar, J.F.; Mrèna, E.; Williamson, R. Involvement of Penicillin-Binding Protein 2 with Other Penicillin-Binding Proteins in Lysis of Escherichia coli by Some β-Lactam Antibiotics Alone and in Synergistic Lytic Effect of Amdinocillin (Mecillinam). Antimicrob. Agents Chemother. 1986, 30, 906–912. [Google Scholar] [CrossRef] [Green Version]
- Paech, F.; Messner, S.; Spickermann, J.; Wind, M.; Schmitt-Hoffmann, A.H.; Witschi, A.T.; Howell, B.A.; Church, R.J.; Woodhead, J.; Engelhardt, M.; et al. Mechanisms of Hepatotoxicity Associated with the Monocyclic β-Lactam Antibiotic BAL30072. Arch. Toxicol. 2017, 91, 3647–3662. [Google Scholar] [CrossRef] [PubMed]
- Prasad, N.K.; Seiple, I.B.; Cirz, R.T.; Rosenberg, O.S. Leaks in the Pipeline: A Failure Analysis of Gram-Negative Antibiotic Development from 2010 to 2020. Antimicrob. Agents Chemother. 2022, 66, e00054-22. [Google Scholar] [CrossRef]
- Li, J.; Milne, R.W.; Nation, R.L.; Turnidge, J.D.; Coulthard, K. Stability of Colistin and Colistin Methanesulfonate in Aqueous Media and Plasma as Determined by High-Performance Liquid Chromatography. Antimicrob. Agents Chemother. 2003, 47, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aeschlimann, J.R.; Hershberger, E.; Rybak, M.J. Analysis of Vancomycin Population Susceptibility Profiles, Killing Activity, and Postantibiotic Effect against Vancomycin-Intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 1999, 43, 1914–1918. [Google Scholar] [CrossRef] [Green Version]
- Baddour, L.M.; Wilson, W.R.; Bayer, A.S.; Fowler, V.G.; Tleyjeh, I.M.; Rybak, M.J.; Barsic, B.; Lockhart, P.B.; Gewitz, M.H.; Levison, M.E.; et al. Infective Endocarditis in Adults: Diagnosis, Antimicrobial Therapy, and Management of Complications: A Scientific Statement for Healthcare Professionals from the American Heart Association. Circulation 2015, 132, 1435–1486. [Google Scholar] [CrossRef]
- Stevens, D.L.; Bisno, A.L.; Chambers, H.F.; Everett, E.D.; Dellinger, P.; Goldstein, E.J.C.; Gorbach, S.L.; Hirschmann, J.V.; Kaplan, E.L.; Montoya, J.G.; et al. Practice Guidelines for the Diagnosis and Management of Skin and Soft-Tissue Infections. Clin. Infect. Dis. 2005, 41, 1373–1406. [Google Scholar] [CrossRef]
- Gemmell, C.G.; Edwards, D.I.; Fraise, A.P.; Gould, F.K.; Ridgway, G.L.; Warren, R.E. Guidelines for the Prophylaxis and Treatment of Methicillin-Resistant Staphylococcus aureus (MRSA) Infections in the UK. J. Antimicrob. Chemother. 2006, 57, 589–608. [Google Scholar] [CrossRef]
- Mermel, L.A.; Farr, B.M.; Sherertz, R.J.; Raad, I.I.; O’Grady, N.; Harris, J.S.; Craven, D.E. Guidelines for the Management of Intravascular Catheter-Related Infections. Clin. Infect. Dis. 2001, 32, 1249–1272. [Google Scholar] [CrossRef] [Green Version]
- Horstkotte, D.; Follath, F.; Gutschik, E.; Lengyel, M.; Oto, A.; Pavie, A.; Soler-Soler, J.; Thiene, G.; Von Graevenitz, A. Guidelines on Prevention, Diagnosis and Treatment of Infective Endocarditis Executive Summary: The Task Force on Infective Endocarditis of the European Society of Cardiology. Eur. Heart J. 2004, 25, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Elliott, T.S.J.; Foweraker, J.; Gould, F.K.; Perry, J.D.; Sandoe, J.A.T. Guidelines for the Antibiotic Treatment of Endocarditis in Adults: Report of the Working Party of the British Society for Antimicrobial Chemotherapy. J. Antimicrob. Chemother. 2004, 54, 971–981. [Google Scholar] [CrossRef]
