
Roles of Bromodomain Extra Terminal Proteins in Metabolic Signaling and Diseases

Abstract
:1. Introduction
2. Overview of BET Bromodomain
3. BET Bromodomain and Metabolic Signaling
3.1. Adenosine Monophosphate-Activated Protein Kinase (AMPK) and Autophagy
3.2. Yes-Associated Protein (YAP) and Transcriptional Coactivator with a PDZ-Binding Domain (TAZ)
3.3. PGC1A
4. BET Bromodomain Functions in Fat Tissue Biology
4.1. Adipogenesis
4.2. Lipolysis
4.3. Thermogenesis
4.4. Obesity
5. BET Bromodomains in Hepatic Biology
5.1. Hepatic Steatosis
5.2. Hepatic Fibrosis
5.3. HDL Biology
5.4. Fatty Acid Oxidation, Gluconeogenesis, and Fasting Biology
6. BET Bromodomains and Cardiovascular Diseases
6.1. Cardiac Metabolism and Heart Failure
6.2. BET Bromodomain in Metaflammation and Atherosclerosis
7. BET Bromodomain in Diabetes
7.1. Pancreatic β Cells and Type 1 Diabetes
7.2. Insulin Resistant and Type 2 Diabetes
7.3. Clinical Evaluations of BET Inhibitors for the Treatment of Diabetes
8. Conclusion and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, J.; Tsai, L.H. Histone acetylation: Molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 2013, 14, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Rickards, B.; Flint, S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532.e9. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Alexanian, M.; Linares-Saldana, R.; Gonzalez-Teran, B.; Andreoletti, G.; Huang, Y.; Connolly, A.J.; Kim, W.; Hsu, A.; Duan, Q.; et al. BRD4 (Bromodomain-Containing Protein 4) Interacts with GATA4 (GATA Binding Protein 4) to Govern Mitochondrial Homeostasis in Adult Cardiomyocytes. Circulation 2020, 142, 2338–2355. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, D.A.; Andrieu, G.; Strissel, K.J.; Nikolajczyk, B.S.; Denis, G.V. BET bromodomain proteins and epigenetic regulation of inflammation: Implications for type 2 diabetes and breast cancer. Cell. Mol. Life Sci. 2017, 74, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borck, P.C.; Guo, L.W.; Plutzky, J. BET Epigenetic Reader Proteins in Cardiovascular Transcriptional Programs. Circ. Res. 2020, 126, 1190–1208. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [Green Version]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J., 3rd; Singer, D.S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Yang, X.D.; Zhou, M.M.; Ozato, K.; Chen, L.F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.D.; Feldman, Z.B.; Doherty, S.P.; Reyes, J.M.; Rahl, P.B.; Lin, C.Y.; Sheng, Q.; Duan, Q.; Federation, A.J.; Kung, A.L.; et al. BET bromodomain proteins regulate enhancer function during adipogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 2144–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Y.; Lee, A.Y.; Lai, H.T.; Zhang, H.; Chiang, C.M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.L.; Li, F.; Zhang, W.J.; Li, S.J.; Yang, Z.H.; Yang, T.L.; Qi, J.; Duan, Q.; Li, C.Q. Brd4 participates in epigenetic regulation of the extinction of remote auditory fear memory. Neurobiol. Learn. Mem. 2021, 179, 107383. [Google Scholar] [CrossRef] [PubMed]
