
Discovery of Triple Inhibitors of Both SARS-CoV-2 Proteases and Human Cathepsin L

 , , , and
, , , and
Abstract
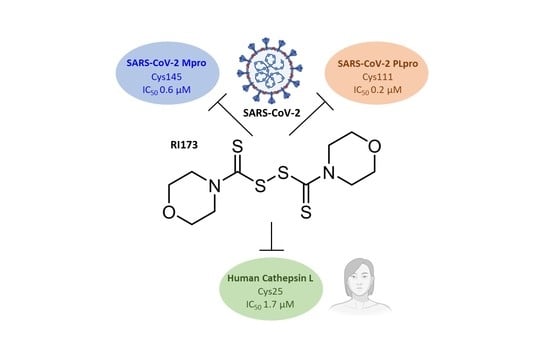
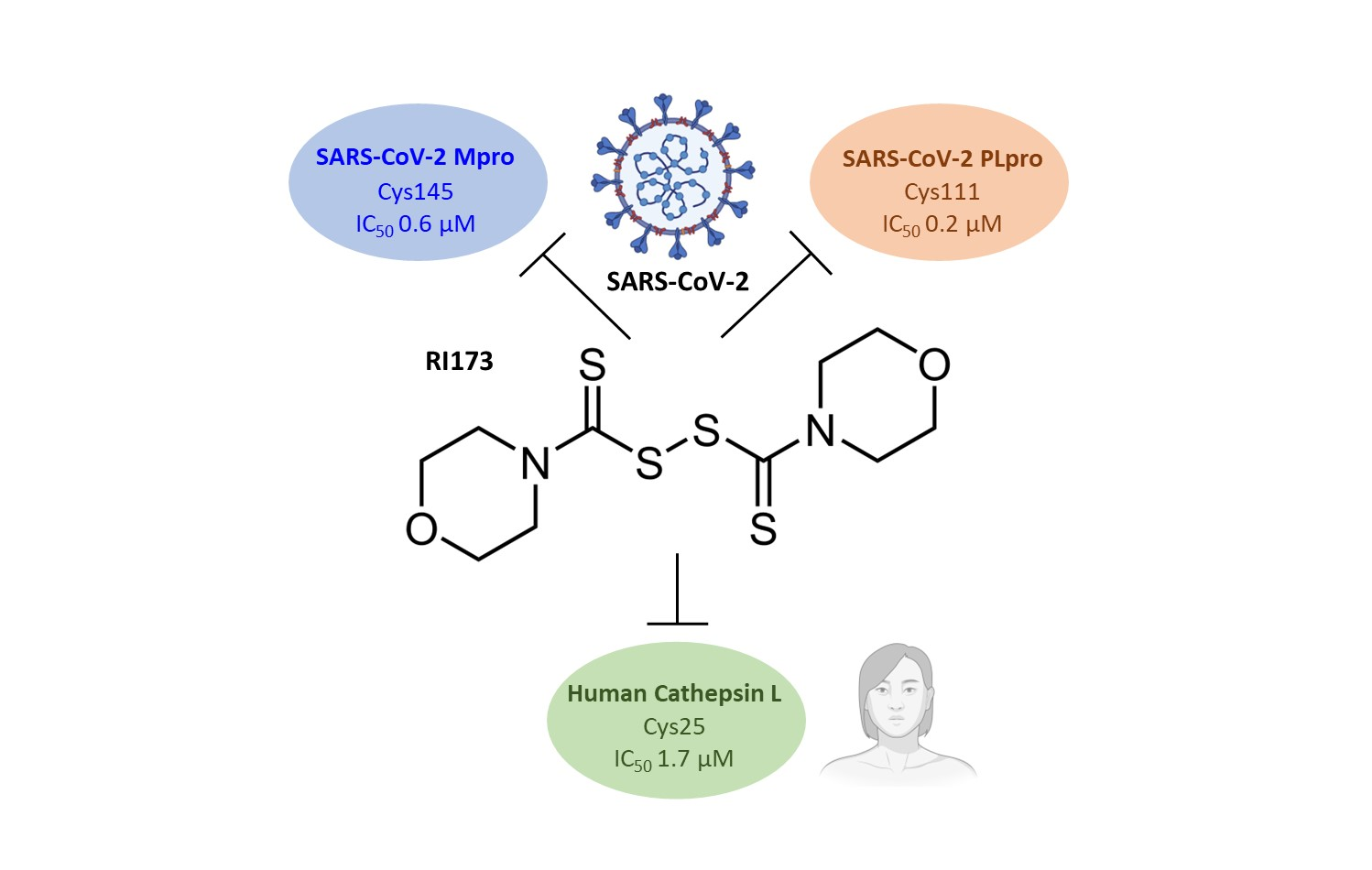
:
1. Introduction
2. Results
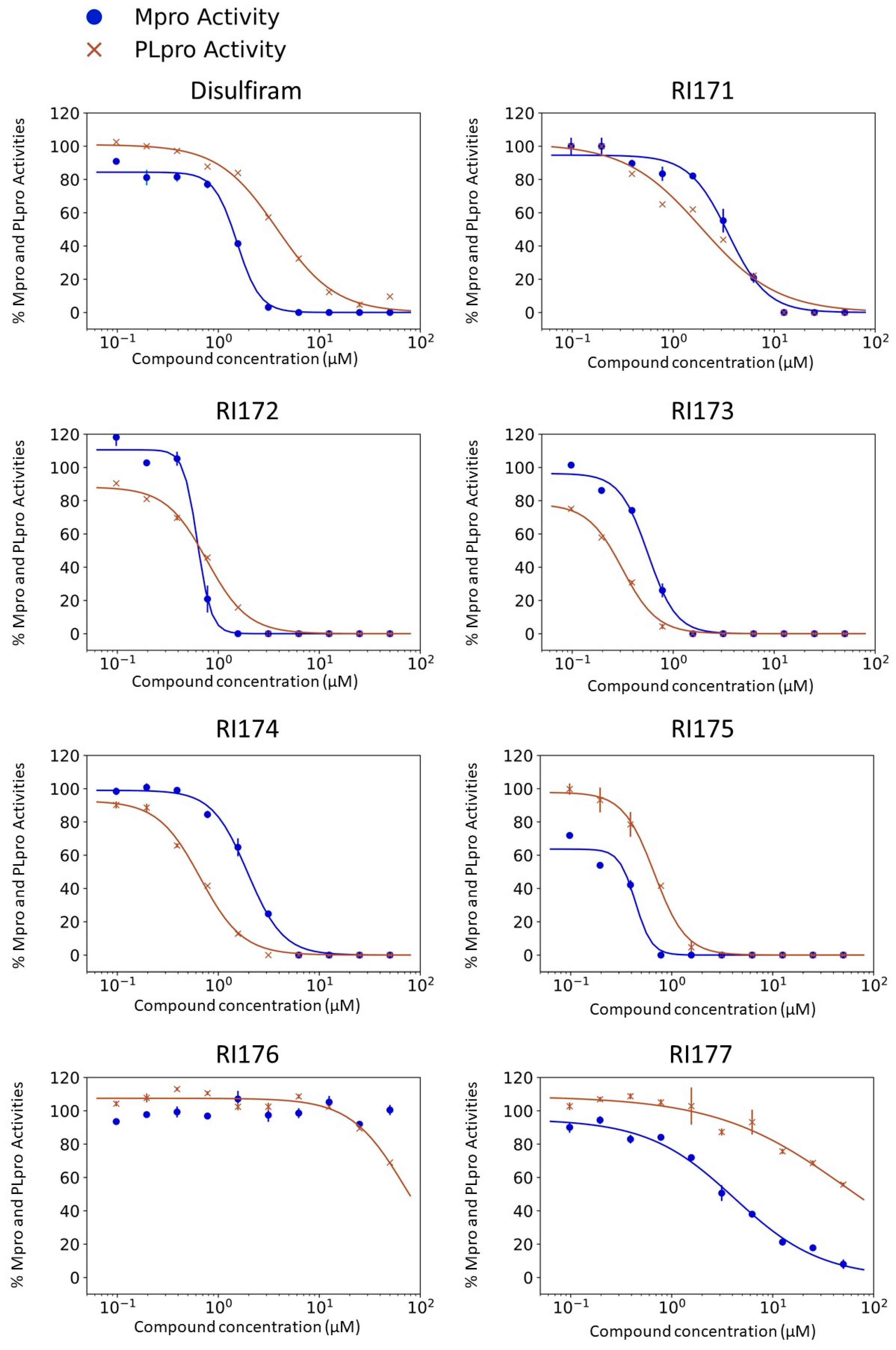
