
The Future of Tissue-Targeted Lipid Nanoparticle-Mediated Nucleic Acid Delivery


Abstract
:1. Introduction
2. LNP Chemistry, Formulation and Background

3. Inherent LNP Liver Tropism

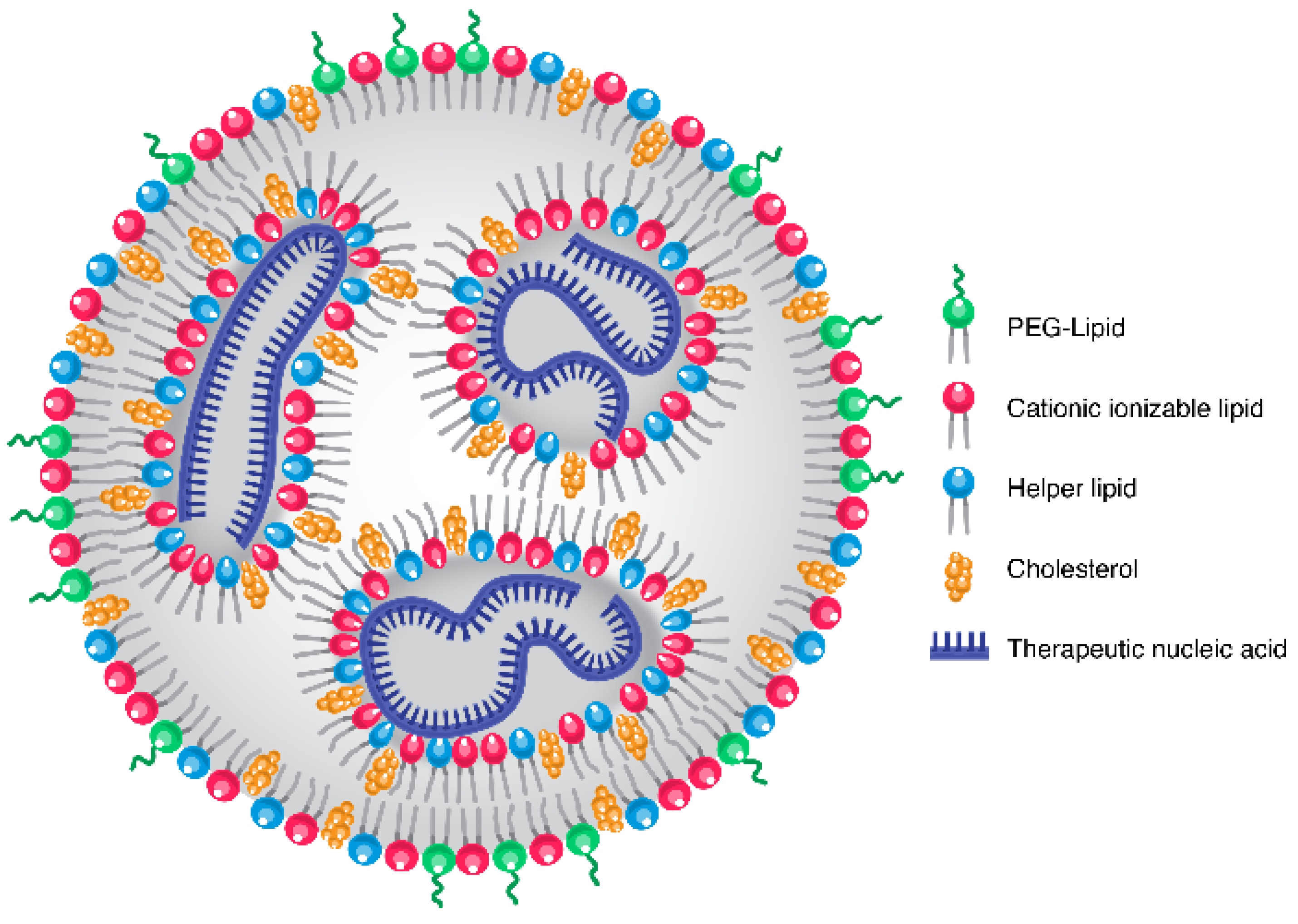{kind=link}
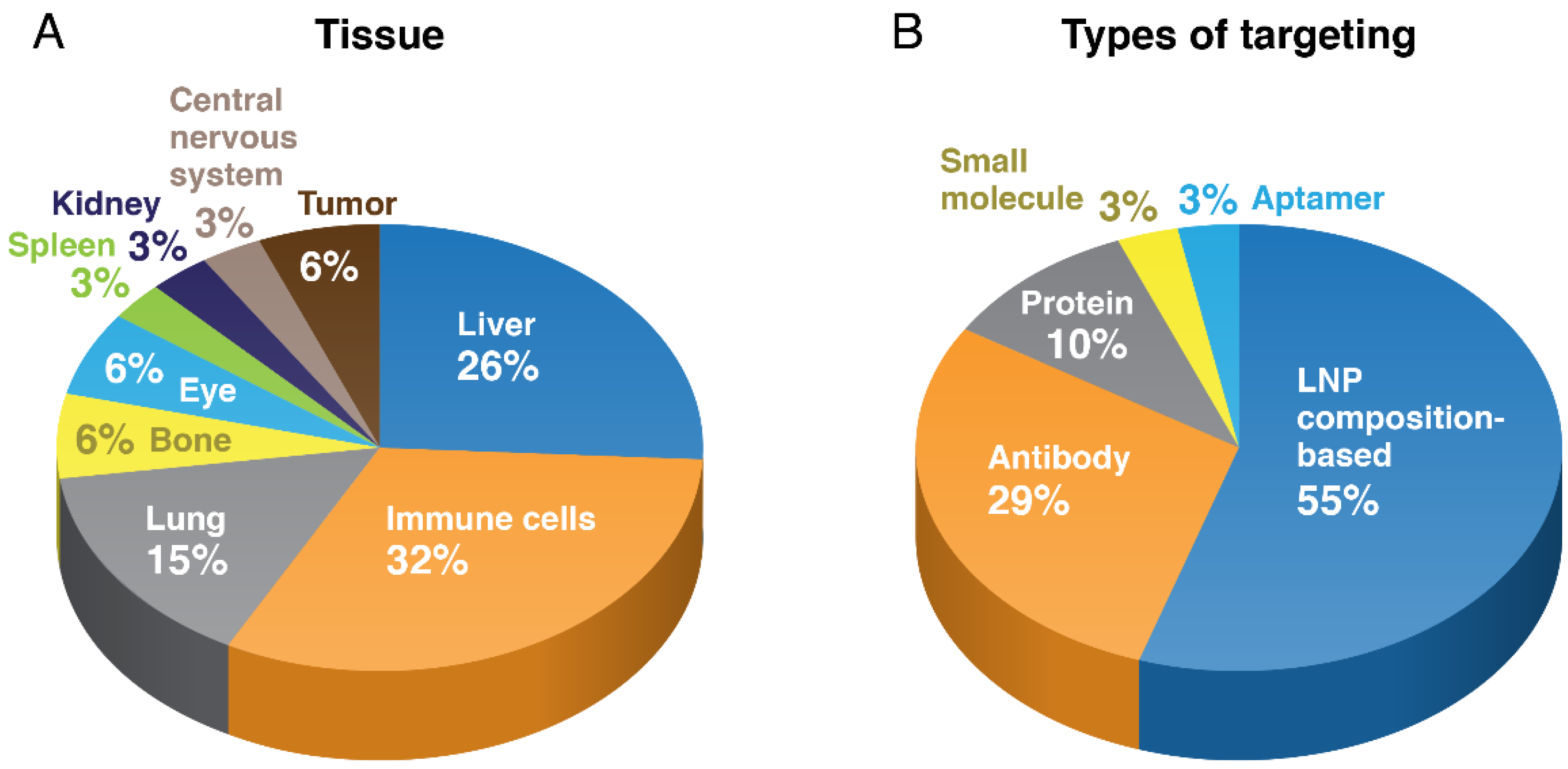{kind=link}
| LNP Targeting Components or Properties | Physicochemical Properties (DLS Size, PDI and Zeta Potential) | Route of Administration | Payload and Indication | Model | Tissue/Cell Type Specificity | Ref. |
|---|---|---|---|---|---|---|
| 50 mol% 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) | 113 nm 0.22 PDI −0.52 mV | i.v. | human Erythropoietin, mouse Interleukin-10, mouse Klotho, Luciferase and Cre mRNA, and Cas9 mRNA/sgTom1 * | 18–20 g male C57BL/6 mice; (age and sex not specified) B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J mice (also known as Ai9 or Ai9(RCL-tdT) mice | hepatocyte uptake | [17] |
| 30 molar% 1,2-dioleoyl-sn-glycero-3-phosphate (18PA) | 142 nm 0.13 PDI −2.11 mV | i.v. | human Erythropoietin, mouse Interleukin-10, mouse Klotho, Luciferase and Cre mRNA, and Cas9 mRNA/sgPTEN and Cas9 mRNA/sgTom1 * | 18–20 g male C57BL/6 mice; (age and sex not specified) B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J mice (also known as Ai9 or Ai9(RCL-tdT) mice | hepatocyte uptake | [17] |
| 20 molar% 1,2-dioleoyl-3-dimethylammonium-propane (DODAP) | 12 nm 0.18 PDI (ZP not specified) | i.v. | human Erythropoietin, mouse Interleukin-10, mouse Klotho, Luciferase and Cre mRNA, and Cas9 mRNA/sgPCSK9 and Cas9 mRNA/sgTom1 * | 18–20 g male C57BL/6 mice; (age and sex not specified) B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J mice (also known as Ai9 or Ai9(RCL-tdT) mice | hepatocyte uptake | [17] |
| 50:10:38.5:1.5% mole ratios DLin-KC2 DMA:DSPC:Cholesterol: DMG-PEG2000, with N:P molar ratio = 2, imparting negative charge | 75 nm (PDI not specified) −10 mV | i.m. | Luciferase mRNA * | 8-week-old female Balb/c mice | greater hepatic distribution following i.m. administration | [4] |
| 50:23.5:6.5:20% mole ratios 7C1 **:C14PEG2K:18:1 Lyso PC 60:10:25:5% mole ratios 7C1 **:C14PEG2K:DOPE | 20–200 nm (PDI not specified) (ZP not specified) | i.v. | ICAM-2 siRNA, Cre mRNA, CRISPR-Cas9 mRNA and ICAM-2 sgRNA * | 5–12-week-old (sex not specified) LSL-Tomato, C57BL/6J, and constitutive SpCas9 mice | lung, spleen, liver and kidney endothelial cell uptake | [37] |
| Endogenous absorption of apoE to neutral LNP | 64.5 nm (PDI not specified) (ZP not specified) | i.v. | Factor VII siRNA * | 6–8-week-old female C57Bl/6, ApoE−/− and LDLR−/− mice | hepatocyte uptake | [29] |