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkina, A.C.; Nikolajczyk, B.S.; Denis, G.V. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J. Immunol. 2013, 190, 3670–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.; McMahon, S.; Anand, P.; Shah, H.; Thomas, S.; Salunga, H.T.; Huang, Y.; Zhang, R.; Sahadevan, A.; Lemieux, M.E.; et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017, 9, eaah5084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiltoir, J.I.; Stratton, M.S.; Cavasin, M.A.; Demos-Davies, K.; Reid, B.G.; Qi, J.; Bradner, J.E.; McKinsey, T.A. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2013, 63, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Brown, J.D.; Lin, C.Y.; Qi, J.; Zhang, R.; Artero, P.C.; Alaiti, M.A.; Bullard, J.; Alazem, K.; Margulies, K.B.; et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013, 154, 569–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozuka, C.; Efthymiou, V.; Sales, V.M.; Zhou, L.; Osataphan, S.; Yuchi, Y.; Chimene-Weiss, J.; Mulla, C.; Isganaitis, E.; Desmond, J.; et al. Bromodomain Inhibition Reveals FGF15/19 as a Target of Epigenetic Regulation and Metabolic Control. Diabetes 2022, 71, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaeloudes, C.; Mercado, N.; Clarke, C.; Bhavsar, P.K.; Adcock, I.M.; Barnes, P.J.; Chung, K.F. Bromodomain and extraterminal proteins suppress NF-E2-related factor 2-mediated antioxidant gene expression. J. Immunol. 2014, 192, 4913–4920. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [Green Version]
- Abedin, S.M.; Boddy, C.S.; Munshi, H.G. BET inhibitors in the treatment of hematologic malignancies: Current insights and future prospects. OncoTargets Ther. 2016, 9, 5943–5953. [Google Scholar]
- Gao, Z.; Yuan, T.; Zhou, X.; Ni, P.; Sun, G.; Li, P.; Cheng, Z.; Wang, X. Targeting BRD4 proteins suppresses the growth of NSCLC through downregulation of eIF4E expression. Cancer Biol. Ther. 2018, 19, 407–415. [Google Scholar] [CrossRef]
- Ocana, A.; Nieto-Jimenez, C.; Pandiella, A. BET inhibitors as novel therapeutic agents in breast cancer. Oncotarget 2017, 8, 71285–71291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tian, S.; Xiong, J.; Zhou, Y.; Song, H.; Liu, C. JQ-1 Inhibits Colon Cancer Proliferation via Suppressing Wnt/beta-Catenin Signaling and miR-21. Chem. Res. Toxicol. 2018, 31, 302–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, B.; Juiz, N.A.; Gayet, O.; Bigonnet, M.; Brandone, N.; Roques, J.; Cros, J.; Wang, N.; Dusetti, N.; Iovanna, J. Pancreatic Cancer Organoids for Determining Sensitivity to Bromodomain and Extra-Terminal Inhibitors (BETi). Front. Oncol. 2019, 9, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Yang, X.; Lin, J.; Cao, Z.; Lu, C.; Yang, Z.; Zheng, M.; Pan, R.; Cai, W. Super-Enhancer Induced IL-20RA Promotes Proliferation/Metastasis and Immune Evasion in Colorectal Cancer. Front. Oncol. 2021, 11, 724655. [Google Scholar] [CrossRef]
- Li, G.Q.; Guo, W.Z.; Zhang, Y.; Seng, J.J.; Zhang, H.P.; Ma, X.X.; Zhang, G.; Li, J.; Yan, B.; Tang, H.W.; et al. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 2016, 7, 2462–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLure, K.G.; Gesner, E.M.; Tsujikawa, L.; Kharenko, O.A.; Attwell, S.; Campeau, E.; Wasiak, S.; Stein, A.; White, A.; Fontano, E.; et al. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS ONE 2013, 8, e83190. [Google Scholar] [CrossRef]
- Siebel, A.L.; Trinh, S.K.; Formosa, M.F.; Mundra, P.A.; Natoli, A.K.; Reddy-Luthmoodoo, M.; Huynh, K.; Khan, A.A.; Carey, A.L.; van Hall, G.; et al. Effects of the BET-inhibitor, RVX-208 on the HDL lipidome and glucose metabolism in individuals with prediabetes: A randomized controlled trial. Metabolism 2016, 65, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Kulikowski, E.; Halliday, C.; Johansson, J.; Sweeney, M.; Lebioda, K.; Wong, N.; Haarhaus, M.; Brandenburg, V.; Beddhu, S.; Tonelli, M.; et al. Apabetalone Mediated Epigenetic Modulation is Associated with Favorable Kidney Function and Alkaline Phosphatase Profile in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2018, 43, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Cousin, S.; Blay, J.Y.; Garcia, I.B.; de Bono, J.S.; le Tourneau, C.; Moreno, V.; Trigo, J.; Hann, C.L.; Azad, A.A.; Im, S.A.; et al. Safety, pharmacokinetic, pharmacodynamic and clinical activity of molibresib for the treatment of nuclear protein in testis carcinoma and other cancers: Results of a Phase I/II open-label, dose escalation study. Int. J. Cancer 2022, 150, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.K.; Bae, H.J.; Kwon, W.S.; Kim, T.S.; Kim, K.H.; Park, S.; Yu, S.Y.; Hwang, J.; Park, J.; Chung, H.C.; et al. Inhibition of the bromodomain and extra-terminal family of epigenetic regulators as a promising therapeutic approach for gastric cancer. Cell. Oncol. 2021, 44, 1387–1403. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Trotta, A.; Wang, Z.; Shersher, E.; Li, B.; Long, J.; Lohse, I.; Wahlestedt, C.; El-Rifai, W.; Robbins, D.J.; Capobianco, A.J. The bromodomain inhibitor IBET-151 attenuates vismodegib-resistant esophageal adenocarcinoma growth through reduction of GLI signaling. Oncotarget 2020, 11, 3174–3187. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Mijatov, B.; Gunatilake, D.; Tiffen, J.C.; Gowrishankar, K.; Jin, L.; Pupo, G.M.; Cullinane, C.; Prinjha, R.K.; Smithers, N.; et al. The epigenetic regulator I-BET151 induces BIM-dependent apoptosis and cell cycle arrest of human melanoma cells. J. Investig. Dermatol. 2014, 134, 2795–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaidos, A.; Caputo, V.; Gouvedenou, K.; Liu, B.; Marigo, I.; Chaudhry, M.S.; Rotolo, A.; Tough, D.F.; Smithers, N.N.; Bassil, A.K.; et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood 2014, 123, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Sachdev, J.C.; Barve, M.; LoRusso, P.; Szmulewitz, R.; Patel, S.P.; Lara, P.N., Jr.; Chen, X.; Hu, B.; Freise, K.J.; et al. First-in-Human Study of Mivebresib (ABBV-075), an Oral Pan-Inhibitor of Bromodomain and Extra Terminal Proteins, in Patients with Relapsed/Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 6309–6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthakur, G.; Odenike, O.; Aldoss, I.; Rizzieri, D.A.; Prebet, T.; Chen, C.; Popovic, R.; Modi, D.A.; Joshi, R.H.; Wolff, J.E.; et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer 2021, 127, 2943–2953. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, G.; Mu, H.; Ma, X.; Wang, Z.; Lv, Y.; Zhang, T.; Xu, J.; Wang, J.; Li, Y.; et al. Bromodomain Inhibition Attenuates the Progression and Sensitizes the Chemosensitivity of Osteosarcoma by Repressing GP130/STAT3 Signaling. Front. Oncol. 2021, 11, 642134. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; da Costa, D.; Thanasopoulou, A.; Filippakopoulos, P.; Fish, P.V.; Philpott, M.; Fedorov, O.; Brennan, P.; Bunnage, M.E.; Owen, D.R.; et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013, 73, 3336–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef]
- Roboz, G.J.; Desai, P.; Lee, S.; Ritchie, E.K.; Winer, E.S.; DeMario, M.; Brennan, B.; Nuesch, E.; Chesne, E.; Brennan, L.; et al. A dose escalation study of RO6870810/TEN-10 in patients with acute myeloid leukemia and myelodysplastic syndrome. Leuk. Lymphoma 2021, 62, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; LoRusso, P.; Dowlati, A.; TDo, K.; Jacobson, C.A.; Vaishampayan, U.; Weise, A.; Caimi, P.F.; Eder, J.P.; French, C.A.; et al. A Phase 1 study of RO6870810, a novel bromodomain and extra-terminal protein inhibitor, in patients with NUT carcinoma, other solid tumours, or diffuse large B-cell lymphoma. Br. J. Cancer 2021, 124, 744–753. [Google Scholar] [CrossRef]
