
The N-Methyl-D-Aspartate Receptor Blocker REL-1017 (Esmethadone) Reduces Calcium Influx Induced by Glutamate, Quinolinic Acid, and Gentamicin

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. QA Acts as a Low-Potency Subtype-Selective NMDAR Partial Agonist, and REL-1017 Reduces QA-Induced Calcium Entry
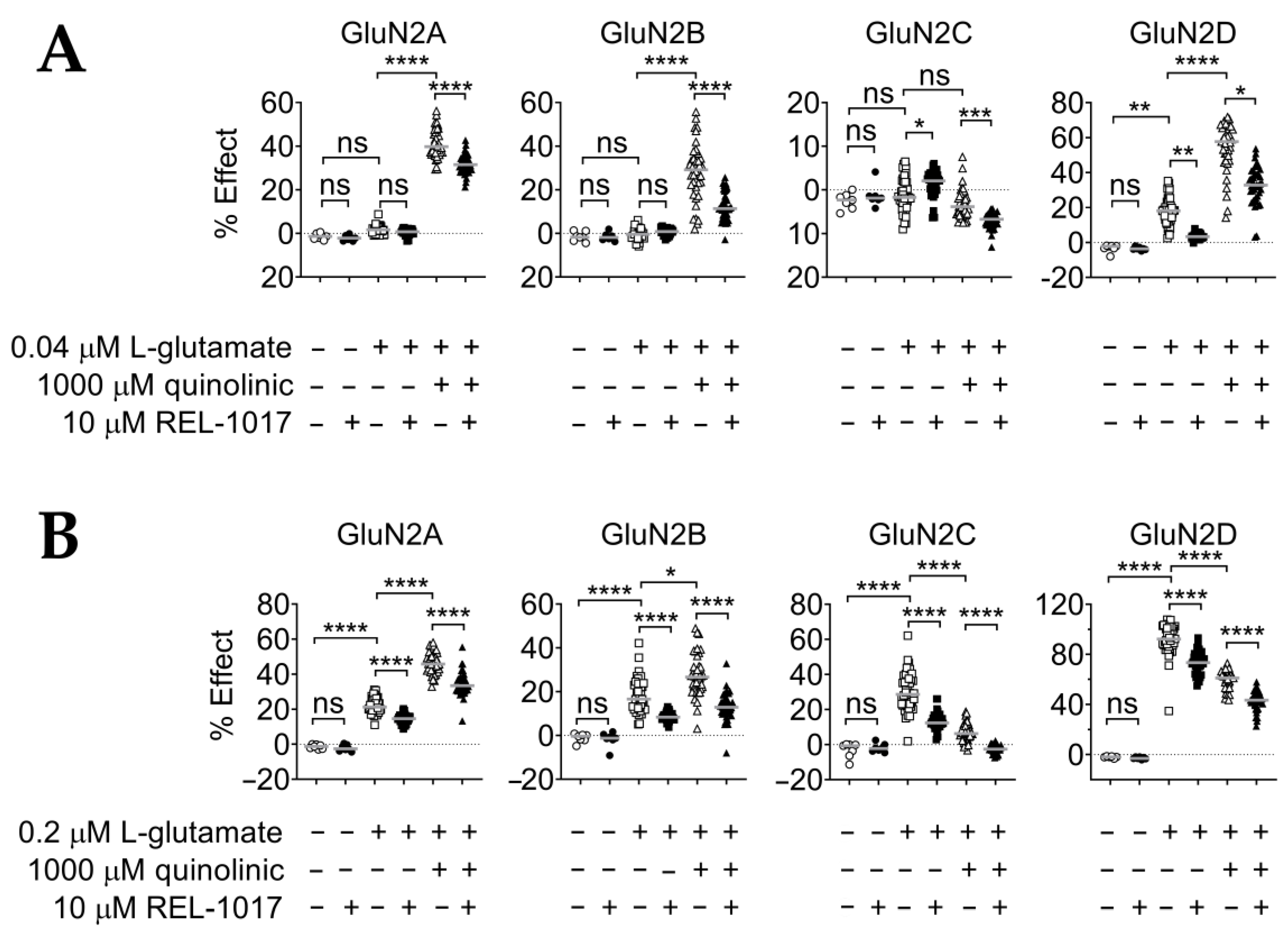
2.2. REL-1017 Reduces Calcium Entry in the Presence of 0.04 μM or 0.2 μM L-Glutamate Plus 1000 μM QA
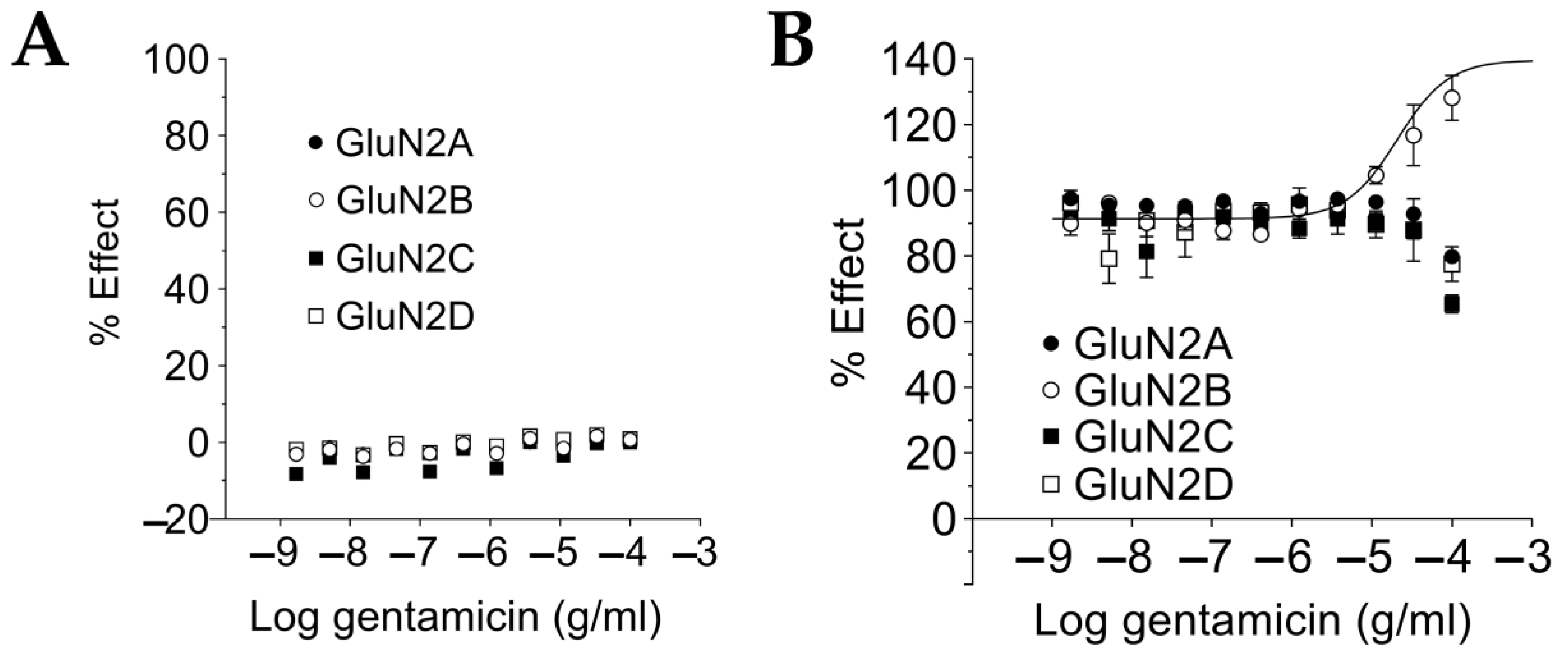