| N-acetylgalactosamine (GalNAc) ligand | 69.4 nm (PDI not specified) (ZP not specified) | i.v. | Factor VII siRNA * | 6–8-week-old female C57Bl/6 and ASGR2−/− mice | hepatocyte uptake | [29] |
| Plasmalemma vesicle-associated protein (PV1) | 70 nm, 0.104 PDI and 160 nm, 0.150–0.240 PDI (ZP not specified) | i.v. | Luciferase mRNA, Cy5-mRNA * | 5-week-old female Balb-c mice | lung uptake | [7] |
| Anti-Ly6c mAbs | 70 nm (PDI not specified) (ZP not specified) | i.v. | Luciferase or IL-10 mRNA; treatment of inflammatory bowel disease | Colitis was induced in: 12-week-old female C57BL/6 mice using dextran sodium sulfate | leukocyte uptake | [38] |
| 15–20 mol% C18PEG2000:80 mol% 7C1 **:0.1–10 mol% cholesterol | 45–50 nm <0.2 PDI (ZP not specified) | i.v. | ICAM-2 siRNA, ICAM-2 targeting sgRNA * | 5–12-week-old (sex not specified) C57BL6/j and constitutive SpCas9 mice | bone marrow endothelial cell | [39] |
| Anti-CD4 antibody | 129 nm 0.12 PDI −10 mV | i.v. | Cy5-labeled siRNA and CD45 siRNA * | 6–8-week-old (sex not specified) C57BL6/j mice | T cells | [9] |
| Anti-CD4 antibody | 88 nm 0.1 PDI (ZP not specified) | i.v. | Cre recombinase-encoding mRNA * | (age not specified) (sex not specified) Ai6 (RCL-ZsGreen) mice on C57BL/6J | Splenic and lymph node T cells | [10] |
| Adamantane-constrained lipid | 20–200 nm 0.20–0.23 PID (ZP not specified) | i.v. | GFP siRNA * | 5–8-week-old female C57BL/6-Tg(UBC-GFP)30Scha/J, ‘GFP mice’ | splenic T cells | [40] |
| Anti-CD29 antibody | 66–75 nm 0.10–0.16 PDI (ZP not specified) | i.v. | PLK1 siRNA; treatment of disseminated bone marrow mantle cell lymphoma xenograft | 8-week-old female C.B-17/IcrHsd-Prkdc scid mice | mantle cell lymphoma | [8] |
| Cholesterol oleate | 22–115 nm (PDI not specified) (ZP not specified) | i.v. | ICAM-2 siRNA, GFP-targeted sgRNA * | 5–8-week-old female C57BL/6J and C57BL/6-Tg(UBC-GFP)30Scha/J, ‘GFP mice’ | hepatic endothelial cells | [41] |
| ~30 nm, negatively charged LNP | 34 nm 0.242 PDI −12 mV | s.c. | DiD-labeled LNP (no nucleic acid) * | 7–9-week-old female C57BL/6J mice | CD8+ dendritic cells/lymph node | [14] |
| 35:5:55:5% mole ratios 7C1 **:Cholesterol:C14PEG2000:DOTAP | 40 nm (PDI not specified) (ZP not specified) | nebulization | Therapeutic membrane-anchored FI6 antibody mRNA, H1N1 influenza model | 6–8-week-old female BALBc mice | lung | [42] |
| ~150 nm size, ~0.5% PEG density | 150 nm <0.1 PD (ZP not specified) | intravitreal and subretinal injection | Cre, mCherry, luciferase mRNA * | 1–6 months old male and female Albino BALB/c, Ai9, apoE−/−, Mertk−/− and C57BL6 mice | optic nerve, trabecular meshwork, retinal pigment epithelium, Muller glia | [43] |