- Ramasamy, K.; Nooka, A.; Quach, H.; Htut, M.; Popat, R.; Liedtke, M.; Tuchman, S.A.; Laubach, J.; Gasparetto, C.; Chanan-Khan, A.; et al. A phase 1b dose-escalation/expansion study of BET inhibitor RO6870810 in patients with advanced multiple myeloma. Blood Cancer J. 2021, 11, 149. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Herbschleb, K.; Massard, C.; Woodcock, V.; Soria, J.C.; Walter, A.O.; Ewerton, F.; Poelman, M.; Benson, N.; Ocker, M.; et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: Emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur. J. Cancer 2019, 109, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Jauset, T.; Masso-Valles, D.; Martinez-Martin, S.; Beaulieu, M.E.; Foradada, L.; Fiorentino, F.P.; Yokota, J.; Haendler, B.; Siegel, S.; Whitfield, J.R.; et al. BET inhibition is an effective approach against KRAS-driven PDAC and NSCLC. Oncotarget 2018, 9, 18734–18746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernasconi, E.; Gaudio, E.; Lejeune, P.; Tarantelli, C.; Cascione, L.; Kwee, I.; Spriano, F.; Rinaldi, A.; Mensah, A.A.; Chung, E.; et al. Preclinical evaluation of the BET bromodomain inhibitor BAY 1238097 for the treatment of lymphoma. Br. J. Haematol. 2017, 178, 936–948. [Google Scholar] [PubMed] [Green Version]
- Aggarwal, R.R.; Schweizer, M.T.; Nanus, D.M.; Pantuck, A.J.; Heath, E.I.; Campeau, E.; Attwell, S.; Norek, K.; Snyder, M.; Bauman, L.; et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination with Enzalutamide in Patients with Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2020, 26, 5338–5347. [Google Scholar] [CrossRef]
- Falchook, G.; Rosen, S.; LoRusso, P.; Watts, J.; Gupta, S.; Coombs, C.C.; Talpaz, M.; Kurzrock, R.; Mita, M.; Cassaday, R.; et al. Development of 2 Bromodomain and Extraterminal Inhibitors with Distinct Pharmacokinetic and Pharmacodynamic Profiles for the Treatment of Advanced Malignancies. Clin. Cancer Res. 2020, 26, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Ameratunga, M.; Brana, I.; Bono, P.; Postel-Vinay, S.; Plummer, R.; Aspegren, J.; Korjamo, T.; Snapir, A.; de Bono, J.S. First-in-human Phase 1 open label study of the BET inhibitor ODM-207 in patients with selected solid tumours. Br. J. Cancer 2020, 123, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Moreno, V.; Sepulveda, J.M.; Vieito, M.; Hernandez-Guerrero, T.; Doger, B.; Saavedra, O.; Ferrero, O.; Sarmiento, R.; Arias, M.; de Alvaro, J.; et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann. Oncol. 2020, 31, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lee, H.C.; Shirazi, F.; Baladandayuthapani, V.; Lin, H.; Kuiatse, I.; Wang, H.; Jones, R.J.; Berkova, Z.; Singh, R.K.; et al. Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia 2018, 32, 2224–2239. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.T.; Fiskus, W.; Qian, Y.; Manshouri, T.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Mill, C.P.; et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 2017, 31, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Yu, C.; Chen, L.; Zhang, X.; Lei, Q.; Liu, Q.; Cai, G.; Liu, F. ARV-771 Acts as an Inducer of Cell Cycle Arrest and Apoptosis to Suppress Hepatocellular Carcinoma Progression. Front. Pharmacol. 2022, 13, 858901. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Y.; Rao, X.; Zhang, R.; Tang, J.; Zhang, D.; Jie, X.; Zhu, K.; Wang, X.; Xu, Y.; et al. BRD4-IRF1 axis regulates chemoradiotherapy-induced PD-L1 expression and immune evasion in non-small cell lung cancer. Clin. Transl. Med. 2022, 12, e718. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Mill, C.P.; Qian, Y.; Raina, K.; Rajapakshe, K.; Coarfa, C.; Soldi, R.; Bose, P.; et al. Targeting nuclear beta-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leukemia 2019, 33, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Hu, Y.; Zhou, B.; Fernandez-Salas, E.; Yang, C.Y.; Liu, L.; McEachern, D.; Przybranowski, S.; Wang, M.; Stuckey, J.; et al. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem. 2018, 61, 6685–6704. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhou, B.; Yang, C.Y.; Ji, J.; McEachern, D.; Przybranowski, S.; Jiang, H.; Hu, J.; Xu, F.; Zhao, Y.; et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2476–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafran, J.S.; Jafari, N.; Casey, A.N.; Gyorffy, B.; Denis, G.V. BRD4 regulates key transcription factors that drive epithelial-mesenchymal transition in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Hu, T.; Wang, T.; Shi, X.; Kitano, A.; Eagle, K.; Hoegenauer, K.A.; Konopleva, M.Y.; Lin, C.Y.; Young, N.L.; et al. AMP-activated protein kinase links acetyl-CoA homeostasis to BRD4 recruitment in acute myeloid leukemia. Blood 2019, 134, 2183–2194. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.H.; Guan, K.L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.M.; Avantaggiati, M.L.; Nemazanyy, I.; di Poto, C.; Yang, Y.; Pende, M.; Gibney, G.T.; Ressom, H.W.; Field, J.; Atkins, M.B.; et al. YAP/TAZ Inhibition Induces Metabolic and Signaling Rewiring Resulting in Targetable Vulnerabilities in NF2-Deficient Tumor Cells. Dev. Cell 2019, 49, 425–443.e9. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhang, Y.; Park, H.W.; Jewell, J.L.; Chen, Q.; Deng, Y.; Pan, D.; Taylor, S.S.; Lai, Z.C.; Guan, K.L. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013, 27, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tharp, K.M.; Kang, M.S.; Timblin, G.A.; Dempersmier, J.; Dempsey, G.E.; Zushin, P.H.; Benavides, J.; Choi, C.; Li, C.X.; Jha, A.K.; et al. Actomyosin-Mediated Tension Orchestrates Uncoupled Respiration in Adipose Tissues. Cell Metab. 2018, 27, 602–615.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cheng, X.; Chen, J.; Wang, Y.; Wu, X.; Tian, R.; Liu, B.; Ding, X.; Sun, Q.; Gong, W. Suppression of YAP/TAZ-Notch1-NICD axis by bromodomain and extraterminal protein inhibition impairs liver regeneration. Theranostics 2019, 9, 3840–3852. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Barrow, J.J.; Balsa, E.; Verdeguer, F.; Tavares, C.D.; Soustek, M.S.; Hollingsworth, L.R.T.; Jedrychowski, M.; Vogel, R.; Paulo, J.A.; Smeitink, J.; et al. Bromodomain Inhibitors Correct Bioenergetic Deficiency Caused by Mitochondrial Disease Complex I Mutations. Mol. Cell 2016, 64, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Sarjeant, K.; Stephens, J.M. Adipogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.J.; Belaghzal, H.; Hsiao, W.Y.; Qi, J.; Bradner, J.E.; Guertin, D.A.; Sif, S.; Imbalzano, A.N. Transcriptional and post-transcriptional control of adipocyte differentiation by Jumonji domain-containing protein 6. Nucleic Acids Res. 2015, 43, 7790–7804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.; Wu, P.; Liu, Z.; Xia, F.; Zhu, L.; Zheng, Z.; Yang, T.; Qi, J. BET bromodomain inhibition suppresses adipogenesis in mice. Endocrine 2020, 67, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Park, Y.K.; Park, S.; Jang, Y.; Waring, N.; Dey, A.; Ozato, K.; Lai, B.; Peng, W.; Ge, K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017, 8, 2217. [Google Scholar] [CrossRef] [PubMed]