2.3. Gentamicin Is a Selective Positive Modulator at NMDAR GluN1-GluN2B Subtypes for Saturating L-Glutamate Concentration
2.4. Gentamicin Increases NMDAR Affinity for Glutamate at Sub-Saturating L-Glutamate Concentrations in a Glutamate Concentration- and GluN1-GluN2 Subtype-Dependent Manner
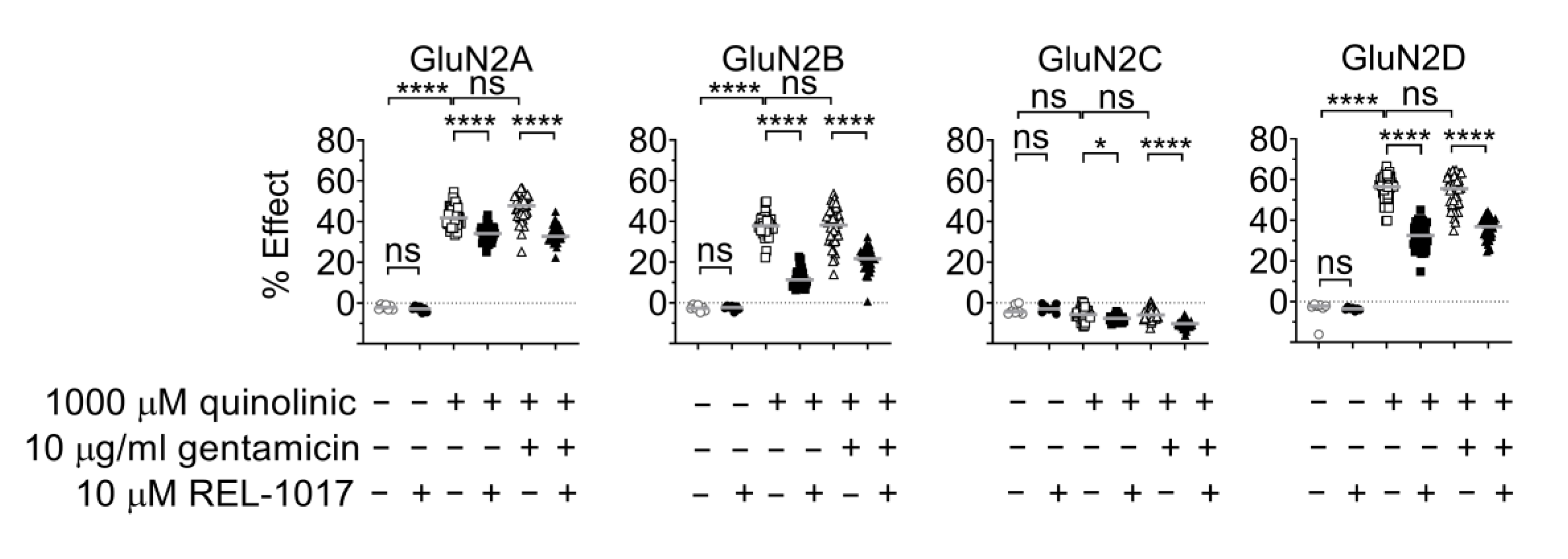
2.5. Gentamicin Is Not a Positive Modulator at NMDARs Activated by Quinolinic Acid
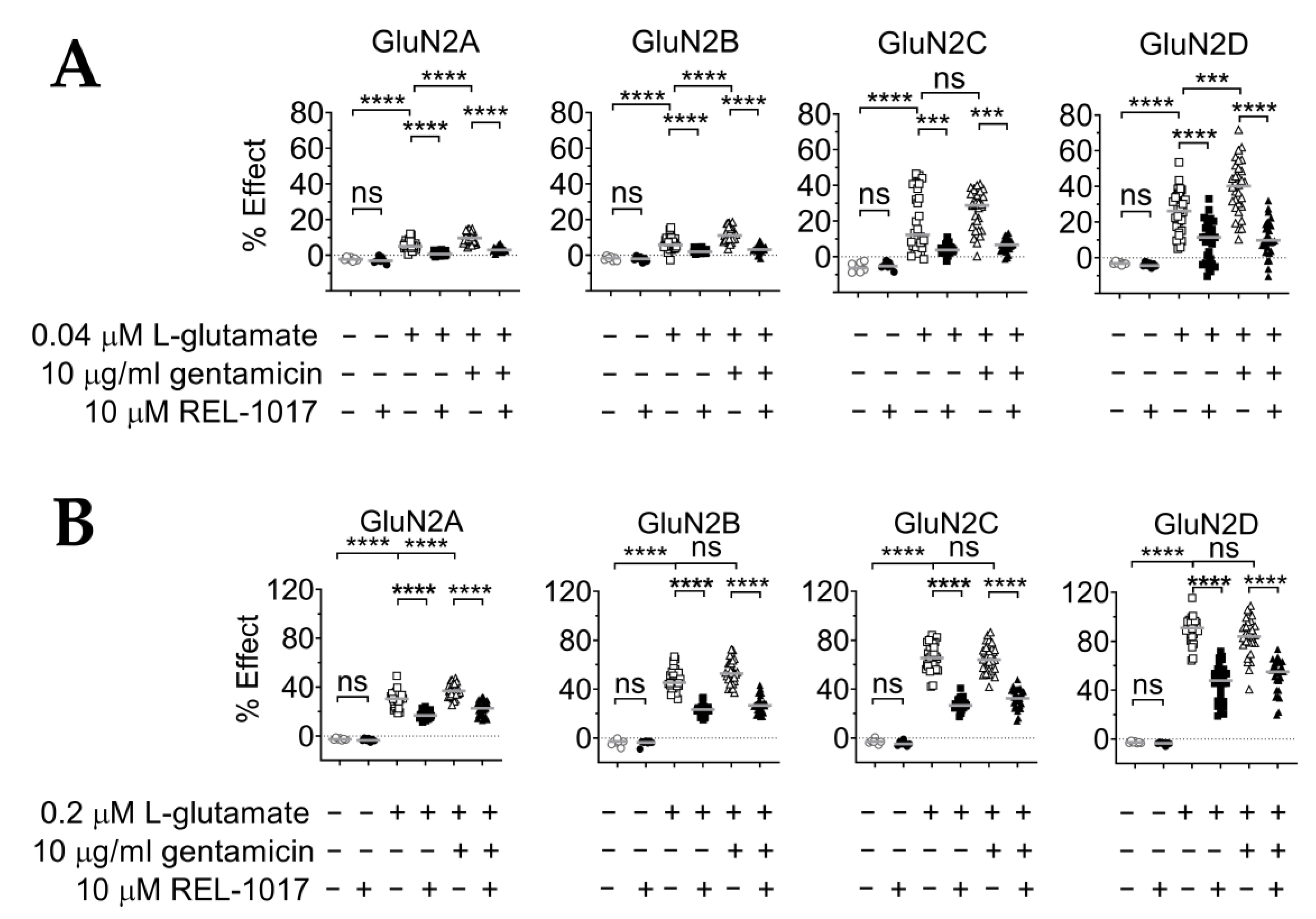
2.6. REL-1017 Lowered Intracellular Calcium Levels in Presence of 10 μM Gentamicin and Low L-Glutamate
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Cell Lines
4.3. FLIPR Assay
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemberg, K.; Kontinen, V.K.; Viljakka, K.; Kylänlahti, I.; Yli-Kauhaluoma, J.; Kalso, E. Morphine, oxycodone, methadone and its enantiomers in different models of nociception in the rat. Anesth. Analg. 2006, 102, 1768–1774. [Google Scholar] [CrossRef]
- Drug Enforcement Administration. Methadone. 2019. Available online: https://www.dea.gov/factsheets/methadone (accessed on 6 July 2022).