| Ionizable lipids with low pKa and unsaturated hydrocarbon chains | 83–229 nm 0.09–0.28 PDI (ZP not specified) | subretinal injection | Luciferase, EGFP, mCherry mRNA * | 1–4 months old male and female Albino BALB/c mice | retinal pigment epithelium | [44] |
| Oxidized cholesterol | ~80 nm 0.16 PDI (ZP not specified) | i.v. | Cre mRNA * | 5–8-week-old (sex not specified) Ai14 Lox-Stop-Lox-tdTomato and C57BL/6J mice | hepatic endothelial and Kupffer cells | [30] |
| Anti PECAM-1 antibody | 103 nm 0.195 PDI −4.1 mV | i.v. | Luciferase mRNA * | (age not specified) (sex not specified) C57BL/6 mice | lung vascular endothelial and immune cells | [11] |
| Adamantyl-constrained lipid | 100 nm (PDI not specified) (ZP not specified) | i.v. | Cre mRNA * | (age not specified) (sex not specified) Ai14 Lox-Stop-Lox-tdTomato mice | hepatic Kupffer cells | [31] |
| ApoE opsonization | 55 nm 0.058 PDI (ZP not specified) | intracranial | PTEN, luciferase and GRIN1 siRNA * | 26–30-day-old (sex not specified) Sprague Dawley rats | CNS neurons | [45] |
| CH6 osteoblast-specific aptamer | 84 nm (PDI not specified) (ZP not specified) | i.v. |
osteogenic pleckstrin homology domain-containing family O member 1 (Plekho1) siRNA; treatment of impaired bone formation (e.g., osteoporosis) | 6-month-old female Sprague Dawley rats | osteoblasts | [46] |
| Mannose-cholesterol | ~140 nm >0.2 PDI (ZP not specified) | i.d. | Influenze hemagglutanin saRNA; H1N1 influenza vaccine | 6−8-week-old female BALB/c mice | dendritic cells | [12] |
| Mannose-PEG-DSPE | ~100 nm (PDI not specified) (ZP not specified) | i.v. | Cre mRNA and FVIII siRNA * | 7–10-week-old female C57BL/6 mice; 8-week-old (sex not specified) Lox-Stop-Lox-tdTomato | hepatic endothelial cells | [13] |
| EGFR-antibody | 79 nm 0.085 PDI 7.7 mV | i.p. | Cas9 mRNA, (polo-like kinase) PLK1 sgRNA; disseminated ovarian cancer | 8-week-old female Hsd: Athymic Nude-Foxn1nu mice with OV8 ovarian cancer peritoneal xenograft | disseminated ovarian cancer | [47] |
| DEC205-antibody | 90–130 nm 0.12–0.20 PDI (ZP not specified) | r.o. | CD40, CD80 and CD86 siRNA | 6–12-week-old (sex not specified) C57BL/6 mice, inhibition of mixed lymphocyte response to LPS | CD8 alpha+ dendritic cells | [48] |
| CD4-antibody | 88 nm 0.1 PDI (ZP not specified) | i.v. | Luciferase and Cre mRNA * |
(age not specified) male and female C57BL/6 and Ai6 (RCL-ZsGreen) mice on C57BL/6J background | CD4+ T cells | [10] |
| CD5-antibody | 80 nm 0.02–0.06 PDI (ZP not specified) | i.v. | CAR mRNA against fibroblast activation protein and Cre mRNA, cardiac fibrosis prevention | (age not specified) (sex not specified) C57BL/6NAi6 Cre-reporter mice (Rosa26CAG-LSL-ZsGreen) | CD5+ T cells | [49] |