- Zang, K.; Wang, J.; Dong, M.; Sun, R.; Wang, Y.; Huang, Y.; Liu, X.; Li, Y.; Wang, F.; Yu, M. Brd2 inhibits adipogenesis via the ERK1/2 signaling pathway in 3T3-L1 adipocytes. PLoS ONE 2013, 8, e78536. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, H.; Blanton, W.P.; Belkina, A.; Lebrasseur, N.K.; Denis, G.V. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem. J. 2009, 425, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, J.; Li, S.; Wang, Y.; Mo, W.; Sun, R.; Yu, M. Bromodomain-containing protein 2 promotes lipolysis via ERK/HSL signalling pathway in white adipose tissue of mice. Gen. Comp. Endocrinol. 2019, 281, 105–116. [Google Scholar] [CrossRef]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G.; Bernson, V.S.; Heaton, G.M. The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Eff. Thermogenesis 1978, 32, 89–93. [Google Scholar]
- Bagchi, R.A.; Ferguson, B.S.; Stratton, M.S.; Hu, T.; Cavasin, M.A.; Sun, L.; Lin, Y.H.; Liu, D.; Londono, P.; Song, K.; et al. HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight 2018, 3, e120159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Hu, X.; Dong, X.; Li, G.; Chen, Y.; Chen, J.; He, X.; Sun, H.; Kim, D.H.; Kemper, J.K.; Chen, L.F. Brd4 modulates diet-induced obesity via PPARgamma-dependent Gdf3 expression in adipose tissue macrophages. JCI Insight 2021, 6, e143379. [Google Scholar] [CrossRef]
- Baiceanu, A.; Mesdom, P.; Lagouge, M.; Foufelle, F. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat. Rev. Endocrinol. 2016, 12, 710–722. [Google Scholar] [CrossRef]
- Yamada, A.; Honma, K.; Mochizuki, K.; Goda, T. BRD4 regulates fructose-inducible lipid accumulation-related genes in the mouse liver. Metabolism 2016, 65, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Cheng, D.; Peng, Y.; Li, Y.; Fu, C.; Wang, Y.; Fu, L.; Peng, S.; Ni, X. Hepatic BRD4 Is Upregulated in Liver Fibrosis of Various Etiologies and Positively Correlated to Fibrotic Severity. Front. Med. 2021, 8, 683506. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Hah, N.; Yu, R.T.; Sherman, M.H.; Benner, C.; Leblanc, M.; He, M.; Liddle, C.; Downes, M.; Evans, R.M. BRD4 is a novel therapeutic target for liver fibrosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15713–15718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wei, B.; Liu, M.; Hirsova, P.; Sehrawat, T.S.; Cao, S.; Hu, X.; Xue, F.; Yaqoob, U.; Kang, N.; et al. Endothelial p300 Promotes Portal Hypertension and Hepatic Fibrosis Through C-C Motif Chemokine Ligand 2-Mediated Angiocrine Signaling. Hepatology 2021, 73, 2468–2483. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Yang, X.; Tian, J.; Wang, X.; Ji, Y.; El-Ashram, S.; Ren, C.; Shen, J.; Liu, M. JQ-1 ameliorates schistosomiasis liver fibrosis by suppressing JAK2 and STAT3 activation. Biomed. Pharmacother. 2021, 144, 112281. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Zu, S.J.; Liu, Y.J.; Li, J.C.; Dang, W.Z.; Liao, L.P.; Liu, L.P.; Chen, P.Y.; Huang, H.M.; Wu, K.H.; et al. Selective bromodomain and extra-terminal bromodomain inhibitor inactivates macrophages and hepatic stellate cells to inhibit liver inflammation and fibrosis. Bioengineered 2022, 13, 10914–10930. [Google Scholar] [CrossRef]