- Eliopoulos, G.M. Antimicrobial Agents for Treatment of Serious Infections Caused by Resistant Staphylococcus aureus and Enterococci. Eur. J. Clin. Microbiol. Infect. Dis. 2005, 24, 826–831. [Google Scholar] [CrossRef]
- FDA. FDA SSSI Guidance for Industry Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment. In US Food and Drug Administration. Guidance for Industry. Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment; FDA: Washington, DC, USA, 2013. [Google Scholar]
- Tally, F.P.; DeBruin, M.F. Development of Daptomycin for Gram-Positive Infections. J. Antimicrob. Chemother. 2000, 46, 523–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weems, J.J. The Many Faces of Staphylococcus aureus Infection: Recognizing and Managing Its Life-Threatening Manifestations. Postgraduate Med. 2001, 110, 24–36. [Google Scholar] [CrossRef] [PubMed]
- King, M.D.; Humphrey, B.J.; Wang, Y.F.; Kourbatova, E.V.; Ray, S.M. Annals of Internal Medicine Article Staphylococcus aureus USA 300 Clone as the Predominant Cause of Skin and Soft-Tissue Infections. Ann. Intern. Med. 2006, 144, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Rayner, C.R.; Nation, R.L.; Owen, R.J.; Spelman, D.; Tan, K.E.; Liolios, L. Heteroresistance to Colistin in Multidrug-Resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2006, 50, 2946–2950. [Google Scholar] [CrossRef] [Green Version]
- Cathy, A.; Petti, M.D.A.; Vance, G.; Fowler, M.D., Jr. Optimal Treatment of Complicated Skin and Skin Structure Infections. Infect. Dis. Clin. N. Am. 2002, 16, 413–435. [Google Scholar]
- Eisenstein, B.I.; Oleson, F.B., Jr.; Baltz, R.H. Daptomycin: From the Mountain to the Clinic, with Essential Help from Francis Tally, MD. Clin. Infect. Dis. 2010, 50, S10–S15. [Google Scholar] [CrossRef] [Green Version]
- Fenton, C.; Keating, G.M.; Curran, M.P. Daptomycin. Drugs 2004, 64, 445–455. [Google Scholar] [CrossRef]
- Streit, J.M.; Jones, R.N.; Sader, H.S. Daptomycin Activity and Spectrum: A Worldwide Sample of 6737 Clinical Gram-Positive Organisms. J. Antimicrob. Chemother. 2004, 53, 669–674. [Google Scholar] [CrossRef] [Green Version]
- Niccolai, D.; Tarsi, L.; Thomas, R.J. The Renewed Challenge of Antibacterial Chemotherapy. Chem. Commun. 1997, 24, 2333–2342. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the Clinical Antibacterial and Antituberculosis Pipeline. Lancet Infect. Dis. 2019, 19, e40–e50. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The Global Preclinical Antibacterial Pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Buchy, P.; Ascioglu, S.; Buisson, Y.; Datta, S.; Nissen, M.; Tambyah, P.A.; Vong, S. Impact of Vaccines on Antimicrobial Resistance. Int. J. Infect. Dis. 2020, 90, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Micoli, F.; Bagnoli, F.; Rappuoli, R.; Serruto, D. The Role of Vaccines in Combatting Antimicrobial Resistance. Nat. Rev. Microbiol. 2021, 19, 287–302. [Google Scholar] [CrossRef]