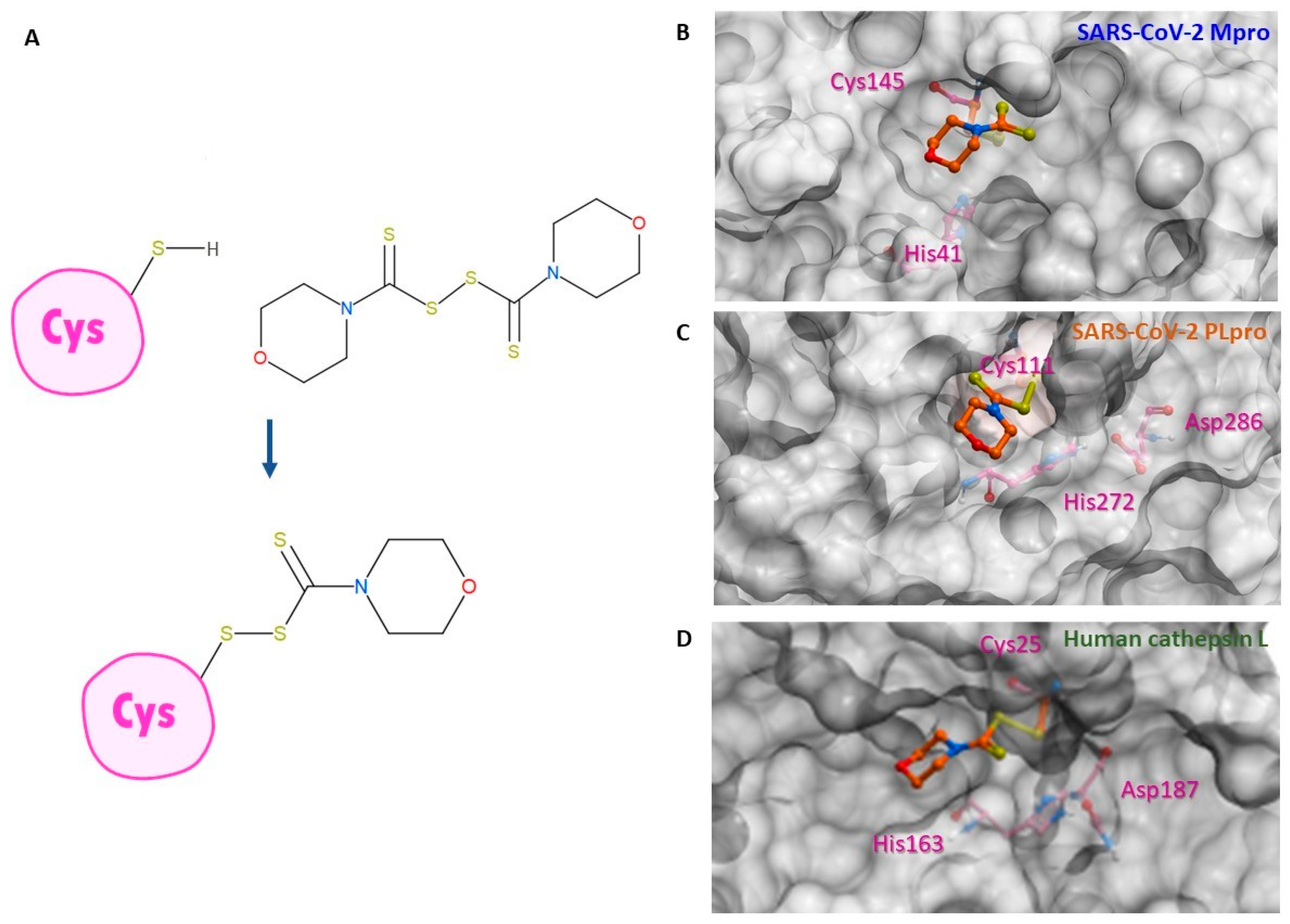
2.1. SARS-CoV-2 Mpro and PLpro Activity Assay
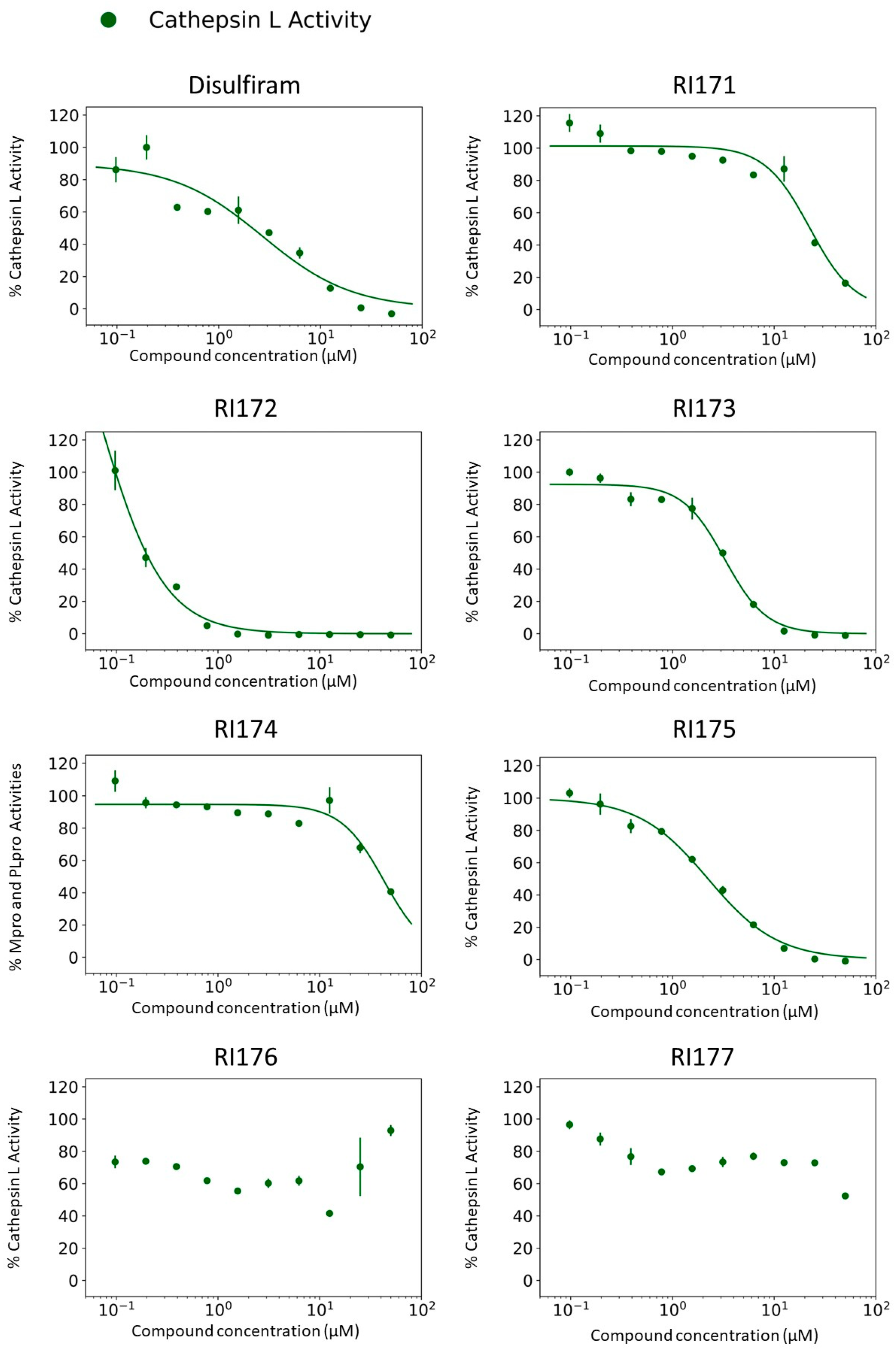
2.2. Inhibition of Human Cathepsin L Protease with Identified Inhibitors of Viral Proteases
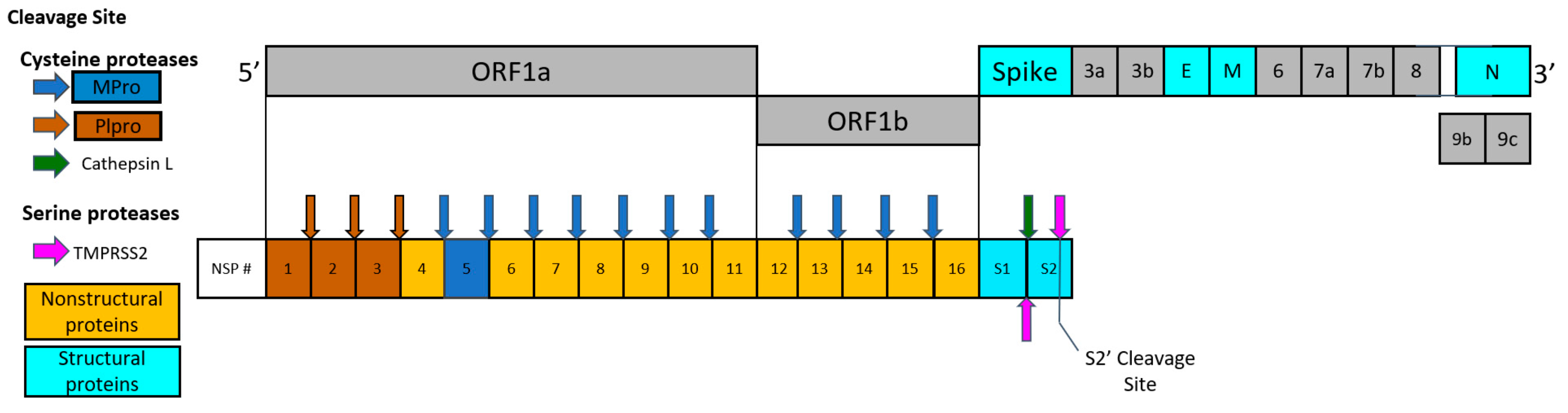
2.3. Indirect Effect in COVID-19 Treatment through Inhibition of Thrombin
