
Identification of NAPRT Inhibitors with Anti-Cancer Properties by In Silico Drug Discovery


 , , ,
, , ,  , , , ,
, , , ,
Abstract
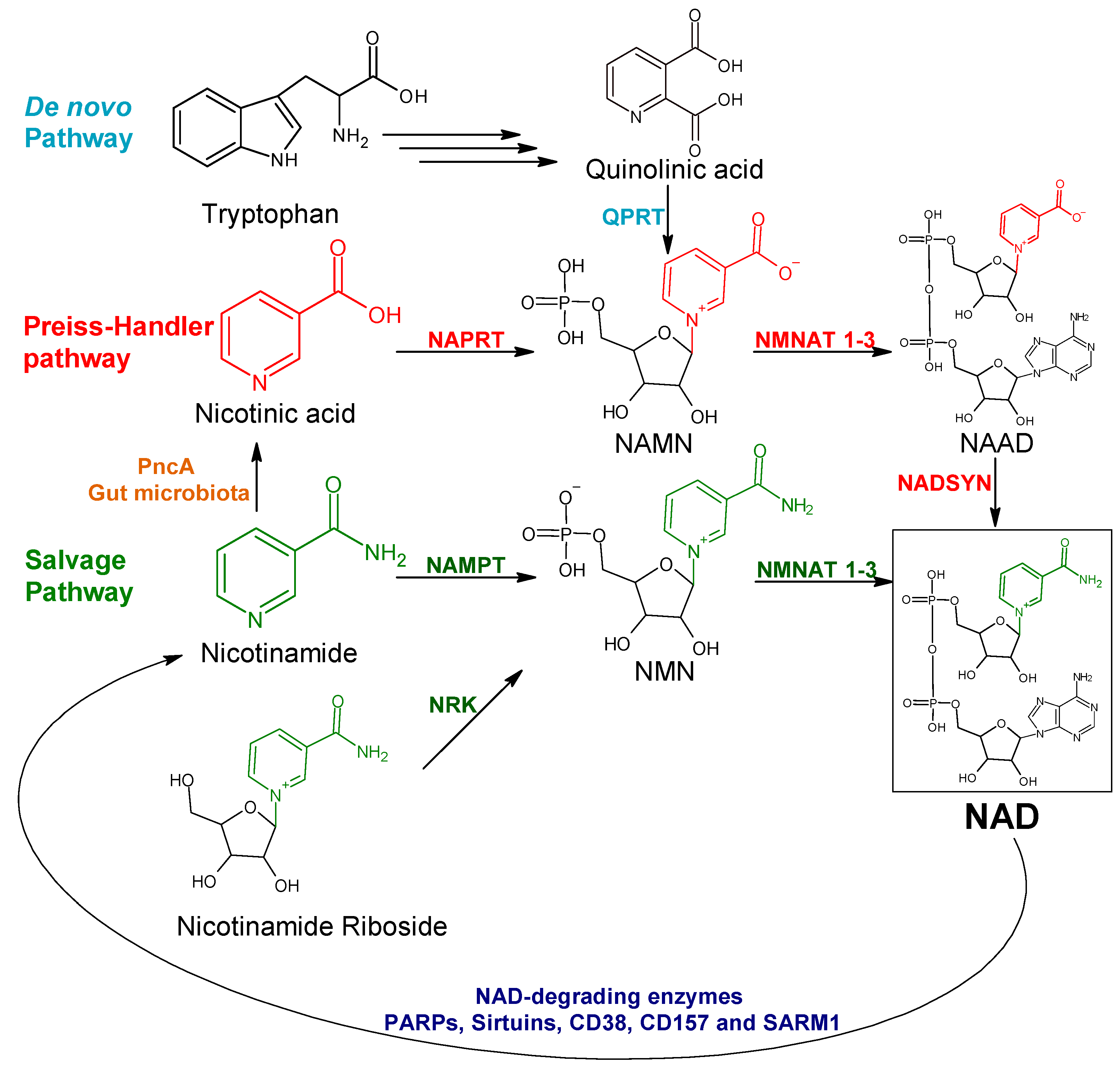
:1. Introduction
2. Results
2.1. Structure-Based Virtual High-Throughput Screening
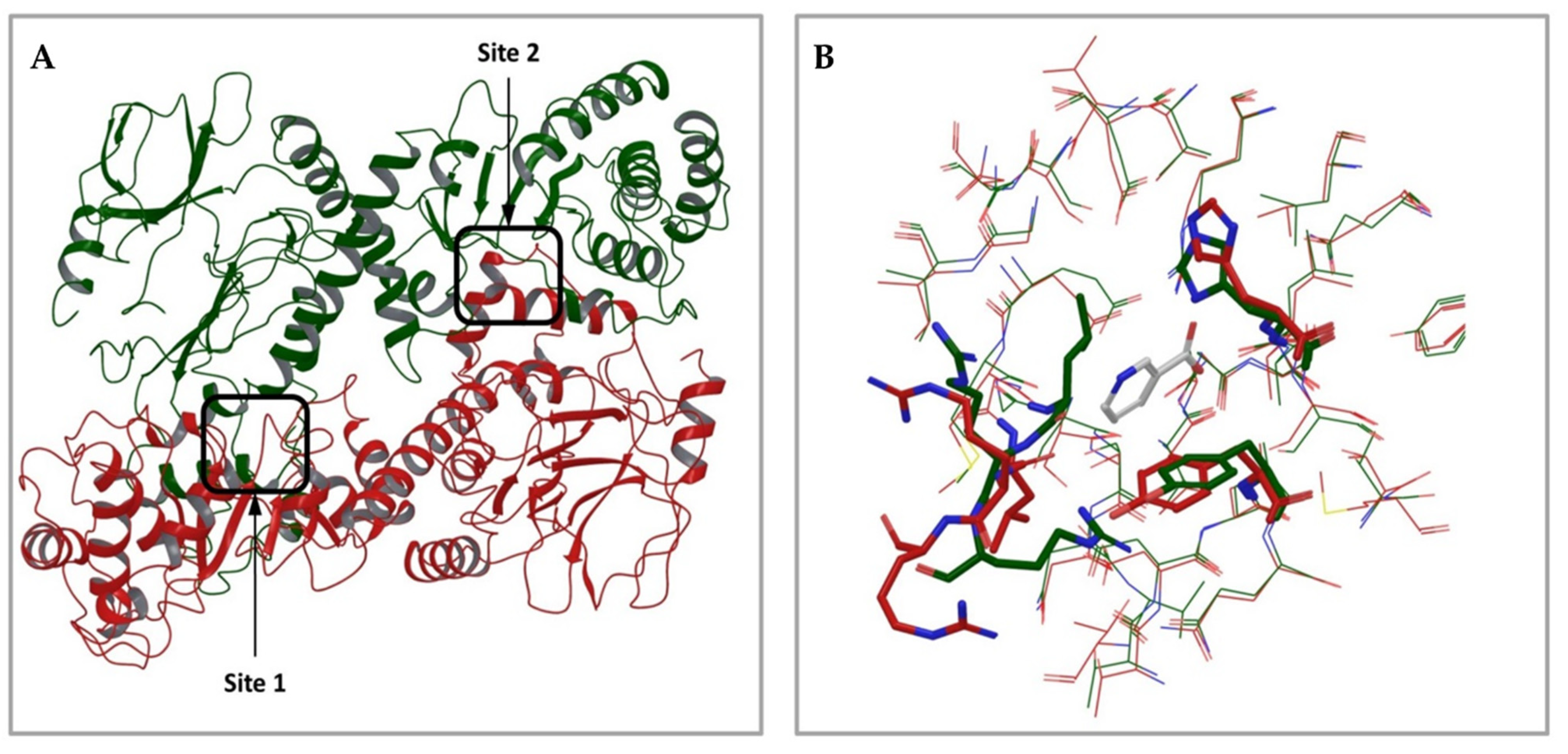
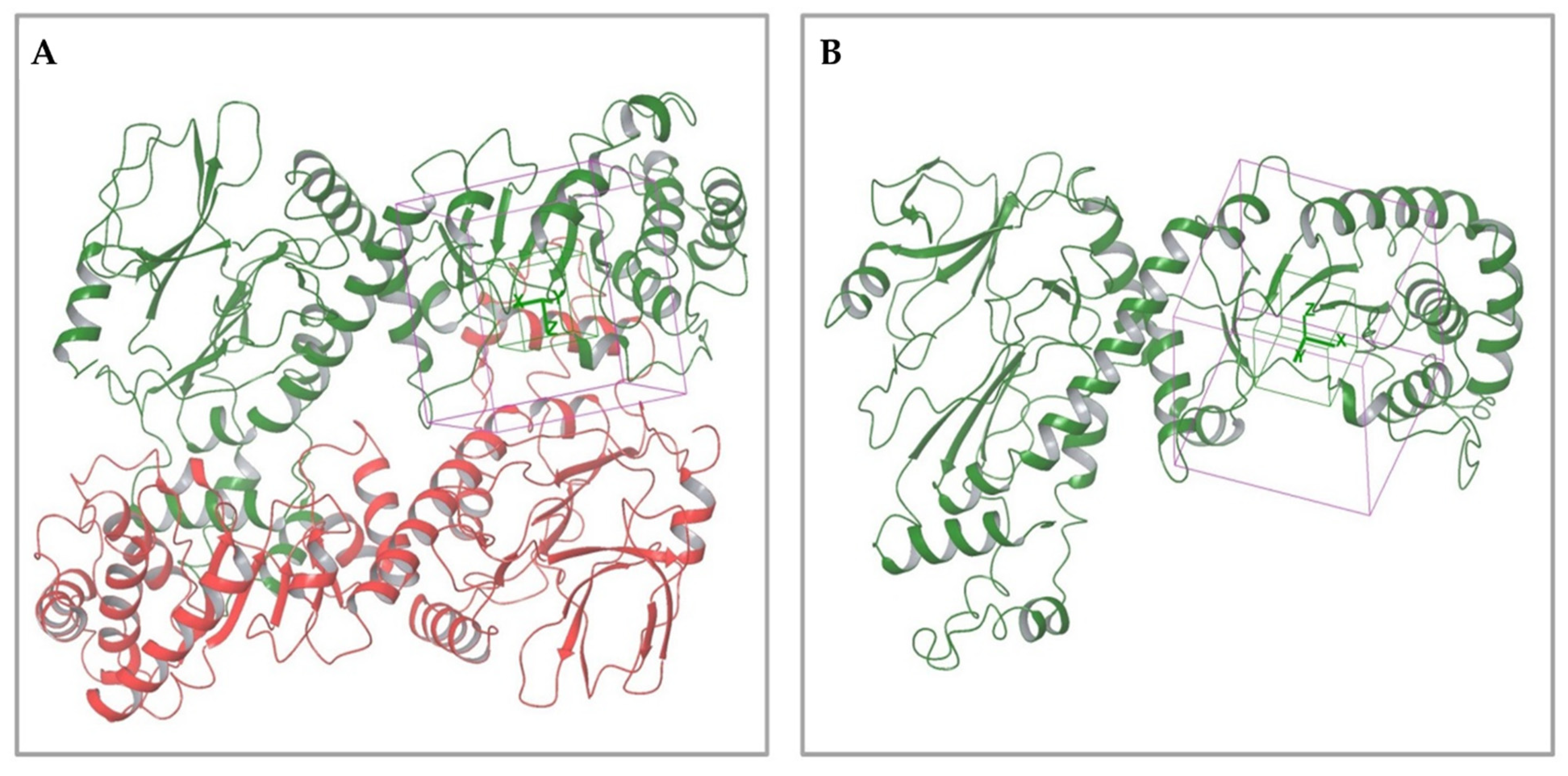
2.1.1. Analysis of Available 3D Structures of NAPRT
2.1.2. Virtual Screening Procedure
2.2. Biological Annotation of the Selected Compounds
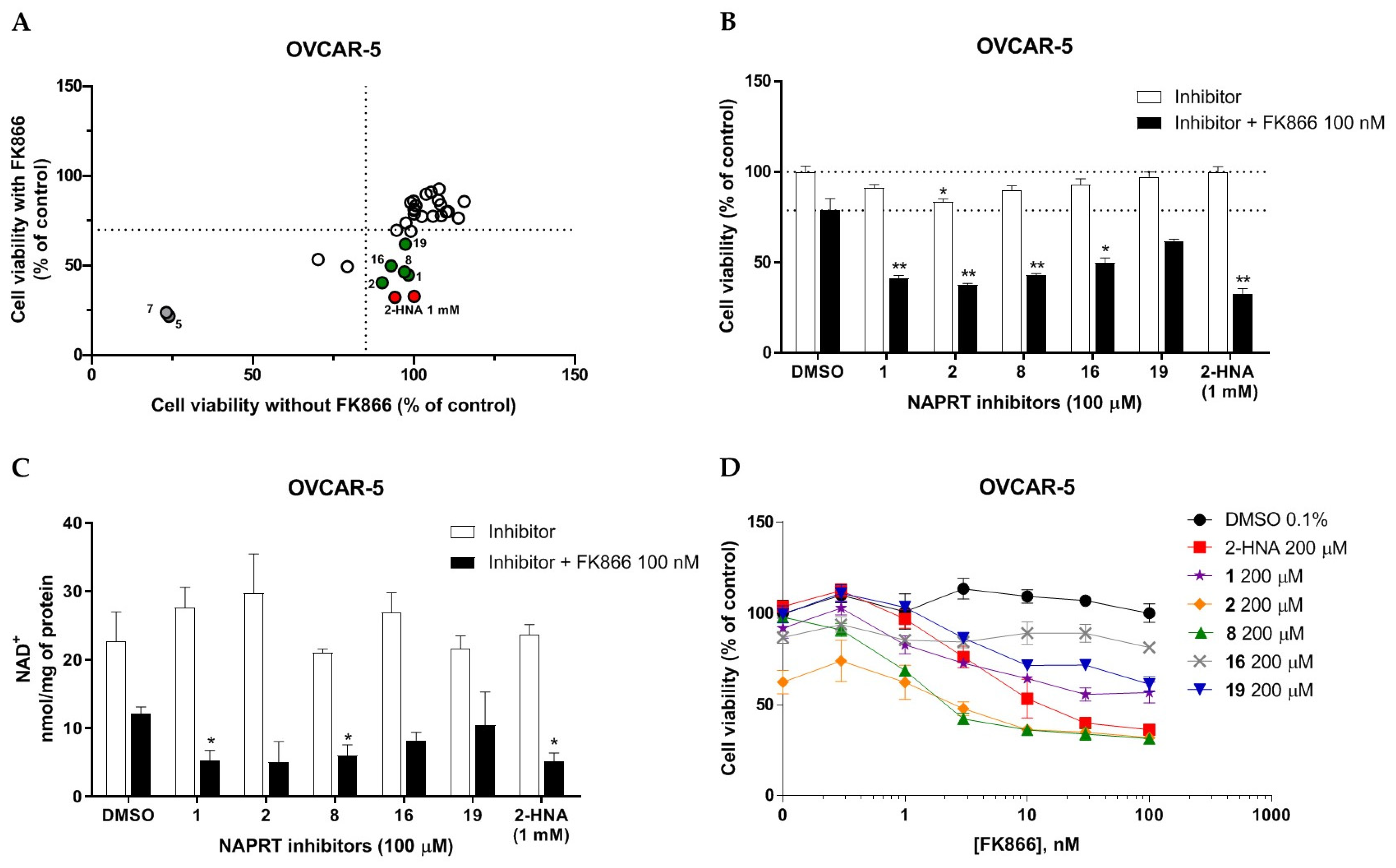
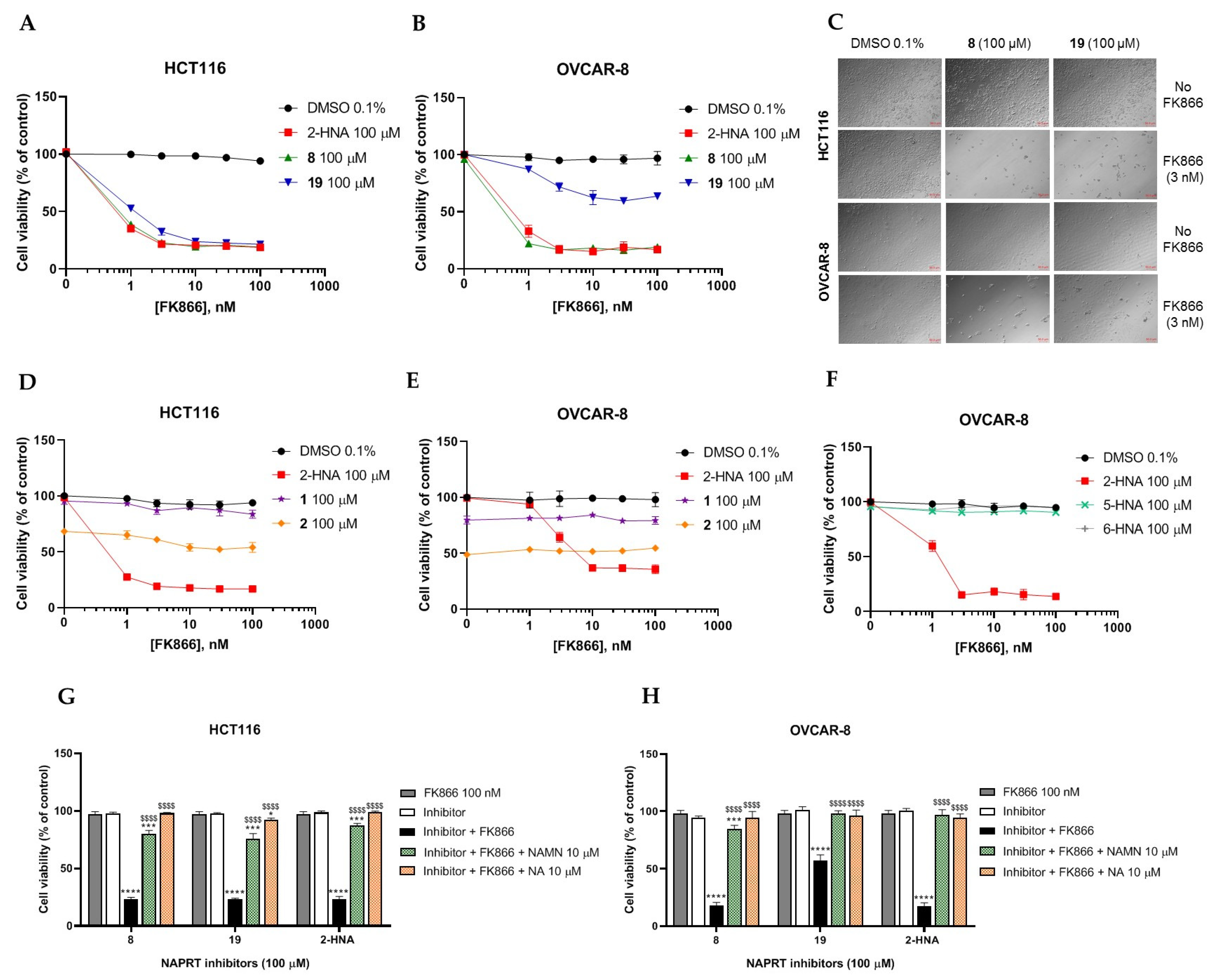
2.2.1. In Vitro Compound Screening
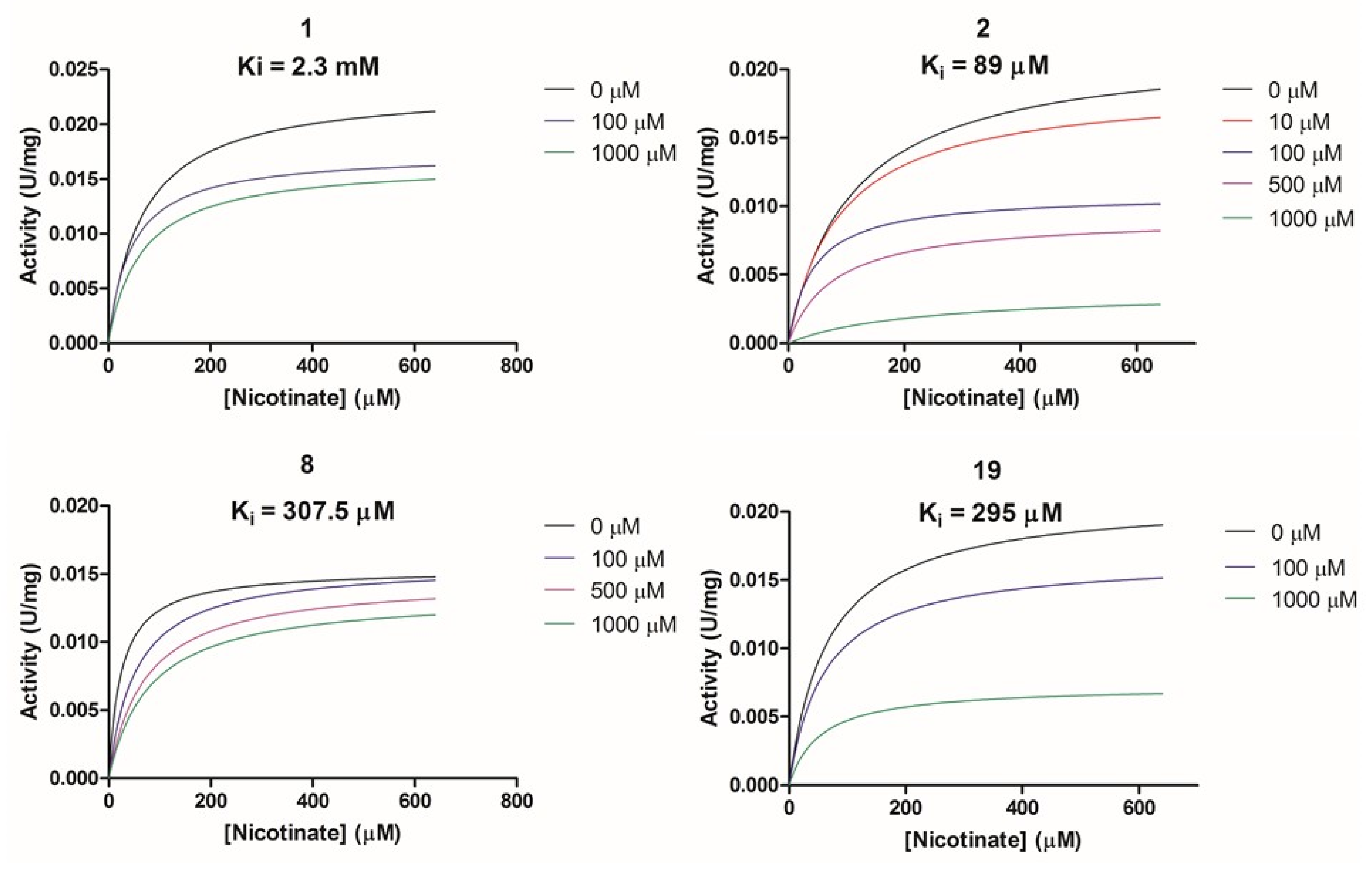
2.2.2. Biochemical Activity on Recombinant Human NAPRT
2.3. In Silico Solubility Prediction and Pharmacokinetic Characterization
3. Discussion
4. Materials and Methods
4.1. Reagents and Cell Lines
4.2. Sulforhodamine B (SRB)Assay
4.3. Intracellular NAD Levels Measurements
4.4. Recombinant Human NAPRT Production and Purification
4.5. Enzymatic Activity Assays and Ki Calculation
4.6. In Silico Screening of the Life Chemicals HTS Compound Collection
4.7. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD Metabolome—A Key Determinant of Cancer Cell Biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD + Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 1–37. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ Metabolism, Stemness, the Immune Response, and Cancer. Signal Transduct. Target. Ther. 2021, 6, 1–20. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ Metabolism and Its Roles in Cellular Processes during Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Preiss, J.; Handler, P. Biosynthesis of Diphosphopyridine Nucleotide I. Identification of intermediates. J. Biol. Chem. 1958, 233, 488–492. [Google Scholar] [CrossRef]