- Gorman, A.L.; Elliott, K.J.; Inturrisi, C.E. The d- and l-isomers of methadone bind to the non-competitive site on the N-methyl-D-aspartate (NMDA) receptor in rat forebrain and spinal cord. Neurosci. Lett. 1997, 223, 5–8. [Google Scholar] [CrossRef]
- Hanania, T.; Manfredi, P.; Inturrisi, C.; Vitolo, O.V. The N-methyl-D-aspartate receptor antagonist d-methadone acutely improves depressive-like behavior in the forced swim test performance of rats. Exp. Clin. Psychopharmacol. 2020, 28, 196–201. [Google Scholar] [CrossRef]
- Fogaça, M.V.; Fukumoto, K.; Franklin, T.; Liu, R.-J.; Duman, C.H.; Vitolo, O.V.; Duman, R.S. N-Methyl-D-aspartate receptor antagonist d-methadone produces rapid, mTORC1-dependent antidepressant effects. Neuropsychopharmacology 2019, 44, 2230–2238. [Google Scholar] [CrossRef]
- Fava, M.; Stahl, S.; Pani, L.; De Martin, S.; Pappagallo, M.; Guidetti, C.; Alimonti, A.; Bettini, E.; Mangano, R.M.; Wessel, T.; et al. REL-1017 (Esmethadone) as Adjunctive Treatment in Patients with Major Depressive Disorder: A Phase 2a Randomized Double-Blind Trial. Am. J. Psychiatry 2022, 179, 122–131. [Google Scholar] [CrossRef]
- Bernstein, G.; Davis, K.; Mills, C.; Wang, L.; McDonnell, M.; Oldenhof, J.; Inturrisi, C.; Manfredi, P.L.; Vitolo, O.V. Characterization of the Safety and Pharmacokinetic Profile of D-Methadone, a Novel N-Methyl-D-Aspartate Receptor Antagonist in Healthy, Opioid-Naive Subjects: Results of Two Phase 1 Studies. J. Clin. Psychopharmacol. 2019, 39, 226–237. [Google Scholar] [CrossRef]
- Olney, J.W.; Labruyere, J.; Price, M.T. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 1989, 244, 1360–1362. [Google Scholar] [CrossRef] [PubMed]
- Bifari, F.; Pappagallo, M.; Bleavins, M.; Traversa, S.; Folli, F.; Manfredi, P.L. REL-1017 (esmethadone), a novel NMDAR blocker for the treatment of MDD is not neurotoxic in Sprague-Dawley rats. Front. Pharmacol. 2022, 13, 863959. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.P.; Lackner, A.; et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992, 115, 1249–1273. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic Acid: An Endogenous Neurotoxin with Multiple Targets. Oxid. Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [Green Version]
- Bansal, Y.; Singh, R.; Parhar, I.; Kuhad, A.; Soga, T. Quinolinic Acid and Nuclear Factor Erythroid 2-Related Factor 2 in Depression: Role in Neuroprogression. Front. Pharmacol. 2019, 10, 452. [Google Scholar] [CrossRef]
- Erhardt, S.; Lim, C.K.; Linderholm, K.R.; Janelidze, S.; Lindqvist, D.; Samuelsson, M.; Lundberg, K.; Postolache, T.T.; Träskman-Bendz, L.; Guillemin, G.J.; et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 2013, 38, 743–752. [Google Scholar] [CrossRef]
- Kindler, J.; Lim, C.K.; Weickert, C.S.; Boerrigter, D.; Galletly, C.; Liu, D.; Jacobs, K.R.; Balzan, R.; Bruggemann, J.; O’Donnell, M.; et al. Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol. Psychiatry 2020, 25, 2860–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, W.; McGuinness, A.J.; Rocks, T.; Ruusunen, A.; Cleminson, J.; Walker, A.J.; Gomes-da-Costa, S.; Lane, M.; Sanches, M.; Diaz, A.P.; et al. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: A meta-analysis of 101 studies. Mol. Psychiatry 2021, 26, 4158–4178. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Ford, B.N.; Yeh, H.W.; Akeman, E.; Cosgrove, K.; Clausen, A.N.; Martell, C.; Kirlic, N.; Santiago, J.; Teague, T.K.; et al. Behavioral activation therapy for depression is associated with a reduction in the concentration of circulating quinolinic acid. Psychol. Med. 2020, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, F.; Petit, A.C.; Abdel-Ahad, P.; Vinckier, F.; Jouvion, G.; de Maricourt, P.; De Medeiros, G.F.; Danckaert, A.; Van Steenwinckel, J.; Blatzer, M.; et al. Microglial production of quinolinic acid as a target and a biomarker of the antidepressant effect of ketamine. Brain Behav. Immun. 