- Dixon, J.L.; Ginsberg, H.N. Hepatic synthesis of lipoproteins and apolipoproteins. Semin. Liver Dis. 1992, 12, 364–372. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Gordon, A.; Johansson, J.; Wolski, K.; Ballantyne, C.M.; Kastelein, J.J.; Taylor, A.; Borgman, M.; Nissen, S.E. Efficacy and safety of a novel oral inducer of apolipoprotein a-I synthesis in statin-treated patients with stable coronary artery disease a randomized controlled trial. J. Am. Coll. Cardiol. 2011, 57, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Gilham, D.; Wasiak, S.; Tsujikawa, L.M.; Halliday, C.; Norek, K.; Patel, R.G.; Kulikowski, E.; Johansson, J.; Sweeney, M.; Wong, N.C.; et al. RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 2016, 247, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267 Pt 2, H742–H750. [Google Scholar] [CrossRef]
- Kim, S.Y.; Zhang, X.; Schiattarella, G.G.; Altamirano, F.; Ramos, T.A.R.; French, K.M.; Jiang, N.; Szweda, P.A.; Evers, B.M.; May, H.I.; et al. Epigenetic Reader BRD4 (Bromodomain-Containing Protein 4) Governs Nucleus-Encoded Mitochondrial Transcriptome to Regulate Cardiac Function. Circulation 2020, 142, 2356–2370. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Christ, A.; Lauterbach, M.; Latz, E. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019, 51, 794–811. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin. Epigenetics 2019, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Brewer, H.B.; Kastelein, J.J.; Hu, B.; Uno, K.; Kataoka, Y.; et al. Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial. Am. J. Cardiovasc. Drugs 2016, 16, 55–65. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Deeney, J.T.; Belkina, A.C.; Shirihai, O.S.; Corkey, B.E.; Denis, G.V. BET Bromodomain Proteins Brd2, Brd3 and Brd4 Selectively Regulate Metabolic Pathways in the Pancreatic beta-Cell. PLoS ONE 2016, 11, e0151329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, P.J.; Shah, A.; Apostolopolou, H.; Bhushan, A. BET Proteins Are Required for Transcriptional Activation of the Senescent Islet Cell Secretome in Type 1 Diabetes. Int. J. Mol. Sci. 2019, 20, 4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Lu, Y.; Zhou, Z.; Li, Q.; Chen, X.; Wang, W.; Jin, Y.; Hu, Z.; Chen, G.; Deng, Q.; et al. Human expandable pancreatic progenitor-derived beta cells ameliorate diabetes. Sci. Adv. 2022, 8, eabk1826. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wu, Y.; Hou, W.; Sun, Z.; Wang, Y.; Wei, H.; Mo, W.; Yu, M. Bromodomain-containing protein 2 induces insulin resistance via the mTOR/Akt signaling pathway and an inflammatory response in adipose tissue. Cell. Signal. 2017, 30, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Ray, K.K.; Johansson, J.O.; Gordon, A.; Sweeney, M.; Halliday, C.; Kulikowski, E.; Wong, N.; Kim, S.W.; Schwartz, G.G. Selective BET Protein Inhibition with Apabetalone and Cardiovascular Events: A Pooled Analysis of Trials in Patients with Coronary Artery Disease. Am. J. Cardiovasc. Drugs 2018, 18, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Schwartz, G.G.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 2021, 20, 13. [Google Scholar] [CrossRef] [PubMed]



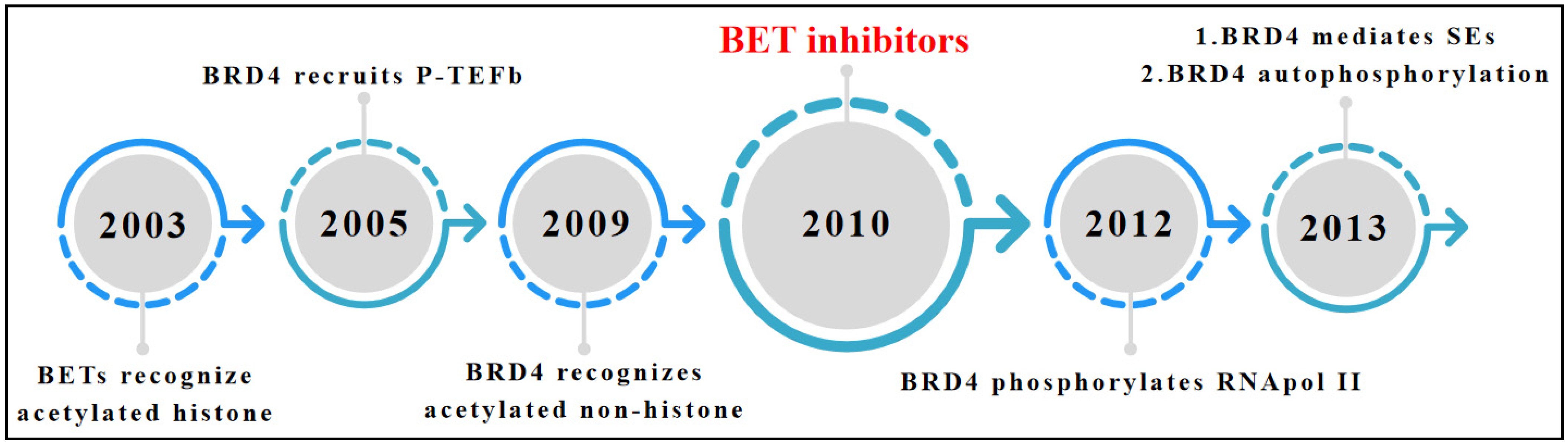{kind=link}
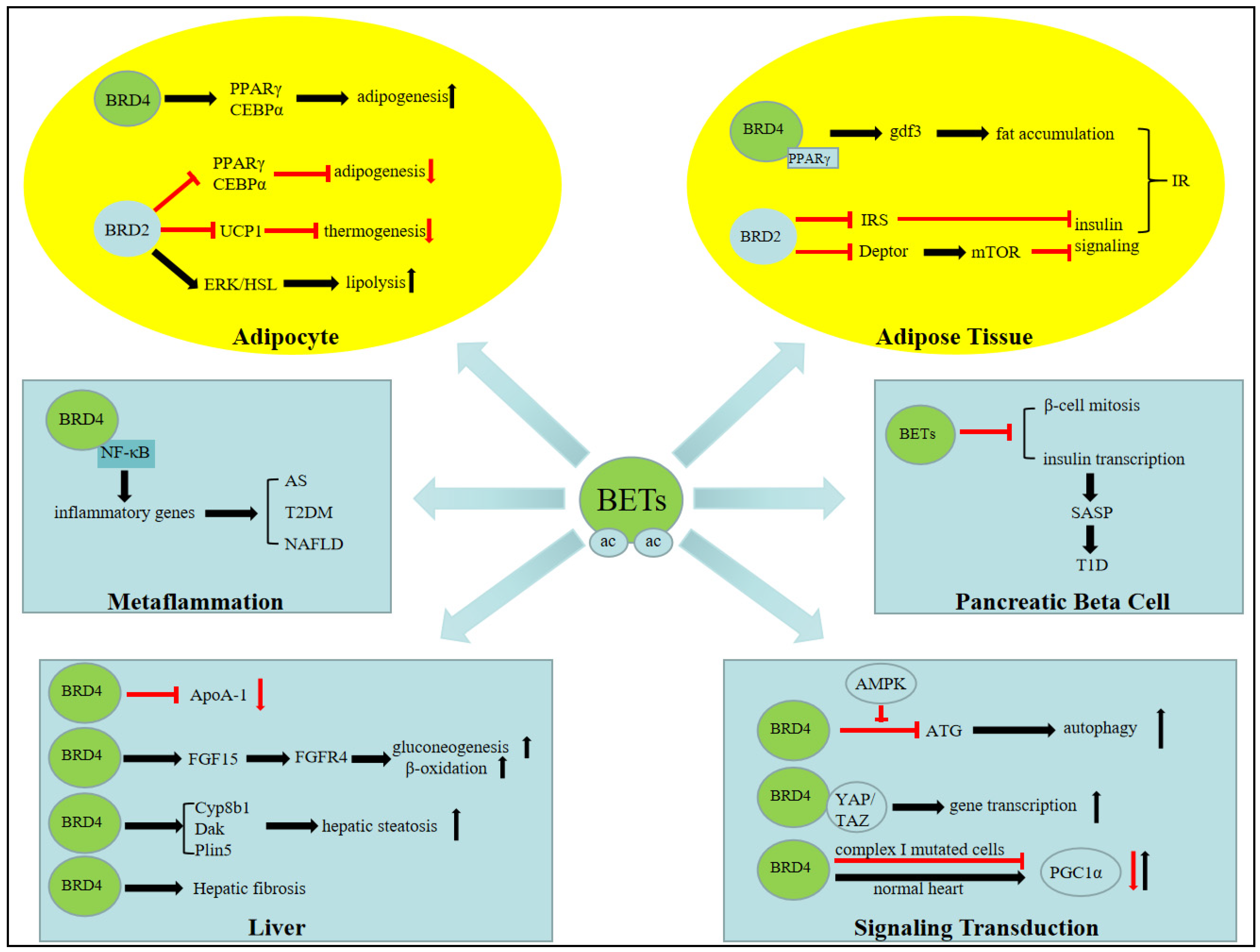{kind=link}
| Compound | Selectivity | Indication | Reference | |
|---|---|---|---|---|
| BET inhibitors | JQ1 | BD1 and BD2 from BRD2/3/4 and BRDT | Chronic obstructive pulmonary disease | [33] |
| NUT midline carcinoma | [6] | |||
| Multiple myeloma | [34] | |||
| Acute myeloid leukemia | [35] | |||
| Diffuse large B-cell lymphoma | [36] | |||
| Hematologic malignancies | [37] | |||
| Lung cancer | [38] | |||
| Breast cancer | [39] | |||
| Colon cancer | [40] | |||
| Pancreatic ductal adenocarcinoma | [41] | |||
| Colorectal cancer | [42] | |||
| Hepatocellular cancer | [43] | |||
| RVX-208 | BD2 from BRD2,3,4 | Atherosclerosis | [44] | |
| Diabetes | [45] | |||
| Fabry disease | [12] | |||