- Marchetti, F.; Prato, R.; Viale, P. Survey among Italian Experts on Existing Vaccines’ Role in Limiting Antibiotic Resistance. Hum. Vaccines Immunother. 2021, 17, 4283–4290. [Google Scholar] [CrossRef]
- Jansen, K.U.; Anderson, A.S. The Role of Vaccines in Fighting Antimicrobial Resistance (AMR). Hum. Vaccines Immunother. 2018, 14, 2142–2149. [Google Scholar] [CrossRef] [Green Version]
- Lobanovska, M.; Pilla, G. Penicillin’s Discovery and Antibiotic Resistance: Lessons for the Future? Yale J. Biol. Med. 2017, 90, 135–145. [Google Scholar]
- Guevara Salazar, J.A.; Morán Díaz, J.R.; Ramírez Segura, E.; Trujillo Ferrara, J.G. What Are the Origins of Growing Microbial Resistance? Both Lamarck and Darwin Were Right. Expert Rev. Anti-Infect. Ther. 2021, 19, 563–569. [Google Scholar] [CrossRef]
- Rosas, N.C.; Lithgow, T. Targeting Bacterial Outer-Membrane Remodelling to Impact Antimicrobial Drug Resistance. Trends Microbiol. 2021, 30, 544–552. [Google Scholar] [CrossRef]
- Reygaert, W.C. An Overview of the Antimicrobial Resistance Mechanisms of Bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Li, X.-Z.; Plésiat, P.; Nikaido, H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [Green Version]
- Ghai, I.; Ghai, S. Understanding Antibiotic Resistance via Outer Membrane Permeability. Infect. Drug Resist. 2018, 11, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.P.; Uttley, A.H.C.; Woodford, N.; George, R.C. Resistance to Vancomycin and Teicoplanin: An Emerging Clinical Problem. Clin. Microbiol. Rev. 1990, 3, 280–291. [Google Scholar] [CrossRef]
- Capeletti, L.B.; de Oliveira, J.F.A.; Loiola, L.M.D.; Galdino, F.E.; da Silva Santos, D.E.; Soares, T.A.; de Oliveira Freitas, R.; Cardoso, M.B. Gram-Negative Bacteria Targeting Mediated by Carbohydrate–Carbohydrate Interactions Induced by Surface-Modified Nanoparticles. Adv. Funct. Mater. 2019, 29, 1–11. [Google Scholar] [CrossRef]
- Plotkin, S. History of Vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 12283–12287. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, A. Immunology of Bacterial Polysaccharide Antigens. Carbohydr. Res. 2003, 338, 2539–2547. [Google Scholar] [CrossRef]
- Sun, L.; Middleton, D.R.; Wantuch, P.L.; Ozdilek, A.; Avci, F.Y. Carbohydrates as T-Cell Antigens with Implications in Health and Disease. Glycobiology 2016, 26, 1029–1040. [Google Scholar] [CrossRef] [Green Version]
- Werdelin, O.; Meldal, M.; Jensen, T. Processing of Glycans on Glycoprotein and Glycopeptide Antigens in Antigen-Presenting Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9611–9613. [Google Scholar] [CrossRef] [Green Version]
- Khatun, F.; Toth, I.; Stephenson, R.J. Immunology of Carbohydrate-Based Vaccines. Adv. Drug Deliv. Rev. 2020, 165–166, 117–126. [Google Scholar] [CrossRef]
- Gallorini, S.; Berti, F.; Mancuso, G.; Cozzi, R.; Tortoli, M.; Volpini, G.; Telford, J.L.; Beninati, C.; Maione, D.; Wack, A. Toll-like Receptor 2 Dependent Immunogenicity of Glycoconjugate Vaccines Containing Chemically Derived Zwitterionic Polysaccharides. Proc. Natl. Acad. Sci. USA 2009, 106, 17481–17486. [Google Scholar] [CrossRef] [Green Version]