2019, 81, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Budac, D.P.; Bisulco, S.; Lee, A.W.; Smith, R.A.; Beenders, B.; Kelley, K.W.; Dantzer, R. NMDA Receptor Blockade by Ketamine Abrogates Lipopolysaccharide-Induced Depressive-Like Behavior in C57BL/6J Mice. Neuropsychopharmacology 2013, 38, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Wurfel, B.E.; Drevets, W.C.; Bliss, S.A.; McMillin, J.R.; Suzuki, H.; Ford, B.N.; Morris, H.M.; Teague, T.K.; Dantzer, R.; Savitz, J.B. Serum kynurenic acid is reduced in affective psychosis. Transl. Psychiatry 2017, 7, e1115. [Google Scholar] [CrossRef] [Green Version]
- Yellon, R.F.; Rose, E.; Kenna, M.A.; Doyle, W.J.; Casselbrant, M.; Diven, W.F.; Whiteside, T.L.; Swarts, J.D.; Heyes, M.P. Sensorineural hearing loss from quinolinic acid: A neurotoxin in middle ear effusions. Laryngoscope 1994, 104, 176–181. [Google Scholar] [CrossRef]
- Xiao, H.; Yang, C.; He, Y.; Zheng, N. Neurotoxicity of quinolinic acid to spiral ganglion cells in rats. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 397–402. [Google Scholar] [CrossRef]
- Boldrini, M.; Canoll, P.D.; Klein, R.S. How COVID-19 Affects the Brain. JAMA Psychiatry 2021, 78, 682–683. [Google Scholar] [CrossRef] [PubMed]
- Collier, M.E.W.; Zhang, S.; Scrutton, N.S.; Giorgini, F. Inflammation control and improvement of cognitive function in COVID-19 infections: Is there a role for kynurenine 3-monooxygenase inhibition? Drug Discov. Today 2021, 26, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Behan, W.M.; McDonald, M.; Darlington, L.G.; Stone, T.W. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: Protection by melatonin and deprenyl. Br. J. Pharmacol. 1999, 128, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Perkins, M.N. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Moroni, F. Tryptophan metabolism and brain function: Focus on kynurenine and other indole metabolites. Eur. J. Pharmacol. 1999, 375, 87–100. [Google Scholar] [CrossRef]
- Monaghan, D.T.; Beaton, J.A. Quinolinate differentiates between forebrain and cerebellar NMDA receptors. Eur. J. Pharmacol. 1991, 194, 123–125. [Google Scholar] [CrossRef]
- de Carvalho, L.P.; Bochet, P.; Rossier, J. The endogenous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem. Int. 1996, 28, 445–452. [Google Scholar] [CrossRef]
- Banke, T.G.; Traynelis, S.F. Activation of NR1/NR2B NMDA receptors. Nat. Neurosci. 2003, 6, 144–152. [Google Scholar] [CrossRef]
- Kussius, C.L.; Popescu, G.K. Kinetic basis of partial agonism at NMDA receptors. Nat. Neurosci. 2009, 12, 1114–1120. [Google Scholar] [CrossRef]
- Kussius, C.L.; Popescu, A.M.; Popescu, G.K. Agonist-specific gating of NMDA receptors. Channels 2010, 4, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.; Bonus, M.; Gohlke, H.; Klöcker, N. Isoform-specific Inhibition of N-methyl-D-aspartate Receptors by Bile Salts. Sci. Rep. 2019, 9, 10068. [Google Scholar] [CrossRef]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef]
- Basile, A.S.; Saito, K.; al-Mardini, H.; Record, C.O.; Hughes, R.D.; Harrison, P.; Williams, R.; Li, Y.; Heyes, M.P. The relationship between plasma and brain quinolinic acid levels and the severity of hepatic encephalopathy. Gastroenterology 1995, 108, 818–823. [Google Scholar] [CrossRef]