- Preiss, J.; Handler, P. Biosynthesis of Diphosphopyridine Nucleotide. II. Enzymatic Aspects. J. Biol. Chem. 1958, 233, 493–500. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of Cellular NAD Levels by Nicotinic Acid and Involvement of Nicotinic Acid Phosphoribosyltransferase in Human Cells. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Su, X.; Quinn, W.J.; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef] [Green Version]
- Bogan, K.L.; Brenner, C. Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD + Precursor Vitamins in Human Nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Bieganowski, P.; Brenner, C. Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, N.; Zhang, G.; Sauve, A.A. NRH Salvage and Conversion to NAD + Requires NRH Kinase Activity by Adenosine Kinase. Nat. Metab. 2020, 2, 364–379. [Google Scholar] [CrossRef] [PubMed]
- Giroud-Gerbetant, J.; Joffraud, M.; Giner, M.P.; Cercillieux, A.; Bartova, S.; Makarov, M.V.; Zapata-Pérez, R.; Sánchez-García, J.L.; Houtkooper, R.H.; Migaud, M.E.; et al. A Reduced Form of Nicotinamide Riboside Defines a New Path for NAD+ Biosynthesis and Acts as an Orally Bioavailable NAD+ Precursor. Mol. Metab. 2019, 30, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasmann, M.; Schemainda, I. FK866, a Highly Specific Noncompetitive Inhibitor of Nicotinamide Phosphoribosyltransferase, Represents a Novel Mechanism for Induction of Tumor Cell Apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Hjarnaa, P.J.; Jonsson, E.; Latini, S.; Dhar, S.; Larsson, R.; Bramm, E.; Skov, T.; Binderup, L. CHS 828, a Novel Pyridyl Cyanoguanidine with Potent Antitumor Activity in Vitro and in Vivo. Cancer Res. 1999, 59, 5751–5757. [Google Scholar]
- Ghanem, M.S.; Monacelli, F.; Nencioni, A. Advances in NAD-Lowering Agents for Cancer Treatment. Nutrients 2021, 13, 1665. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef]