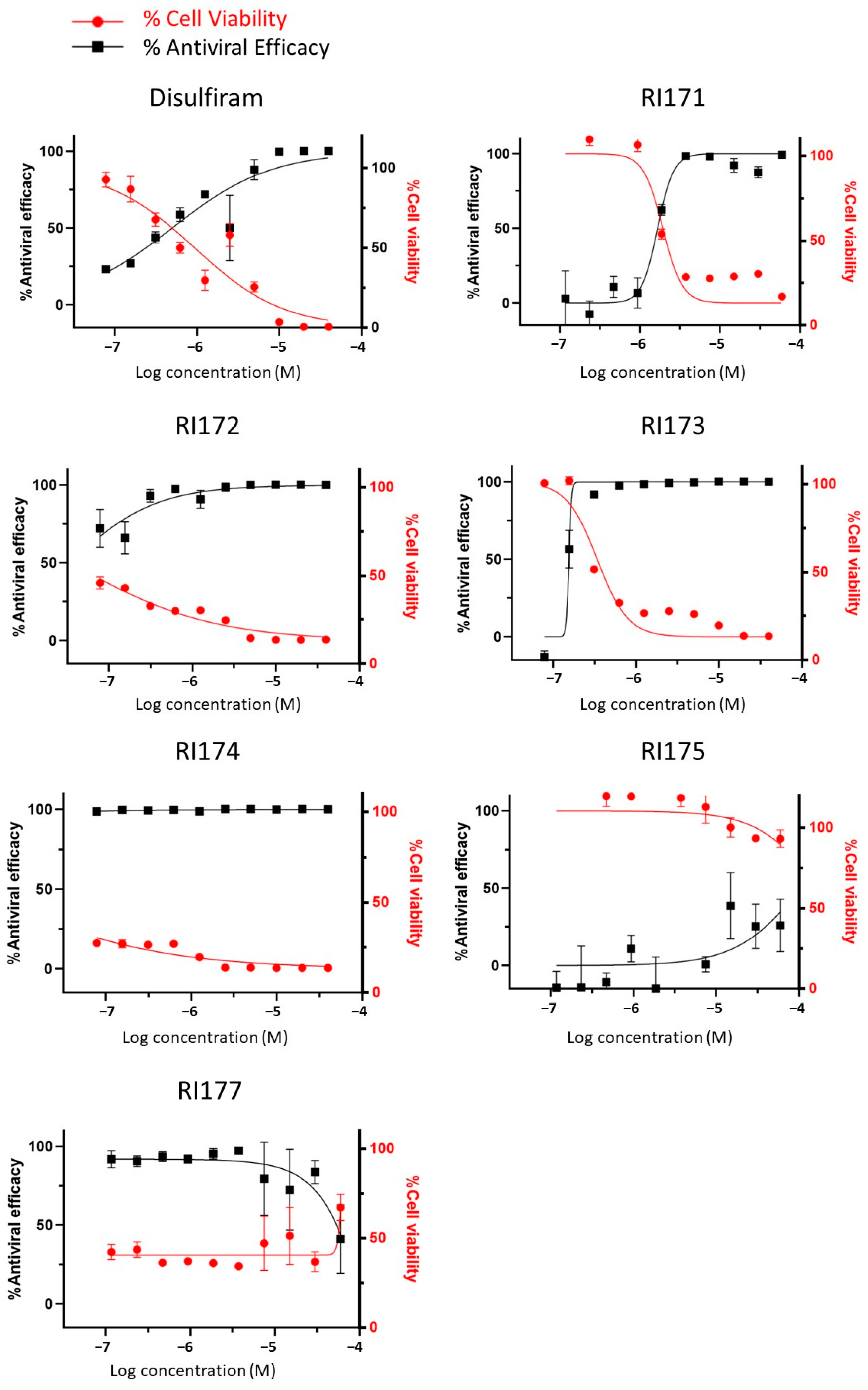
2.4. SARS-CoV-2 Infectivity Cells-Based Assay
3. Discussion
4. Materials and Methods
4.1. Computational Modeling
4.2. Compounds and Reagents
4.3. Recombinant Protein and Substrates
4.4. Enzymatic Inhibition Assay
4.5. Cells Culture and Immunofluorescence Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tu, Y.-F.; Chien, C.-S.; Yarmishyn, A.A.; Lin, Y.-Y.; Luo, Y.-H.; Lin, Y.-T.; Lai, W.-Y.; Yang, D.-M.; Chou, S.-J.; Yang, Y.-P.; et al. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, H.; Ortiz-Ospina, E.; Beltekian, D.; Mathieu, E.; Hasell, J.; Macdonald, B.; Giattino, C.; Appel, C.; Rodés-Guirao, L.; Roser, M. Coronavirus Pandemic (COVID-19). Our World in Data [Internet]. Available online: https://ourworldindata.org/coronavirus (accessed on 19 November 2021).