- Saito, K.; Fujigaki, S.; Heyes, M.P.; Shibata, K.; Takemura, M.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Mechanism of increases in l-kynurenine and quinolinic acid in renal insufficiency. Am. J. Physiol. Renal Physiol. 2000, 279, F565–F572. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, K.; Brzosko, S.; Mysliwiec, M.; Pawlak, D. Kynurenine, quinolinic acid—The new factors linked to carotid atherosclerosis in patients with end-stage renal disease. Atherosclerosis 2009, 204, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Brunton, L.L.; Knollmann, B.R.C.; Hilal-Dandan, R. Goodman & Gilman’s the Pharmacological Basis Of Therapeutics, 13th ed.; McGraw Hill Medical: New York, NY, USA, 2018; p. xiii. ISBN 978-00-7162-442-8. [Google Scholar]
- Huth, M.E.; Ricci, A.J.; Cheng, A.G. Mechanisms of Aminoglycoside Ototoxicity and Targets of Hair Cell Protection. Int. J. Otolaryngol. 2011, 2011, 937861. [Google Scholar] [CrossRef] [Green Version]
- Medić, B.; Stojanović, M.; Rovčanin, B.; Kekić, D.; Škodrić, S.R.; Jovanović, G.B.; Vujović, K.S.; Divac, N.; Stojanović, R.; Radenković, M.; et al. Pioglitazone attenuates kidney injury in an experimental model of gentamicin-induced nephrotoxicity in rats. Sci. Rep. 2019, 9, 13689. [Google Scholar] [CrossRef]
- Basile, A.S.; Huang, J.M.; Xie, C.; Webster, D.; Berlin, C.; Skolnick, P. N-methyl-D-aspartate antagonists limit aminoglycoside antibiotic-induced hearing loss. Nat. Med. 1996, 2, 1338–1343. [Google Scholar] [CrossRef]
- Hong, J.; Chen, Y.; Zhang, Y.; Li, J.; Ren, L.; Yang, L.; Shi, L.; Li, A.; Zhang, T.; Li, H.; et al. N-Methyl-D-Aspartate Receptors Involvement in the Gentamicin-Induced Hearing Loss and Pathological Changes of Ribbon Synapse in the Mouse Cochlear Inner Hair Cells. Neural Plast. 2018, 2018, 3989201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, J.C.; Marphis, T.; Craver, R.D.; Silverstein, D.M. Altered NMDA receptor expression in renal toxicity: Protection with a receptor antagonist. Kidney Int. 2004, 66, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, J.A.; Harris, B.D.; Kustova, Y.; Basile, A.; Skolnick, P. Aminoglycoside neurotoxicity involves NMDA receptor activation. Brain Res. 1999, 815, 270–277. [Google Scholar] [CrossRef]
- Sanchez, J.T.; Ghelani, S.; Otto-Meyer, S. From development to disease: Diverse functions of NMDA-type glutamate receptors in the lower auditory pathway. Neuroscience 2015, 285, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Valdivielso, J.M.; Eritja, À.; Caus, M.; Bozic, M. Glutamate-Gated NMDA Receptors: Insights into the Function and Signaling in the Kidney. Biomolecules 2020, 10, 1051. [Google Scholar] [CrossRef]
- Masuko, T.; Kuno, T.; Kashiwagi, K.; Kusama, T.; Williams, K.; Igarashi, K. Stimulatory and inhibitory properties of aminoglycoside antibiotics at N-methyl-D-aspartate receptors. J. Pharmacol. Exp. Ther. 1999, 290, 1026–1033. [Google Scholar]
- Segal, J.A.; Skolnick, P. Polyamine-like actions of aminoglycosides and aminoglycoside derivatives at NMDA receptors. Eur. J. Pharmacol. 1998, 347, 311–317. [Google Scholar] [CrossRef]
- Harvey, S.C.; Skolnick, P. Polyamine-like actions of aminoglycosides at recombinant N-methyl-D-aspartate receptors. J. Pharmacol. Exp. Ther. 1999, 291, 285–291. [Google Scholar]
- Tang, W.; Liu, D.; Traynelis, S.F.; Yuan, H. Positive allosteric modulators that target NMDA receptors rectify loss-of-function GRIN variants associated with neurological and neuropsychiatric disorders. Neuropharmacology 2020, 177, 108247. [Google Scholar] [CrossRef]
- Nicoll, R.A. A Brief History of Long-Term Potentiation. Neuron 2017, 93, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Narducci, R.; Mannaioni, G. Harnessing ionic mechanisms to achieve disease modification in neurodegenerative disorders. Pharmacol. Res. 2019, 147, 104343. [Google Scholar] [CrossRef]