- Goldinger, S.M.; Gobbi Bischof, S.; Fink-Puches, R.; Klemke, C.-D.; Dréno, B.; Bagot, M.; Dummer, R. Efficacy and Safety of APO866 in Patients With Refractory or Relapsed Cutaneous T-Cell Lymphoma: A Phase 2 Clinical Trial. JAMA Dermatol. 2016, 152, 837–839. [Google Scholar] [CrossRef] [Green Version]
- Hovstadius, P.; Larsson, R.; Jonsson, E.; Skov, T.; Kissmeyer, A.-M.; Krasilnikoff, K.; Bergh, J.; Karlsson, M.O.; Lönnebo, A.; Ahlgren, J. A Phase I Study of CHS 828 in Patients with Solid Tumor Malignancy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 2843–2850. [Google Scholar]
- von Heideman, A.; Berglund, A.; Larsson, R.; Nygren, P. Safety and Efficacy of NAD Depleting Cancer Drugs: Results of a Phase I Clinical Trial of CHS 828 and Overview of Published Data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holen, K.; Saltz, L.B.; Hollywood, E.; Burk, K.; Hanauske, A.-R. The Pharmacokinetics, Toxicities, and Biologic Effects of FK866, a Nicotinamide Adenine Dinucleotide Biosynthesis Inhibitor. Invest. New Drugs 2008, 26, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Piacente, F.; Caffa, I.; Ravera, S.; Sociali, G.; Passalacqua, M.; Vellone, V.G.; Becherini, P.; Reverberi, D.; Monacelli, F.; Ballestrero, A.; et al. Nicotinic Acid Phosphoribosyltransferase Regulates Cancer Cell Metabolism, Susceptibility to NAMPT Inhibitors, and DNA Repair. Cancer Res. 2017, 77, 3857–3869. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD Metabolic Dependency in Cancer Is Shaped by Gene Amplification and Enhancer Remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Andris, F.; Van Gool, F.; Leo, O. Reconstructing Eukaryotic NAD Metabolism. BioEssays News Rev. Mol. Cell. Dev. Biol. 2003, 25, 683–690. [Google Scholar] [CrossRef]