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.-C.; Ke, Y.-Y.; Huang, S.Y.; Huang, P.-N.; Kung, Y.-A.; Chang, T.-Y.; Yen, K.-J.; Peng, T.-T.; Chang, S.-E.; Huang, C.-T.; et al. Discovery of M Protease Inhibitors Encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00872-20. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Xia, Z.; Lambrinidis, G.; Townsend, J.A.; Hu, Y.; Meng, X.; Szeto, T.; Ba, M.; Zhang, X.; et al. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 2021, 7, 1245–1260. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Mahmoud, I.S.; Jarrar, Y.B.; Alshaer, W.; Ismail, S. SARS-CoV-2 entry in host cells-multiple targets for treatment and prevention. Biochimie 2020, 175, 93–98. [Google Scholar] [CrossRef]
- Murgolo, N.; Therien, A.G.; Howell, B.; Klein, D.; Koeplinger, K.; Lieberman, L.A.; Adam, G.C.; Flynn, J.; McKenna, P.; Swaminathan, G.; et al. SARS-CoV-2 tropism, entry, replication, and propagation: Considerations for drug discovery and development. PLoS Pathog. 2021, 17, e1009225. [Google Scholar] [CrossRef]
- Ou, T.; Mou, H.; Zhang, L.; Ojha, A.; Choe, H.; Farzan, M. Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog. 2021, 17, e1009212. [Google Scholar] [CrossRef]
- Zhao, M.-M.; Yang, W.-L.; Yang, F.-Y.; Zhang, L.; Huang, W.-J.; Hou, W.; Fan, C.-F.; Jin, R.-H.; Feng, Y.-M.; Wang, Y.-C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef]
- Mellott, D.M.; Tseng, C.-T.; Drelich, A.; Fajtová, P.; Chenna, B.C.; Kostomiris, D.H.; Hsu, J.; Zhu, J.; Taylor, Z.W.; Kocurek, K.I.; et al. A Clinical-Stage Cysteine Protease Inhibitor blocks SARS-CoV-2 Infection of Human and Monkey Cells. ACS Chem. Biol. 2021, 16, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef] [PubMed]
- Bakowski, M.A.; Beutler, N.; Wolff, K.C.; Kirkpatrick, M.G.; Chen, E.; Nguyen, T.-T.H.; Riva, L.; Shaabani, N.; Parren, M.; Ricketts, J.; et al. Drug repurposing screens identify chemical entities for the development of COVID-19 interventions. Nat. Commun. 2021, 12, 3309. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.; Abdijadid, S. Disulfiram; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sargsyan, K.; Lin, C.-C.; Chen, T.; Grauffel, C.; Chen, Y.-P.; Yang, W.-Z.; Yuan, H.S.; Lim, C. Multi-targeting of functional cysteines in multiple conserved SARS-CoV-2 domains by clinically safe Zn-ejectors. Chem. Sci. 2020, 11, 9904–9909. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M (pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.-H.; Moses, D.C.; Hsieh, C.-H.; Cheng, S.-C.; Chen, Y.-H.; Sun, C.-Y.; Chou, C.-Y. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antivir. Res. 2018, 150, 155–163. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine Metabolism Supports Macrophage IL-1β Production. Cell Metab. 2019, 29, 1003–1011.e4. [Google Scholar] [CrossRef] [Green Version]
- Nobel, C.S.; Kimland, M.; Nicholson, D.W.; Orrenius, S.; Slater, A.F. Disulfiram is a potent inhibitor of proteases of the caspase family. Chem. Res. Toxicol. 1997, 10, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. Disulfiram for COVID-19 (DISCO) Trial (DISCO). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04485130 (accessed on 19 November 2021).