4. Non-Hepatic LNP Targeting
5. Oncology and Immuno-Oncology
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Probst, J.; Weide, B.; Scheel, B.; Pichler, B.J.; Hoerr, I.; Rammensee, H.G.; Pascolo, S. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Therapy 2007, 14, 1175–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eygeris, Y.; Gupta, M.; Kim, J.; Sahay, G. Chemistry of Lipid Nanoparticles for RNA Delivery. Acc. Chem. Res. 2022, 55, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, M.J.; Alishetty, S.; Alameh, M.G.; Said, H.; Wright, L.; Paige, M.; Soliman, O.; Weissman, D.; Cleveland, T.E.T.; Grishaev, A.; et al. Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Commun. Biol. 2021, 4, 956. [Google Scholar] [CrossRef]
- Stern, S.T.; McNeil, S.E. Nanotechnology safety concerns revisited. Toxicol. Sci. 2008, 101, 4–21. [Google Scholar] [CrossRef]
- Shobaki, N.; Sato, Y.; Harashima, H. Mixing lipids to manipulate the ionization status of lipid nanoparticles for specific tissue targeting. Int. J. Nanomed. 2018, 13, 8395–8410. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Chan, C.; Peterson, N.; Hanna, R.N.; Alfaro, A.; Allen, K.L.; Wu, H.; Dall’Acqua, W.F.; Borrok, M.J.; Santos, J.L. Engineering Caveolae-Targeted Lipid Nanoparticles To Deliver mRNA to the Lungs. ACS Chem. Biol. 2020, 15, 830–836. [Google Scholar] [CrossRef]
- Kedmi, R.; Veiga, N.; Ramishetti, S.; Goldsmith, M.; Rosenblum, D.; Dammes, N.; Hazan-Halevy, I.; Nahary, L.; Leviatan-Ben-Arye, S.; Harlev, M.; et al. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol. 2018, 13, 214–219. [Google Scholar] [CrossRef]
- Ramishetti, S.; Kedmi, R.; Goldsmith, M.; Leonard, F.; Sprague, A.G.; Godin, B.; Gozin, M.; Cullis, P.R.; Dykxhoorn, D.M.; Peer, D. Systemic Gene Silencing in Primary T Lymphocytes Using Targeted Lipid Nanoparticles. ACS Nano 2015, 9, 6706–6716. [Google Scholar] [CrossRef]
- Tombacz, I.; Laczko, D.; Shahnawaz, H.; Muramatsu, H.; Natesan, A.; Yadegari, A.; Papp, T.E.; Alameh, M.G.; Shuvaev, V.; Mui, B.L.; et al. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNPs. Mol. Therapy 2021, 29, 3293–3304. [Google Scholar] [CrossRef]
- Parhiz, H.; Shuvaev, V.V.; Pardi, N.; Khoshnejad, M.; Kiseleva, R.Y.; Brenner, J.S.; Uhler, T.; Tuyishime, S.; Mui, B.L.; Tam, Y.K.; et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control. Release 2018, 291, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Chatzikleanthous, D.; Lou, G.; Giusti, F.; Bonci, A.; Taccone, M.; Brazzoli, M.; Gallorini, S.; Ferlenghi, I.; Berti, F.; et al. Mannosylation of LNP Results in Improved Potency for Self-Amplifying RNA (SAM) Vaccines. ACS Infect. Dis. 2019, 5, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jeong, M.; Hur, S.; Cho, Y.; Park, J.; Jung, H.; Seo, Y.; Woo, H.A.; Nam, K.T.; Lee, K.; et al. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci Adv. 2021, 7, eabf4398. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kawai, M.; Sato, Y.; Maeki, M.; Tokeshi, M.; Harashima, H. The Effect of Size and Charge of Lipid Nanoparticles Prepared by Microfluidic Mixing on Their Lymph Node Transitivity and Distribution. Mol. Pharm. 2020, 17, 944–953. [Google Scholar] [CrossRef]
- Zukancic, D.; Suys, E.J.A.; Pilkington, E.H.; Algarni, A.; Al-Wassiti, H.; Truong, N.P. The Importance of Poly(ethylene glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles. Pharmaceutics 2020, 12, 1068. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Liu, S.; Cheng, Q.; Wei, T.; Yu, X.; Johnson, L.T.; Farbiak, L.; Siegwart, D.J. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing. Nat. Mater. 2021, 20, 701–710. [Google Scholar] [CrossRef]
- Alvarez-Benedicto, E.; Farbiak, L.; Marquez Ramirez, M.; Wang, X.; Johnson, L.T.; Mian, O.; Guerrero, E.D.; Siegwart, D.J. Optimization of phospholipid chemistry for improved lipid nanoparticle (LNP) delivery of messenger RNA (mRNA). Biomater. Sci. 2022, 10, 549–559. [Google Scholar] [CrossRef]
- Lee, S.M.; Cheng, Q.; Yu, X.; Liu, S.; Johnson, L.T.; Siegwart, D.J. A Systematic Study of Unsaturation in Lipid Nanoparticles Leads to Improved mRNA Transfection In Vivo. Angew. Chem. Int. Ed. Engl. 2021, 60, 5848–5853. [Google Scholar] [CrossRef]
- Kimura, S.; Khalil, I.A.; Elewa, Y.H.A.; Harashima, H. Novel lipid combination for delivery of plasmid DNA to immune cells in the spleen. J. Control. Release 2021, 330, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Algarni, A.; Pilkington, E.H.; Suys, E.J.A.; Al-Wassiti, H.; Pouton, C.W.; Truong, N.P. In vivo delivery of plasmid DNA by lipid nanoparticles: The influence of ionizable cationic lipids on organ-selective gene expression. Biomater. Sci. 2022, 10, 2940–2952. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Amiji, M. Clinical approval of nanotechnology-based SARS-CoV-2 mRNA vaccines: Impact on translational nanomedicine. Drug Deliv. Transl. Res. 2021, 11, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Surging Nanomedicine Investments Improve Global Healthcare and Pandemic Protection. Global Health & Pharma. Available online: https://www.ghp-news.com/surging-nanomedicine-investments-improve-global-healthcare-and-pandemic-protection/ (accessed on 13 June 2022).