- Shames, D.S.; Elkins, K.; Walter, K.; Holcomb, T.; Du, P.; Mohl, D.; Xiao, Y.; Pham, T.; Haverty, P.M.; Liederer, B.; et al. Loss of NAPRT1 Expression by Tumor-Specific Promoter Methylation Provides a Novel Predictive Biomarker for NAMPT Inhibitors. Clin. Cancer Res. 2013, 19, 6912–6923. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.; Roulston, A.; Bélec, L.; Billot, X.; Marcellus, R.; Bédard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The Small Molecule GMX1778 Is a Potent Inhibitor of NAD+ Biosynthesis: Strategy for Enhanced Therapy in Nicotinic Acid Phosphoribosyltransferase 1-Deficient Tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef] [Green Version]
- Peterse, E.F.P.; van den Akker, B.E.W.M.; Niessen, B.; Oosting, J.; Suijker, J.; de Jong, Y.; Danen, E.H.J.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. NAD Synthesis Pathway Interference Is a Viable Therapeutic Strategy for Chondrosarcoma. Mol. Cancer Res. 2017, 15, 1714–1721. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, H.; Lee, J.E.; Shin, S.-J.; Oh, S.; Kwon, G.; Kim, H.; Choi, Y.Y.; White, M.A.; Paik, S.; et al. Selective Cytotoxicity of the NAMPT Inhibitor FK866 Toward Gastric Cancer Cells With Markers of the Epithelial-Mesenchymal Transition, Due to Loss of NAPRT. Gastroenterology 2018, 155, 799–814.e13. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Fons, N.R.; Sundaram, R.K.; Breuer, G.A.; Peng, S.; McLean, R.L.; Kalathil, A.N.; Schmidt, M.S.; Carvalho, D.M.; Mackay, A.; Jones, C.; et al. PPM1D Mutations Silence NAPRT Gene Expression and Confer NAMPT Inhibitor Sensitivity in Glioma. Nat. Commun. 2019, 10, 3790. [Google Scholar] [CrossRef] [Green Version]