- Adrover, J.M.; Carrau, L.; Daßler-Plenker, J.; Bram, Y.; Chandar, V.; Houghton, S.; Redmond, D.; Merrill, J.R.; Shevik, M.; tenOever, B.R.; et al. Disulfiram inhibits neutrophil extracellular trap formation protecting rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight 2022, 7, e157342. [Google Scholar] [CrossRef] [PubMed]
- Florvall, L.; Corrodi, H. Dopamine beta-hydroxylase inhibitors. The preparation and the dopamine beta-hydroxylase inhibitory activity of some compounds related to dithiocarbamic acid and thiuramdisulfide. Acta Pharm. Suec. 1970, 7, 7–22. [Google Scholar] [PubMed]
- Sun, W.; Xie, Z.; Liu, Y.; Zhao, D.; Wu, Z.; Zhang, D.; Lv, H.; Tang, S.; Jin, N.; Jiang, H.; et al. JX06 Selectively Inhibits Pyruvate Dehydrogenase Kinase PDK1 by a Covalent Cysteine Modification. Cancer Res. 2015, 75, 4923–4936. [Google Scholar] [CrossRef] [Green Version]
- Brahemi, G.; Kona, F.R.; Fiasella, A.; Buac, D.; Soukupová, J.; Brancale, A.; Burger, A.M.; Westwell, A.D. Exploring the structural requirements for inhibition of the ubiquitin E3 ligase breast cancer associated protein 2 (BCA2) as a treatment for breast cancer. J. Med. Chem. 2010, 53, 2757–2765. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.J., Sr. Dangerous Properties of Industrial Materials, 9th ed.; Van Nostrand Reinhold: New York, NY, USA, 1996; Volume 1–3. [Google Scholar]
- Kapanda, C.N.; Muccioli, G.G.; Labar, G.; Poupaert, J.H.; Lambert, D.M. Bis (dialkylaminethiocarbonyl) disulfides as potent and selective monoglyceride lipase inhibitors. J. Med. Chem. 2009, 52, 7310–7314. [Google Scholar] [CrossRef] [Green Version]
- Heymans Institute of Pharmacology. Archives Internationales de Pharmacodynamie et de Thérapie; Heymans Institute of Pharmacology: Brussels, Belgium, 1957; p. 36. [Google Scholar]
- Korablev, M.V. Toxicity of dithiocarbamic acid derivatives and of structurally similar compounds. Farmakol. Toksikol. 1965, 28, 230–233. [Google Scholar]
- Arkhangel’skaya, L.N.; Roshchina, T.A. Toxicological characterization of furfuramide, a new vulcanization accelerator. Gig Sanit 1964, 29, 37–42. [Google Scholar]
- De Sousa, A. Disulfiram: Pharmacology and Mechanism of Action. In Disulfiram: Its Use in Alcohol Dependence and Other Disorders; De Sousa, A., Ed.; Springer: Singapore, 2019; pp. 9–20. ISBN 978-981-329-876-7. [Google Scholar]
- Huang, Y.; Xu, Y.; Song, R.; Ni, S.; Liu, J.; Xu, Y.; Ren, Y.; Rao, L.; Wang, Y.; Wei, L.; et al. Identification of the New Covalent Allosteric Binding Site of Fructose-1,6-bisphosphatase with Disulfiram Derivatives toward Glucose Reduction. J. Med. Chem. 2020, 63, 6238–6247. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Sosnowski, P.; Turk, D. Caught in the act: The crystal structure of cleaved cathepsin L bound to the active site of Cathepsin L. FEBS Lett. 2016, 590, 1253–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, G.; Bertram, S.; Glowacka, I.; Steffen, I.; Chaipan, C.; Agudelo, J.; Lu, K.; Rennekamp, A.J.; Hofmann, H.; Bates, P.; et al. Different host cell proteases activate the SARS-coronavirus spike-protein for cell-cell and virus-cell fusion. Virology 2011, 413, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet Lond. Engl. 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Pasquereau, S.; Nehme, Z.; Haidar Ahmad, S.; Daouad, F.; Van Assche, J.; Wallet, C.; Schwartz, C.; Rohr, O.; Morot-Bizot, S.; Herbein, G. Resveratrol Inhibits HCoV-229E and SARS-CoV-2 Coronavirus Replication In Vitro. Viruses 2021, 13, 354. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Horby, P.W.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Emberson, J.; Palfreeman, A.; Raw, J.; Elmahi, E.; Prudon, B.; et al. Lopinavir-ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2020, 396, 1345–1352. [Google Scholar] [CrossRef]