- Evers, M.J.W.; Kulkarni, J.A.; van der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Di, J.; Du, Z.; Wu, K.; Jin, S.; Wang, X.; Li, T.; Xu, Y. Biodistribution and Non-linear Gene Expression of mRNA LNPs Affected by Delivery Route and Particle Size. Pharm. Res. 2022, 39, 105–114. [Google Scholar] [CrossRef]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, O.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Therapy 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Shi, B.; Keough, E.; Matter, A.; Leander, K.; Young, S.; Carlini, E.; Sachs, A.B.; Tao, W.; Abrams, M.; Howell, B.; et al. Biodistribution of small interfering RNA at the organ and cellular levels after lipid nanoparticle-mediated delivery. J. Histochem. Cytochem. 2011, 59, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Therapy 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Paunovska, K.; Da Silva Sanchez, A.J.; Sago, C.D.; Gan, Z.; Lokugamage, M.P.; Islam, F.Z.; Kalathoor, S.; Krupczak, B.R.; Dahlman, J.E. Nanoparticles Containing Oxidized Cholesterol Deliver mRNA to the Liver Microenvironment at Clinically Relevant Doses. Adv. Mater. 2019, 31, e1807748. [Google Scholar] [CrossRef]
- Gan, Z.; Lokugamage, M.P.; Hatit, M.Z.C.; Loughrey, D.; Paunovska, K.; Sato, M.; Cristian, A.; Dahlman, J.E. Nanoparticles containing constrained phospholipids deliver mRNA to liver immune cells in vivo without targeting ligands. Bioeng. Transl. Med. 2020, 5, e10161. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, M.; Griffiths, G. Phagocytosis: Latex leads the way. Curr. Opin. Cell Biol. 2003, 15, 498–503. [Google Scholar] [CrossRef]
- Basha, G.; Novobrantseva, T.I.; Rosin, N.; Tam, Y.Y.; Hafez, I.M.; Wong, M.K.; Sugo, T.; Ruda, V.M.; Qin, J.; Klebanov, B.; et al. Influence of cationic lipid composition on gene silencing properties of lipid nanoparticle formulations of siRNA in antigen-presenting cells. Mol. Therapy 2011, 19, 2186–2200. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.M.; Paolini, M.S.; Fenton, O.S.; Poul, L.; Devalliere, J.; Mpambani, F.; Darmon, A.; Bergere, M.; Jibault, O.; Germain, M.; et al. A Nanoprimer To Improve the Systemic Delivery of siRNA and mRNA. Nano Lett. 2020, 20, 4264–4269. [Google Scholar] [CrossRef]
- Jain, R.; Frederick, J.P.; Huang, E.Y.; Burke, K.E.; Mauger, D.M.; Andrianova, E.A.; Farlow, S.J.; Siddiqui, S.; Pimentel, J.; Cheung-Ong, K.; et al. MicroRNAs Enable mRNA Therapeutics to Selectively Program Cancer Cells to Self-Destruct. Nucleic Acid Ther. 2018, 28, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Kurian, A.A.; Chepurko, E.; Sassi, Y.; Hajjar, R.J.; Zangi, L. Specific Modified mRNA Translation System. Circulation 2020, 142, 2485–2488. [Google Scholar] [CrossRef] [PubMed]
- Sago, C.D.; Lokugamage, M.P.; Paunovska, K.; Vanover, D.A.; Monaco, C.M.; Shah, N.N.; Gamboa Castro, M.; Anderson, S.E.; Rudoltz, T.G.; Lando, G.N.; et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. USA 2018, 115, E9944–E9952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 4493. [Google Scholar] [CrossRef]
- Sago, C.D.; Lokugamage, M.P.; Islam, F.Z.; Krupczak, B.R.; Sato, M.; Dahlman, J.E. Nanoparticles That Deliver RNA to Bone Marrow Identified by in Vivo Directed Evolution. J. Am. Chem. Soc. 2018, 140, 17095–17105. [Google Scholar] [CrossRef]
- Lokugamage, M.P.; Sago, C.D.; Gan, Z.; Krupczak, B.R.; Dahlman, J.E. Constrained Nanoparticles Deliver siRNA and sgRNA to T Cells In Vivo without Targeting Ligands. Adv. Mater. 2019, 31, e1902251. [Google Scholar] [CrossRef]
- Paunovska, K.; Gil, C.J.; Lokugamage, M.P.; Sago, C.D.; Sato, M.; Lando, G.N.; Gamboa Castro, M.; Bryksin, A.V.; Dahlman, J.E. Analyzing 2000 in Vivo Drug Delivery Data Points Reveals Cholesterol Structure Impacts Nanoparticle Delivery. ACS Nano 2018, 12, 8341–8349. [Google Scholar] [CrossRef]
- Lokugamage, M.P.; Vanover, D.; Beyersdorf, J.; Hatit, M.Z.C.; Rotolo, L.; Echeverri, E.S.; Peck, H.E.; Ni, H.; Yoon, J.K.; Kim, Y.; et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat. Biomed. Eng. 2021, 5, 1059–1068. [Google Scholar] [CrossRef]
- Ryals, R.C.; Patel, S.; Acosta, C.; McKinney, M.; Pennesi, M.E.; Sahay, G. The effects of PEGylation on LNP based mRNA delivery to the eye. PLoS ONE 2020, 15, e0241006. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ryals, R.C.; Weller, K.K.; Pennesi, M.E.; Sahay, G. Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Control. Release 2019, 303, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Rungta, R.L.; Choi, H.B.; Lin, P.J.; Ko, R.W.; Ashby, D.; Nair, J.; Manoharan, M.; Cullis, P.R.; Macvicar, B.A. Lipid Nanoparticle Delivery of siRNA to Silence Neuronal Gene Expression in the Brain. Mol. Therapy Nucleic Acids 2013, 2, e136. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Guo, B.; Wu, H.; Shao, N.; Li, D.; Liu, J.; Dang, L.; Wang, C.; Li, H.; Li, S.; et al. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference-based bone anabolic strategy. Nat. Med. 2015, 21, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef]
- Katakowski, J.A.; Mukherjee, G.; Wilner, S.E.; Maier, K.E.; Harrison, M.T.; DiLorenzo, T.P.; Levy, M.; Palliser, D. Delivery of siRNAs to Dendritic Cells Using DEC205-Targeted Lipid Nanoparticles to Inhibit Immune Responses. Mol. Therapy 2016, 24, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Rurik, J.G.; Tombacz, I.; Yadegari, A.; Mendez Fernandez, P.O.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Khanani, A.M.; Thomas, M.J.; Aziz, A.A.; Weng, C.Y.; Danzig, C.J.; Yiu, G.; Kiss, S.; Waheed, N.K.; Kaiser, P.K. Review of gene therapies for age-related macular degeneration. Eye 2022, 36, 303–311. [Google Scholar] [CrossRef]
- Vance, J.E.; Hayashi, H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim. Biophys. Acta 2010, 1801, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Guevara, M.L.; Persano, F.; Persano, S. Advances in Lipid Nanoparticles for mRNA-Based Cancer Immunotherapy. Front. Chem. 2020, 8, 589959. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- BioNTech SE. Available online: https://www.biontech.com/int/en/home/pipeline-and-products/pipeline.html (accessed on 3 June 2022).