- Gaut, Z.N.; Solomon, H.M. Inhibition of Nicotinate Phosphoribosyltransferase in Human Platelet Lysate by Nicotinic Acid Analogs. Biochem. Pharmacol. 1971, 20, 2903–2906. [Google Scholar] [CrossRef]
- Gaut, Z.N.; Solomon, H.M. Uptake and Metabolism of Nicotinic Acid by Human Blood Platelets. Effects of Structure Analogs and Metabolic Inhibitors. Biochim. Biophys. Acta 1970, 201, 316–322. [Google Scholar] [CrossRef]
- Gaut, Z.N.; Solomon, H.M. Inhibition of Nicotinate Phosphoribosyl Transferase by Nonsteroidal Anti-Inflammatory Drugs: A Possible Mechanism of Action. J. Pharm. Sci. 1971, 60, 1887–1888. [Google Scholar] [CrossRef] [PubMed]
- Galassi, L.; Di Stefano, M.; Brunetti, L.; Orsomando, G.; Amici, A.; Ruggieri, S.; Magni, G. Characterization of Human Nicotinate Phosphoribosyltransferase: Kinetic Studies, Structure Prediction and Functional Analysis by Site-Directed Mutagenesis. Biochimie 2012, 94, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Marletta, A.S.; Massarotti, A.; Orsomando, G.; Magni, G.; Rizzi, M.; Garavaglia, S. Crystal Structure of Human Nicotinic Acid Phosphoribosyltransferase. FEBS Open Bio 2015, 5, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted Inhibitors of P-Glycoprotein Increase Chemotherapeutic-Induced Mortality of Multidrug Resistant Tumor Cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [Green Version]
- Khan, J.A.; Tao, X.; Tong, L. Molecular Basis for the Inhibition of Human NMPRTase, a Novel Target for Anticancer Agents. Nat. Struct. Mol. Biol. 2006, 13, 582–588. [Google Scholar] [CrossRef]
- Kang, G.B.; Bae, M.-H.; Kim, M.-K.; Im, I.; Kim, Y.-C.; Eom, S.H. Crystal Structure of Rattus Norvegicus Visfatin/PBEF/Nampt in Complex with an FK866-Based Inhibitor. Mol. Cells 2009, 27, 667–671. [Google Scholar] [CrossRef] [PubMed]