| Chronic kidney disease | [46] | |||
| IBET-762 (GSK-525762) | BD1 and BD2 from BRD2,3,4,T | Neoplasm | [12] | |
| Testis carcinoma | [47] | |||
| Midline carcinoma | [12] | |||
| IBET-151 | BD1 and BD2 from BRD2,3,4,T | MLL-fusion leukemia | [48] | |
| Colorectal ctumorsancer | [42] | |||
| Gastric cancer | [49] | |||
| Vismodegib-resistant esophageal adenocarcinoma | [50] | |||
| Rheumatoid arthritis | [15] | |||
| Melanoma | [51] | |||
| Myeloma | [52] | |||
| MK8628/OTXO15 | BRD2,3,4 | Lymphoma or multiple myeloma | [53] | |
| Acute leukemia | [54] | |||
| NUT midline carcinoma | [12] | |||
| Triple-negative breast cancer | ||||
| Lung cancer | ||||
| Castration-resistantprostate cancer | ||||
| FT1101 | BRD2,3,4,T | Acute myeloid leukemia | [12] | |
| Non-Hodgkin lymphoma | ||||
| CPI-0610 | BD1 from BRD2,4,T | Multiple myeloma | [12] | |
| ABBV-075 Mivebresib | BRD2,3,4 | Relapsed/Refractory solid tumors. | [55] | |
| Breast cancer | [12] | |||
| Prostate cancer | ||||
| Non-Hodgkin lymphoma | ||||
| Multiple myeloma | ||||
| Relapsed/refractory acute myeloid leukemia. | [56] | |||
| NHWD-870 | BRD4 | Pancreatic ductal adenocarcinoma | [41] | |
| Osteosarcoma | [57] | |||
| BMS-986158 | undisclosed | Advanced tumors | [12] | |
| PFI-1 | BRD2,4 | Acute leukemia | [58] | |
| ABBV-744 | BD2 | prostate cancer | [59] | |
| Acute myeloid leukemia | [59] | |||
| GSK788 | BD1 | Acute myeloid leukemia | [15] | |
| GSK620 | BD2 | Rheumatoid arthritis | [15] | |
| Psoriasis | [15] | |||
| Non–alcoholic fatty liver disease | [15] | |||
| RO6870810/TEN-10 | undisclosed | Acute myeloid leukemia and myelodysplastic syndrome | [60] | |
| NUT carcinoma, other solid tumors, or diffuse large B-cell lymphoma | [61] | |||
| Multiple myeloma | [62] | |||
| BAY 1238097 | BRD4 | Advanced malignancies | [63] | |
| Pancreatic ductal adenocarcinoma Non-small cell lung cancer | [64] | |||
| Lymphoma | [65] | |||
| ZEN-3694 | BD1,BD2 | Metastatic castration-resistant prostate cancer | [66] | |
| INCB054329 | BRD4 | Advanced malignancies | [67] | |
| INCB057643 | BRD4 | Advanced malignancies | [67] | |
| ODM-207 | BRD2,3,4,T | Castration-resistantprostate cancer. | [68] | |
| AZD5153 | BRD4 | Malignant solid tumor and lymphoma | NCT03205176 | |
| CC-90010 | BRD2,4 | Advanced solid tumors and relapsed/refractory Non-Hodgkin’s lymphoma. | [69] | |
| Solid tumor | [69] | |||
| BET degraders | ARV-825 | BRD2,3,4,T | Burkitt’s lymphoma | [70] |
| Multiple myeloma | [71] | |||
| Secondary (s) acute myeloid leukemia | [72] | |||
| dBET1 | BRD2,3,4 | Leukemia | [73] | |
| ARV-763 | BRD4 | Multiple myeloma | [71] | |
| ARV-771 | BRD2,3,4 | Castration-resistant prostate cancer | [74] | |
| Hepatocellular carcinoma | [75] | |||
| Non-small cell lung cancer | [76] | |||
| Post-myeloproliferative neoplasm secondary acute myeloid leukemia | [77] | |||
| QCA570 | BRD4 | Acute leukemia | [78] | |
| BETd-246/BETd-260 | BRD4 | Triple-negative breast cancer | [79] | |
| MZ1 | BRD4 | Castration-resistant prostate cancer | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, D.; Duan, Q. Roles of Bromodomain Extra Terminal Proteins in Metabolic Signaling and Diseases. Pharmaceuticals 2022, 15, 1032. https://doi.org/10.3390/ph15081032
Wu D, Duan Q. Roles of Bromodomain Extra Terminal Proteins in Metabolic Signaling and Diseases. Pharmaceuticals. 2022; 15(8):1032. https://doi.org/10.3390/ph15081032
Chicago/Turabian StyleWu, Dayu, and Qiong Duan. 2022. "Roles of Bromodomain Extra Terminal Proteins in Metabolic Signaling and Diseases" Pharmaceuticals 15, no. 8: 1032. https://doi.org/10.3390/ph15081032