- BioNTech’s Second Act: Can it Transform the Fight against Cancer? Financial Times. Available online: https://www.ft.com/content/12ef99d4-063a-4a45-ae4d-e8115a9c3bb1 (accessed on 15 June 2022).
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Moderna Inc. Available online: https://www.modernatx.com/research/product-pipeline (accessed on 3 June 2022).
- Pfizer Enters into Agreement with Acuitas Therapeutics for Lipid Nanoparticle Delivery System for Use in mRNA Vaccines and Therapeutics. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-enters-agreement-acuitas-therapeutics-lipid (accessed on 13 June 2022).
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Lee, K.; Kim, S.Y.; Seo, Y.; Kim, M.H.; Chang, J.; Lee, H. Adjuvant incorporated lipid nanoparticles for enhanced mRNA-mediated cancer immunotherapy. Biomater. Sci. 2020, 8, 1101–1105. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes Res. 2010, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Petersen, G.H.; Alzghari, S.K.; Chee, W.; Sankari, S.S.; La-Beck, N.M. Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J. Control. Release 2016, 232, 255–264. [Google Scholar] [CrossRef]
- Price, L.S.L.; Stern, S.T.; Deal, A.M.; Kabanov, A.V.; Zamboni, W.C. A reanalysis of nanoparticle tumor delivery using classical pharmacokinetic metrics. Sci. Adv. 2020, 6, eaay9249. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Han, L.; Jiang, C. Evolution of blood-brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm. Sin. B 2021, 11, 2306–2325. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Dong, C.; Fan, W.; Jiang, H.; Xiang, J.; Qiu, N.; Piao, Y.; Xie, T.; Luo, Y.; Li, Z.; et al. Tumor extravasation and infiltration as barriers of nanomedicine for high efficacy: The current status and transcytosis strategy. Biomaterials 2020, 240, 119902. [Google Scholar] [CrossRef] [PubMed]
- Cochran, R.; Cochran, F. Phage display and molecular imaging: Expanding fields of vision in living subjects. Biotechnol. Genet. Eng. Rev. 2010, 27, 57–94. [Google Scholar] [CrossRef]
- Deramchia, K.; Jacobin-Valat, M.J.; Vallet, A.; Bazin, H.; Santarelli, X.; Sanchez, S.; Dos Santos, P.; Franconi, J.M.; Claverol, S.; Bonetto, S.; et al. In vivo phage display to identify new human antibody fragments homing to atherosclerotic endothelial and subendothelial tissues [corrected]. Am. J. Pathol. 2012, 180, 2576–2589. [Google Scholar] [CrossRef]
- Fang, R.H.; Kroll, A.V.; Gao, W.; Zhang, L. Cell Membrane Coating Nanotechnology. Adv. Mater. 2018, 30, e1706759. [Google Scholar] [CrossRef]
- Rao, L.; Meng, Q.F.; Bu, L.L.; Cai, B.; Huang, Q.; Sun, Z.J.; Zhang, W.F.; Li, A.; Guo, S.S.; Liu, W.; et al. Erythrocyte Membrane-Coated Upconversion Nanoparticles with Minimal Protein Adsorption for Enhanced Tumor Imaging. ACS Appl. Mater. Interfaces 2017, 9, 2159–2168. [Google Scholar] [CrossRef]
- Rao, L.; Yu, G.T.; Meng, Q.F.; Bu, L.L.; Tian, R.; Lin, L.S.; Deng, H.Z.; Yang, W.J.; Zan, M.H.; Ding, J.X.; et al. Cancer Cell Membrane-Coated Nanoparticles for Personalized Therapy in Patient-Derived Xenograft Models. Adv. Funct. Mater. 2019, 29, 1905671. [Google Scholar] [CrossRef]
- Wang, D.; Dong, H.; Li, M.; Cao, Y.; Yang, F.; Zhang, K.; Dai, W.; Wang, C.; Zhang, X. Erythrocyte-Cancer Hybrid Membrane Camouflaged Hollow Copper Sulfide Nanoparticles for Prolonged Circulation Life and Homotypic-Targeting Photothermal/Chemotherapy of Melanoma. ACS Nano 2018, 12, 5241–5252. [Google Scholar] [CrossRef]
- Wang, S.; Duan, Y.; Zhang, Q.; Komarla, A.; Gong, H.; Gao, W.; Zhang, L. Drug Targeting via Platelet Membrane-Coated Nanoparticles. Small Struct. 2020, 1, 2000018. [Google Scholar] [CrossRef]