- ElMokh, O.; Matsumoto, S.; Biniecka, P.; Bellotti, A.; Schaeuble, K.; Piacente, F.; Gallart-Ayala, H.; Ivanisevic, J.; Stamenkovic, I.; Nencioni, A.; et al. Gut Microbiota Severely Hampers the Efficacy of NAD-Lowering Therapy in Leukemia. Cell Death Dis. 2022, 13, 320. [Google Scholar] [CrossRef] [PubMed]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e7. [Google Scholar] [CrossRef] [PubMed]
- Gille, A.; Bodor, E.T.; Ahmed, K.; Offermanns, S. Nicotinic Acid: Pharmacological Effects and Mechanisms of Action. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 79–106. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Graeff, R.; Lee, H.C. A Novel Cycling Assay for Cellular CADP-Ribose with Nanomolar Sensitivity. Biochem. J. 2002, 361, 379. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Ropp, P.J.; Spiegel, J.O.; Walker, J.L.; Green, H.; Morales, G.A.; Milliken, K.A.; Ringe, J.J.; Durrant, J.D. Gypsum-DL: An Open-Source Program for Preparing Small-Molecule Libraries for Structure-Based Virtual Screening. J. Cheminformatics 2019, 11, 34. [Google Scholar] [CrossRef]
- Parenti, M.D.; Grozio, A.; Bauer, I.; Galeno, L.; Damonte, P.; Millo, E.; Sociali, G.; Franceschi, C.; Ballestrero, A.; Bruzzone, S.; et al. Discovery of Novel and Selective SIRT6 Inhibitors. J. Med. Chem. 2014, 57, 4796–4804. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Reported Ki * (µM) | Compound | Reported Ki (µM) |
|---|---|---|---|
| Flufenamic acid | 10 | 6-Chloronicotinic acid | 560 |
| Mefenamic acid | 50 | Isonicotinic acid | 750 |
| 2-Pyrazinoic acid | 75 | 3-Pyridylsulfonic acid | 750 |
| Phenylbutazone | 100 | Pyridine | 780 |
| Indomethacin | 150 | 2-Aminonicotinic acid | 820 |
| Salicylic acid | 160 | Acetanilide | 1000 |
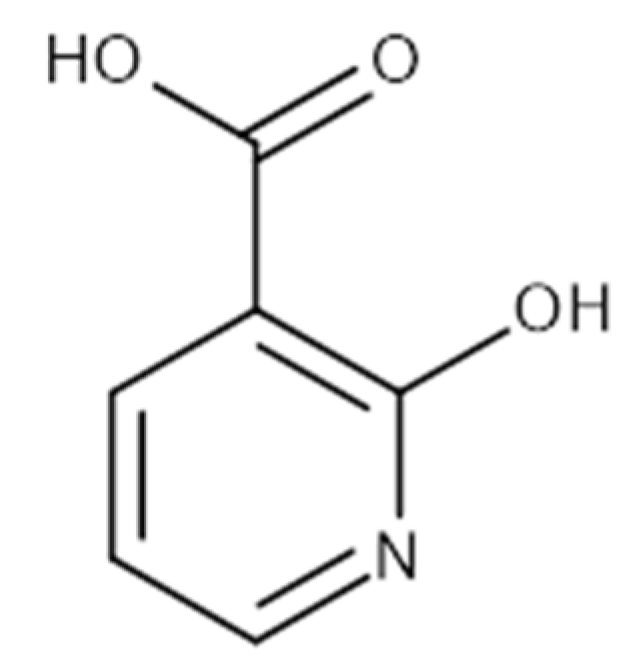
| 2-Hydroxynicotinic acid | 230 | Aminopyrine | 1000 |