- Bolcato, G.; Bissaro, M.; Pavan, M.; Sturlese, M.; Moro, S. Targeting the coronavirus SARS-CoV-2: Computational insights into the mechanism of action of the protease inhibitors lopinavir, ritonavir and nelfinavir. Sci. Rep. 2020, 10, 20927. [Google Scholar] [CrossRef]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie 2021, 180, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillier, Q.; Vertommen, D.; Ravez, S.; Marteau, R.; Thémans, Q.; Corbet, C.; Feron, O.; Wouters, J.; Frédérick, R. Anti-alcohol abuse drug disulfiram inhibits human PHGDH via disruption of its active tetrameric form through a specific cysteine oxidation. Sci. Rep. 2019, 9, 4737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Previously Characterized Targets | SARS-CoV-2 Proteases Targets | Human Protease Target | LD50 Mouse oral in mg/kg | SARS-CoV-2 Infectivity Cells-Based Assay | ||
|---|---|---|---|---|---|---|---|---|
| Mpro IC50 (µM) | PLpro IC50 (µM) | Cathepsin L IC50 (µM) | EC50 (nM) CC50 (nM) CC50/EC50 Ratio | |||||
| 1 | Disulfiram |  | Aldehyde dehydrogenase 2 [16] SARS-CoV-2 Mpro and PLpro [18] Gasdermin D [21] Dopamine beta hydroxylase [26] Pyruvate Dehydrogenase Kinase 1 [27] Ubiquitin E3 Ligase Breast Cancer-Associated Protein 2 [28] | 1.5 ± 0.1 | 3.8 ± 0.1 | 2.2 ± 1.4 | 1980 [29] | 0.5 ± 0.2 0.9 ± 0.3 1.8 |
| 2 | RI173 (JX 06) di-Morpholine-thiuram disulfide |  | Gasdermin D [21] Pyruvate Dehydrogenase Kinase 1 [27] Human Monoacylglycerol Lipase [30] | 0.6 ± 0.1 | 0.2 ± 0.1 | 1.7 ± 0.6 | 3250 [31] | 0.1 ± 0.00 0.3 ± 0.1 3.0 |
| 3 | RI175 O,O-di-Ethyl dithiobis-thioformate |  | 0.3 ± 0.1 | 0.6 ± 0.1 | 1.9 ± 0.6 | 1200 [32] | >60 >60 N/A | |
| 4 | RI172 di-Pyrrolidine-thiuram disulfide |  | Ubiquitin E3 Ligase Breast Cancer-Associated Protein 2 [28] Human Monoacylglycerol Lipase [30] | 0.6 ± 0.1 | 0.7 ± 0.0 | 0.2 ± 0.0 | LD50 not available | <0.08 <0.08 Not available |
| 5 | RI174 (Thiram) tetra-Methyl-thiuram disulfide |  | Pyruvate Dehydrogenase Kinase 1 [27] | 2.0 ± 0.3 | 0.6 ± 0.0 | 16.9 ± 6.9 | 1350 [33] | <0.08 <0.08 Not available |
| 6 | RI171 di-4-Methylpiperazine-thiuram disulfide |  | Ubiquitin E3 Ligase Breast Cancer-Associated Protein 2 [28] Human Monoacylglycerol Lipase [30] | 3.2 ± 0.1 | 1.9 ± 0.1 | 4.5 ± 0.7 | 100 [26] | 1.9 ± 0.2 1.7 ± 0.4 0.9 |
| 7 | RI177 tetra-IsoPropyl-thiuram disulfide |  | 3.8 ± 0.6 | >50 | >50 | LD50 not available | >60 >60 Not available | |
| 8 | RI176 di-Methyl-di-Phenyl-thiuram disulfide |  | >50 | >50 | >50 | LD50 not available | EC50 not tested CC50 not tested Not available | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meewan, I.; Kattoula, J.; Kattoula, J.Y.; Skinner, D.; Fajtová, P.; Giardini, M.A.; Woodworth, B.; McKerrow, J.H.; Lage de Siqueira-Neto, J.; O’Donoghue, A.J.; et al. Discovery of Triple Inhibitors of Both SARS-CoV-2 Proteases and Human Cathepsin L. Pharmaceuticals 2022, 15, 744. https://doi.org/10.3390/ph15060744
Meewan I, Kattoula J, Kattoula JY, Skinner D, Fajtová P, Giardini MA, Woodworth B, McKerrow JH, Lage de Siqueira-Neto J, O’Donoghue AJ, et al. Discovery of Triple Inhibitors of Both SARS-CoV-2 Proteases and Human Cathepsin L. Pharmaceuticals. 2022; 15(6):744. https://doi.org/10.3390/ph15060744
Chicago/Turabian StyleMeewan, Ittipat, Jacob Kattoula, Julius Y. Kattoula, Danielle Skinner, Pavla Fajtová, Miriam A. Giardini, Brendon Woodworth, James H. McKerrow, Jair Lage de Siqueira-Neto, Anthony J. O’Donoghue, and et al. 2022. "Discovery of Triple Inhibitors of Both SARS-CoV-2 Proteases and Human Cathepsin L" Pharmaceuticals 15, no. 6: 744. https://doi.org/10.3390/ph15060744