- Gallorini, S.; Berti, F.; Parente, P.; Baronio, R.; Aprea, S.; D’Oro, U.; Pizza, M.; Telford, J.L.; Wack, A. Introduction of Zwitterionic Motifs into Bacterial Polysaccharides Generates TLR2 Agonists Able to Activate APCs. J. Immunol. 2007, 179, 8208–8215. [Google Scholar] [CrossRef] [Green Version]
- Carreño, L.J.; Saavedra-Ávila, N.A.; Porcelli, S.A. Synthetic Glycolipid Activators of Natural Killer T Cells as Immunotherapeutic Agents. Clin. Transl. Immunol. 2016, 5, e69. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lalani, S.; Parekh, V.V.; Wu, L.; Van Kaer, L. Glycolipid Ligands of Invariant Natural Killer T Cells as Vaccine Adjuvants. Expert Rev. Vaccines 2008, 7, 1519–1532. [Google Scholar] [CrossRef] [Green Version]
- Avci, F.Y.; Li, X.; Tsuji, M.; Kasper, D.L. Carbohydrates and T Cells: A Sweet Twosome. Semin. Immunol. 2013, 25, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Cavallari, M.; Stallforth, P.; Kalinichenko, A.; Rathwell, D.C.K.; Gronewold, T.M.A.; Adibekian, A.; Mori, L.; Landmann, R.; Seeberger, P.H.; De Libero, G. Asemisynthetic Carbohydrate-Lipid Vaccine That Protects against S. Pneumoniae in Mice. Nat. Chem. Biol. 2014, 10, 950–956. [Google Scholar] [CrossRef]
- Oppenheimer, S.B.; Alvarez, M.; Nnoli, J. Carbohydrate-Based Experimental Therapeutics for Cancer, HIV/AIDS and Other Diseases. Acta Histochem. 2008, 110, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Huang, X. Recent Development in Carbohydrate Based Anticancer Vaccines. J. Carbohydr. Chem. 2012, 31, 143–186. [Google Scholar] [CrossRef] [Green Version]
- Hossain, F.; Andreana, P.R. Developments in Carbohydrate-Based Cancer Therapeutics. Pharmaceuticals 2019, 12, 84. [Google Scholar] [CrossRef] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GRAM NEGATIVE | ILLNESSES |
|---|---|
| Cocci | |
| Moraxella catarrhalis | Respiratory tract infections |
| Neisseria gonorrhoeae | Gonorrhea |
| Neisseria meningitidis | Meningococcal Meningitis |
| Chlamydia trachomatis | Chlamydia |
| Cocci-bacilli | |
| Rickettsia prowazekii Rickettsia typhi | Epidemic typhus Murine typhus |
| Cocci-bacilli and bacilli | |
| Brucella | Brucellosis |
| Bordetella | Whooping cough |
| Legionella | Pneumonia |
| Bacilli | |
| Escherichia coli | Gastroenteric and urinary tract infections |
| Salmonella typhi | Typhoid fever |
| Salmonella spp. | Diarrhea |
| Haemophilus influenzae | Otitis, bronchitis, pneumonia, meningitis |
| Flagellated bacilli | |
| Helicobacter pylori | Stomach and duodenal ulcers |
| Vibrions | |
| Vibrio cholerae | Cholera |
| Spirochetes | |
| Borrelia burgdorferi Leptospira | Lyme disease Leptospirosis |
| Non-fermenting Bacilli | |
| Pseudomonas aeruginosa | Lungs, blood or heart valves infections |
| GRAM POSITIVE | ILLNESSES |
| Cocci | |
| Staphylococcus aureus, S. epidermis | Skin, heart valve and bone infections, pneumonia |
| Enterococcus faecalis, E. faecium | Hospital Intestinal infections |
| Streptococcus B: S. agalactiae | Infections of various systems |