- Chen, M.; Chen, M.; He, J. Cancer cell membrane cloaking nanoparticles for targeted co-delivery of doxorubicin and PD-L1 siRNA. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1635–1641. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, X.; Ruan, M.; Liu, W.; Song, R.; Dai, J.; Xue, W. Worm-Like Biomimetic Nanoerythrocyte Carrying siRNA for Melanoma Gene Therapy. Small 2018, 14, e1803002. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Mohapatra, A.; Zhou, J.; Holay, M.; Krishnan, N.; Gao, W.; Fang, R.H.; Zhang, L. Virus-Mimicking Cell Membrane-Coated Nanoparticles for Cytosolic Delivery of mRNA. Angew. Chem. Int. Ed. Engl. 2022, 61, e202113671. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Gong, H.; Zhou, J.; Zhang, Q.; Gao, W.; Fang, R.H.; Zhang, L. Targeted gene silencing in vivo by platelet membrane-coated metal-organic framework nanoparticles. Sci. Adv. 2020, 6, eaaz6108. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, K.; Swidan, S.; El-Nabarawi, M.; Teaima, M. Lipid based nanoparticles as a novel treatment modality for hepatocellular carcinoma: A comprehensive review on targeting and recent advances. J. Nanobiotech. 2022, 20, 109. [Google Scholar] [CrossRef] [PubMed]
- Shinn, J.; Kwon, N.; Lee, S.A.; Lee, Y. Smart pH-responsive nanomedicines for disease therapy. J. Pharm. Investig. 2022, 52, 427–441. [Google Scholar] [CrossRef]
- Heshmati Aghda, N.; Dabbaghianamiri, M.; Tunnell, J.W.; Betancourt, T. Design of smart nanomedicines for effective cancer treatment. Int. J. Pharm. 2022, 621, 121791. [Google Scholar] [CrossRef]
- Krishnan, N.; Fang, R.H.; Zhang, L. Engineering of stimuli-responsive self-assembled biomimetic nanoparticles. Adv. Drug Deliv. Rev. 2021, 179, 114006. [Google Scholar] [CrossRef]
- Salzano, G.; Costa, D.F.; Torchilin, V.P. siRNA Delivery by Stimuli-Sensitive Nanocarriers. Curr. Pharm. Des. 2015, 21, 4566–4573. [Google Scholar] [CrossRef] [Green Version]
- De Maar, J.S.; Suelmann, B.B.M.; Braat, M.; van Diest, P.J.; Vaessen, H.H.B.; Witkamp, A.J.; Linn, S.C.; Moonen, C.T.W.; van der Wall, E.; Deckers, R. Phase I feasibility study of Magnetic Resonance guided High Intensity Focused Ultrasound-induced hyperthermia, Lyso-Thermosensitive Liposomal Doxorubicin and cyclophosphamide in de novo stage IV breast cancer patients: Study protocol of the i-GO study. BMJ Open 2020, 10, e040162. [Google Scholar] [CrossRef]
- Tak, W.Y.; Lin, S.M.; Wang, Y.; Zheng, J.; Vecchione, A.; Park, S.Y.; Chen, M.H.; Wong, S.; Xu, R.; Peng, C.Y.; et al. Phase III HEAT Study Adding Lyso-Thermosensitive Liposomal Doxorubicin to Radiofrequency Ablation in Patients with Unresectable Hepatocellular Carcinoma Lesions. Clin. Cancer Res. 2018, 24, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Lee, J.C.; Chen, M.H.; Zhang, Z.Y.; Bai, X.M.; Yin, S.S.; Cao, K.; Wang, S.; Wu, W.; Yan, K. Thermosensitive liposomal doxorubicin plus radiofrequency ablation increased tumor destruction and improved survival in patients with medium and large hepatocellular carcinoma: A randomized, double-blinded, dummy-controlled clinical trial in a single center. J. Cancer Res. Therapeutics 2019, 15, 773–783. [Google Scholar] [CrossRef]
- Yingyuad, P.; Mevel, M.; Prata, C.; Kontogiorgis, C.; Thanou, M.; Miller, A.D. Enzyme-triggered PEGylated siRNA-nanoparticles for controlled release of siRNA. J. RNAi Gene Silencing 2014, 10, 490–499. [Google Scholar] [PubMed]
- Zhang, J.; Shrivastava, S.; Cleveland, R.O.; Rabbitts, T.H. Lipid-mRNA Nanoparticle Designed to Enhance Intracellular Delivery Mediated by Shock Waves. ACS Appl. Mater. Interfaces 2019, 11, 10481–10491. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kularatne, R.N.; Crist, R.M.; Stern, S.T. The Future of Tissue-Targeted Lipid Nanoparticle-Mediated Nucleic Acid Delivery. Pharmaceuticals 2022, 15, 897. https://doi.org/10.3390/ph15070897
Kularatne RN, Crist RM, Stern ST. The Future of Tissue-Targeted Lipid Nanoparticle-Mediated Nucleic Acid Delivery. Pharmaceuticals. 2022; 15(7):897. https://doi.org/10.3390/ph15070897
Chicago/Turabian StyleKularatne, Ruvanthi N., Rachael M. Crist, and Stephan T. Stern. 2022. "The Future of Tissue-Targeted Lipid Nanoparticle-Mediated Nucleic Acid Delivery" Pharmaceuticals 15, no. 7: 897. https://doi.org/10.3390/ph15070897