- Du, J.; Zhu, M.; Bao, H.; Li, B.; Dong, Y.; Xiao, C.; Zhang, G.Y.; Henter, I.; Rudorfer, M.; Vitiello, B. The Role of Nutrients in Protecting Mitochondrial Function and Neurotransmitter Signaling: Implications for the Treatment of Depression, PTSD, and Suicidal Behaviors. Crit. Rev. Food Sci. Nutr. 2016, 56, 2560–2578. [Google Scholar] [CrossRef] [Green Version]
- Sah, P.; Hestrin, S.; Nicoll, R.A. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 1989, 246, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.; Armbruster, M.; Lau, L.A.; Sommer, M.E.; Klaft, Z.-J.; Swanger, S.A.; Traynelis, S.F.; Moss, S.J.; Noubary, F.; Chadchankar, J.; et al. Tonic Activation of GluN2C/GluN2D-Containing NMDA Receptors by Ambient Glutamate Facilitates Cortical Interneuron Maturation. J. Neurosci. 2019, 39, 3611–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Sutton, M.A.; Taylor, A.M.; Ito, H.T.; Pham, A.; Schuman, E.M. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 2007, 55, 648–661. [Google Scholar] [CrossRef] [Green Version]
- Bettini, E.; Nola, S.; De Martin, S.; Inturrisi, C.; Mattarei, A.; Pappagallo, M.; Stahl, S.; Traversa, S.; Manfredi, P. Esmethadone (REL-1017) Reduces Glutamate-Induced Currents in NMDA Receptors with the GluN2D Subunit. Biol. Psychiatry 2021, 89, S198. [Google Scholar] [CrossRef]
- Hackos, D.H.; Hanson, J.E. Diverse modes of NMDA receptor positive allosteric modulation: Mechanisms and consequences. Neuropharmacology 2017, 112, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Li, X.H.; Li, Y.J. Glutamate in peripheral organs: Biology and pharmacology. Eur. J. Pharmacol. 2016, 784, 42–48. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A. Developmental Origins of Disease Paradigm: A Mechanistic and Evolutionary Perspective. Pediatr. Res. 2004, 56, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Bettini, E.; Sava, A.; Griffante, C.; Carignani, C.; Buson, A.; Capelli, A.M.; Negri, M.; Andreetta, F.; Senar-Sancho, S.A.; Guiral, L.; et al. Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J. Pharmacol. Exp. Ther. 2010, 335, 636–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Multiple Comparisons Test | Tukey’s GluN2A p-Value | Tukey’s GluN2B p-Value | Tukey’s GluN2C p-Value | Tukey’s GluN2D p-Value |
|---|---|---|---|---|
| Buffer vs. 10 μM REL-1017 | 0.9851 | 0.9851 | 0.3757 | 0.9882 |
| Buffer vs. 1000 μM QA | <0.0001 | <0.0001 | 0.0103 | <0.0001 |
| Buffer vs. 1000 μM QA + 10 μM REL-1017 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 10 μM REL-1017 vs. 1000 μM QA | <0.0001 | <0.0001 | 0.7045 | <0.0001 |
| 10 μM REL-1017 vs. 1000 μM QA + 10 μM REL-1017 | <0.0001 | <0.0001 | 0.0054 | <0.0001 |
| 1000 μM QA vs. 1000 μM QA + 10 μM REL-1017 | <0.0001 | <0.0001 | 0.0002 | <0.0001 |
| Fisher value F and degrees of freedom dF1 and dF2 [3,92] | 442 | 266 | 14 | 319 |
| Multiple Comparisons Test | Tukey’s GluN2A p-Value | Tukey’s GluN2B p-Value | Dunn’s GluN2C p-Value | Dunn’s GluN2D p-Value |
|---|---|---|---|---|
| Buffer vs. 10 μM REL-1017 | 0.9991 | >0.9999 | >0.9999 | >0.9999 |
| Buffer vs. 0.04 μM L-glu | 0.4072 | 0.9992 | >0.9999 | 0.0088 |
| 0.04 μM L-glu vs. 0.04 μM L-glu + 10 μM REL-1017 | 0.7378 | 0.9878 | 0.0410 | 0.0014 |
| 0.04 μM L-glu vs. 0.04 μM L-glu + 1000 μM QA | <0.0001 | <0.0001 | 0.5041 | <0.0001 |
| 0.04 μM L-glu + 1000 μM QA vs. 0.04 μM L-glu + 1000 μM QA + 10 μM REL-1017 | <0.0001 | <0.0001 | 0.0002 | 0.0189 |
| Fisher value F and degrees of freedom dF1 and dF2 [5,176] | 775 | 94 |
| Multiple Comparisons Test | Tukey’s GluN2A p-Value | Dunn’s GluN2B p-Value | Tukey’s GluN2C p-Value | Tukey’s GluN2D p-Value |