| Streptococcus A: S. pyogenes | Necrotizing fasciitis, Acute rheumatic fever, Acute glomerulus nephritis, Scarlet fever |
| Streptococcus pneumoniae | Pneumococcal infections |
| Bacilli | |
| Clostridium perfrigens | Food poisoning |
| Clostridium tetani | Tetanus |
| Clostridium botulinum | Botulism |
| Corynebacterium diphtheriae Corynebacterium jeikeium | Diphtheria Severe infections in the hospitalized patient |
| Actinomyces | Actinomycosis |
| Bacillus anthracis | Anthrax |
| Listeria monocytogenes | Listeriosis |
| Acid-fast bacilli | |
| Mycobacterium avium complex | Lung infections |
| Mycobacterium tuberculosis | Tuberculosis |
| Mycobacterium Leprae | Leprosy |
| Nocardia spp. | Nocardiosis |
| Repeating Unit | Modifications |
|---|---|
 | Unmodified unit |
 | N-Deacetylation |
 | Glycolylation |
 | O-Acetylation |
 | MurN-δ-lactam |
 | MurNAc-6-phosphate |
 | Linkage at GlcNAc |
| Pathogen | Vaccine | Manufacturer and Trade Name |
|---|---|---|
| Haemophilus influenzae type b (Hib) | Glycoconjugate, polysaccharide with tetanus toxoid (TT) | Sanofi Pasteur (ActHIB®); GlaxoSmithKline Biologicals (Hiberix®) |
| Diphtheria toxoid (DT), TT and acellular pertussis adsorbed, inactivated poliovirus and Hib–TT conjugate vaccine | Sanofi Pasteur (Pentacel®) | |
| Hib conjugate (meningococcal protein conjugate) | Merck & Co (PedvaxHIB®) | |
| Hib conjugate (meningococcal protein conjugate) and hepatitis B (recombinant) vaccine | Merck & Co (Comvax®) | |
| Neisseria meningitidis A, C, Y and W-135 | Glycoconjugate, meningococcal polysaccharide with DT | Sanofi Pasteur (Menactra®) |
| Meningococcal polysaccharide | Sanofi Pasteur (Menomune-A/C/Y/W-135®) | |
| De-O-acetylated polysaccharide (C11 strain of MenC) | Pfizer (NeisVac-C®) | |
| Capsular oligosaccharide (C11 strain of MenC) | GSK (Menjugate®) | |
| Salmonella typhi | Vi capsular polysaccharide | Sanofi Pasteur (TYPHIm Vi®) |
| Streptococcus pneumoniae 4, 6B, 9V, 14, 18C, 19F and 23F | Pneumococcal polysaccharide 7-valent–CRm197 conjugate | Wyeth Pharmaceuticals (Prevnar®) |
| Streptococcus pneumoniae 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C,19F, 19A, 20, 22F, 23F and 33F | Pneumococcal polysaccharide, 23-valent | Merck & Co (Pneumovax 23®) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riu, F.; Ruda, A.; Ibba, R.; Sestito, S.; Lupinu, I.; Piras, S.; Widmalm, G.; Carta, A. Antibiotics and Carbohydrate-Containing Drugs Targeting Bacterial Cell Envelopes: An Overview. Pharmaceuticals 2022, 15, 942. https://doi.org/10.3390/ph15080942
Riu F, Ruda A, Ibba R, Sestito S, Lupinu I, Piras S, Widmalm G, Carta A. Antibiotics and Carbohydrate-Containing Drugs Targeting Bacterial Cell Envelopes: An Overview. Pharmaceuticals. 2022; 15(8):942. https://doi.org/10.3390/ph15080942
Chicago/Turabian StyleRiu, Federico, Alessandro Ruda, Roberta Ibba, Simona Sestito, Ilenia Lupinu, Sandra Piras, Göran Widmalm, and Antonio Carta. 2022. "Antibiotics and Carbohydrate-Containing Drugs Targeting Bacterial Cell Envelopes: An Overview" Pharmaceuticals 15, no. 8: 942. https://doi.org/10.3390/ph15080942