| 2-Fluoronicotinic acid | 280 | Antipyrine | 1000 |
| Oxyphenbutazone | 300 | Picolinic acid | 1160 |
| Acetylsalicylic acid | 500 | 3-Pyridylacetic acid | 1280 |
| Sulfinpyrazone | 500 | Benzoic acid | 1900 |
| Compound ID | Structure | Vendor ID | M.W. * | Compound ID | Structure | Vendor ID | M.W. |
|---|---|---|---|---|---|---|---|
| 1 |  | F0020-0171 | 333.3407 | 26 |  | F2721-0386 | 337.3709 |
| 2 |  | F0173-0133 | 349.4063 | 27 |  | F2758-0213 | 297.2688 |
| 3 |  | F0648-0699 | 376.3655 | 28 |  | F3188-0088 | 277.2759 |
| 4 |  | F0648-0785 | 391.3769 | 29 |  | F3226-2226 | 397.4427 |
| 5 |  | F1199-0146 | 358.7757 | 30 |  | F3229-0191 | 283.7076 |
| 6 |  | F1260-1693 | 421.4889 | 31 |  | F3295-0007 | 355.3927 |
| 7 |  | F1299-0156 | 462.6037 | 32 |  | F3311-0032 | 471.9565 |
| 8 |  | F1371-0219 | 139.1088 | 33 |  | F3371-0859 | 414.4268 |
| 9 |  | F1566-0988 | 333.3192 | 34 |  | F3385-4161 | 440.9226 |
| 10 |  | F1710-0049 | 460.429 | 35 |  | F5008-0290 | 369.3977 |
| 11 |  | F1811-0048 | 282.2957 | 36 |  | F5857-5354 | 311.3319 |
| 12 |  | F1907-0958 | 230.2194 | 37 |  | F6127-0104 | 303.2769 |
| 13 |  | F1967s-1157 | 180.1607 | 38 |  | F6127-0210 | 333.3574 |
| 14 |  | F2135-0162 | 181.1455 | 39 |  | F6241-0336 | 388.4408 |
| 15 |  | F2135-0875 | 276.2911 | 40 |  | F6252-1248 | 277.3206 |
| 16 |  | F2135-0897 | 228.2466 | 41 |  | F6252-5764 | 436.4869 |
| 17 |  | F2147-0724 | 174.1513 | 42 |  | F6279-0434 | 365.4008 |
| 18 |  | F2168-0001 | 153.1354 | 43 |  | F6372-1828 | 407.3795 |
| 19 |  | F2169-0490 | 130.1252 | 44 |  | F6414-0992 | 404.4584 |
| 20 |  | F2191-0003 | 153.1354 | 45 |  | F6439-7266 | 398.4604 |
| 21 |  | F2278-0232 | 316.3748 | 46 |  | F6465-0031 | 290.2747 |
| 22 |  | F2503-0045 | 331.3497 | 47 |  | F6497-7775 | 383.3796 |
| 23 |  | F2526-0046 | 346.1572 | 48 |  | F6497-8054 | 447.4104 |