|---|---|---|---|---|
| Buffer vs. 10 μM REL-1017 | 0.9998 | >0.9999 | 0.9992 | >0.9999 |
| Buffer vs. 0.2 μM L-glu | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 0.2 μM L-glu vs. 0.2 μM L-glu + 10 μM REL-1017 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 0.2 μM L-glu vs. 0.2 μM L-glu + 1000 μM QA | <0.0001 | 0.0358 | <0.0001 | <0.0001 |
| 0.2 μM L-glu + 1000 μM QA vs. 0.2 μM L-glu + 1000 μM QA + 10 μM REL-1017 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| Fisher value F and degrees of freedom dF1 and dF2 [5,176] | 307 | 128 | 275 |
| Multiple Comparisons Test | Tukey’s GluN2A p-Value | Tukey’s GluN2B p-Value | Dunn’s GluN2C p-Value | Tukey’s GluN2D p-Value |
|---|---|---|---|---|
| Buffer vs. 10 μM REL-1017 | 0.9916 | >0.9999 | >0.9999 | >0.9999 |
| Buffer vs. 0.04 μM L-glu | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 0.04 μM L-glu vs. 0.04 μM L-glu + 10 μM REL-1017 | <0.0001 | <0.0001 | 0.0006 | <0.0001 |
| 0.04 μM L-glu vs. 0.04 μM L-glu + 10 μg/mL gentamicin | <0.0001 | <0.0001 | 0.6481 | 0.0002 |
| 0.04 μM L-glu + 10 μg/mL gentamicin vs. 0.04 μM L-glu + 10 μg/mL gentamicin + 10 μM REL-1017 | <0.0001 | <0.0001 | 0.0002 | <0.0001 |
| Fisher value F and degrees of freedom dF1 and dF2 [5,176] | 87 | 55 | 38 |
| Multiple Comparisons Test | Tukey’s GluN2A p-Value | Dunn’s GluN2B p-Value | Tukey’s GluN2C p-Value | Dunn’s GluN2D p-Value |
|---|---|---|---|---|
| Buffer vs. 10 μM REL-1017 | 0.9998 | >0.9999 | 0.9996 | >0.9999 |
| Buffer vs. 0.2 μM L-glu | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 0.2 μM L-glu vs. 0.2 μM L-glu + 10 μM REL-1017 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 0.2 μM L-glu vs. 0.2 μM L-glu + 10 μg/mL gentamicin | <0.0001 | >0.9999 | 0.9951 | >0.9999 |
| 0.2 μM L-glu + 10 μg/mL gentamicin vs. 0.2 μM L-glu + 10 μg/mL gentamicin + 10 μM REL-1017 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| Fisher value F and degrees of freedom dF1 and dF2 [5,176] | 139 | 186 |
| Multiple Comparisons Test | Dunn‘s GluN2A p-Value | Dunn‘s GluN2B p-Value | Dunn‘s GluN2C p-Value | Dunn‘s GluN2D p-Value |
|---|---|---|---|---|
| Buffer vs. 10 μM REL-1017 | >0.9999 | >0.9999 | >0.9999 | >0.9999 |
| Buffer vs. 1000 μM QA | <0.0001 | <0.0001 | >0.9999 | <0.0001 |
| 1000 μM QA vs. 1000 μM QA + 10 μM REL-1017 | <0.0001 | <0.0001 | 0.0125 | <0.0001 |
| 1000 μM QA vs. 1000 μM QA + 10 μg/mL gentamicin | 0.8007 | >0.9999 | >0.9999 | >0.9999 |
| 1000 μM QA + 10 μg/mL gentamicin vs. 1000 μM QA + 10 μg/mL gentamicin + 10 μM REL-1017 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bettini, E.; De Martin, S.; Mattarei, A.; Pappagallo, M.; Stahl, S.M.; Bifari, F.; Inturrisi, C.E.; Folli, F.; Traversa, S.; Manfredi, P.L. The N-Methyl-D-Aspartate Receptor Blocker REL-1017 (Esmethadone) Reduces Calcium Influx Induced by Glutamate, Quinolinic Acid, and Gentamicin. Pharmaceuticals 2022, 15, 882. https://doi.org/10.3390/ph15070882
Bettini E, De Martin S, Mattarei A, Pappagallo M, Stahl SM, Bifari F, Inturrisi CE, Folli F, Traversa S, Manfredi PL. The N-Methyl-D-Aspartate Receptor Blocker REL-1017 (Esmethadone) Reduces Calcium Influx Induced by Glutamate, Quinolinic Acid, and Gentamicin. Pharmaceuticals. 2022; 15(7):882. https://doi.org/10.3390/ph15070882
Chicago/Turabian StyleBettini, Ezio, Sara De Martin, Andrea Mattarei, Marco Pappagallo, Stephen M. Stahl, Francesco Bifari, Charles E. Inturrisi, Franco Folli, Sergio Traversa, and Paolo L. Manfredi. 2022. "The N-Methyl-D-Aspartate Receptor Blocker REL-1017 (Esmethadone) Reduces Calcium Influx Induced by Glutamate, Quinolinic Acid, and Gentamicin" Pharmaceuticals 15, no. 7: 882. https://doi.org/10.3390/ph15070882