| 24 |  | F2711-0182 | 305.3042 | 49 |  | F6523-1712 | 367.4017 |
| 25 |  | F2721-0331 | 346.2091 | 50 |  | F9994-0201 | 397.4078 |
| Compound ID | Vendor ID | Ki (µM) | Vmax/Km | Proposed Mechanism |
|---|---|---|---|---|
| 1 | F0020-0171 | 2281 | Vmax ↓/Km ↓ | Un-competetive |
| 2 | F0173-0133 | 88.99 | Vmax ↓/Km ↓ | Un-competetive |
| 8 | F1371-0219 | 307.5 | Vmax =/Km ↑ | Competitive |
| 19 | F2169-0490 | 295.1 | Vmax ↓/Km ↓ | Un-competitive |
| Compound ID | Log S (ESOL) | Log S (Ali) | Log S (SILICOS-IT) | GI Absorption | Pgp Substrate | BBB Permeant | Bioavailability Score |
|---|---|---|---|---|---|---|---|
| 2-HNA | −1.65 | −1.97 | −0.8 | High | No | No | 0.85 |
| 8 | −1.44 | −1.62 | −0.8 | High | No | No | 0.85 |
| 19 | −1.18 | −1.77 | −0.26 | High | No | No | 0.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghanem, M.S.; Caffa, I.; Del Rio, A.; Franco, J.; Parenti, M.D.; Monacelli, F.; Cea, M.; Khalifa, A.; Nahimana, A.; Duchosal, M.A.; et al. Identification of NAPRT Inhibitors with Anti-Cancer Properties by In Silico Drug Discovery. Pharmaceuticals 2022, 15, 848. https://doi.org/10.3390/ph15070848
Ghanem MS, Caffa I, Del Rio A, Franco J, Parenti MD, Monacelli F, Cea M, Khalifa A, Nahimana A, Duchosal MA, et al. Identification of NAPRT Inhibitors with Anti-Cancer Properties by In Silico Drug Discovery. Pharmaceuticals. 2022; 15(7):848. https://doi.org/10.3390/ph15070848
Chicago/Turabian StyleGhanem, Moustafa S., Irene Caffa, Alberto Del Rio, Jorge Franco, Marco Daniele Parenti, Fiammetta Monacelli, Michele Cea, Amr Khalifa, Aimable Nahimana, Michel A. Duchosal, and et al. 2022. "Identification of NAPRT Inhibitors with Anti-Cancer Properties by In Silico Drug Discovery" Pharmaceuticals 15, no. 7: 848. https://doi.org/10.3390/ph15070848