
Bisimidazolium Salt Glycosyltransferase Inhibitors Suppress Hepatocellular Carcinoma Progression In Vitro and In Vivo



Abstract
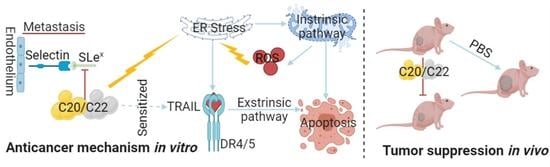
:
{kind=link}
{kind=link}
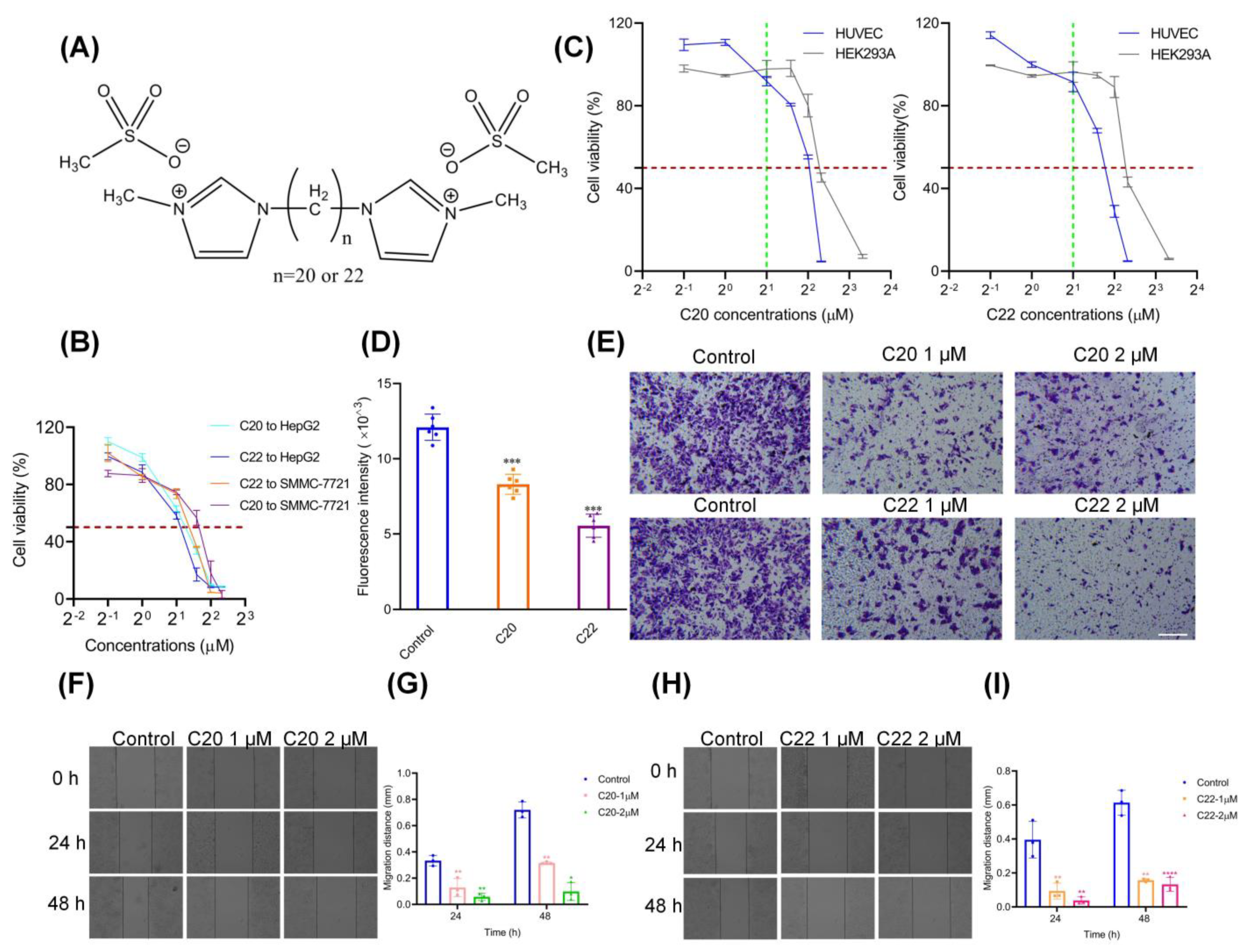{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
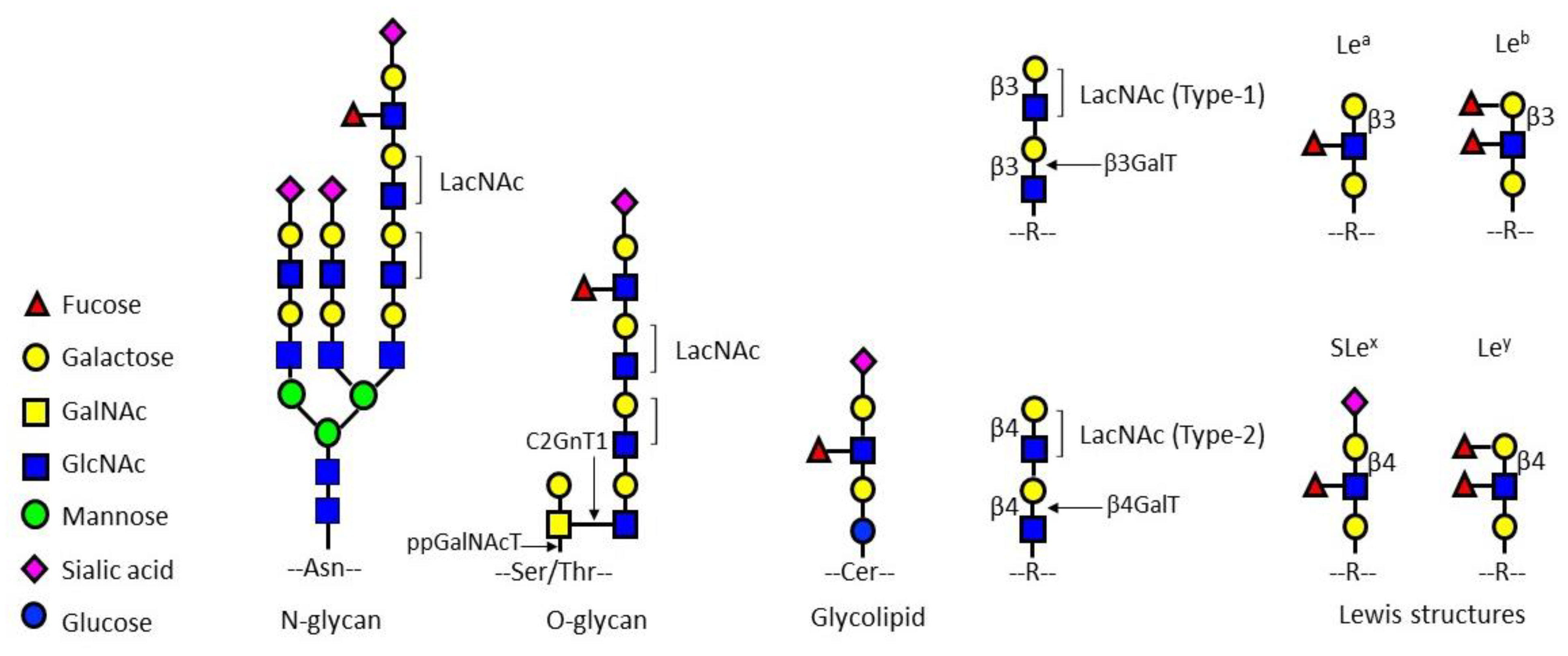
2.1. C20/C22 Inhibited HepG2 Cell Proliferation, Adhesion, Migration, and Invasion
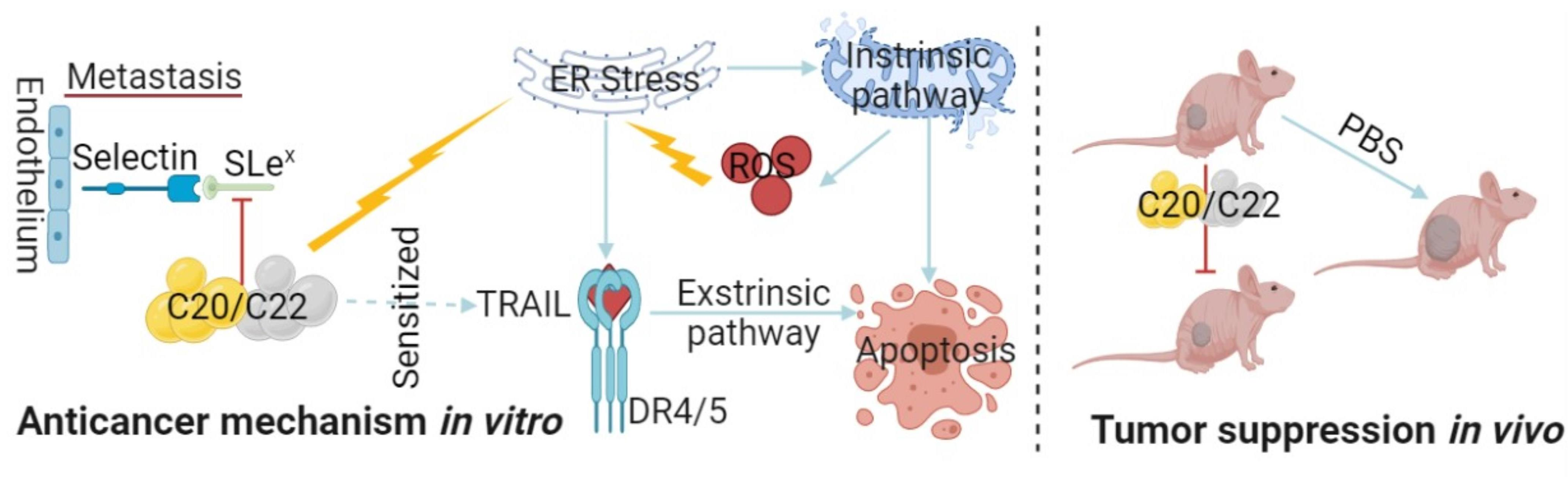
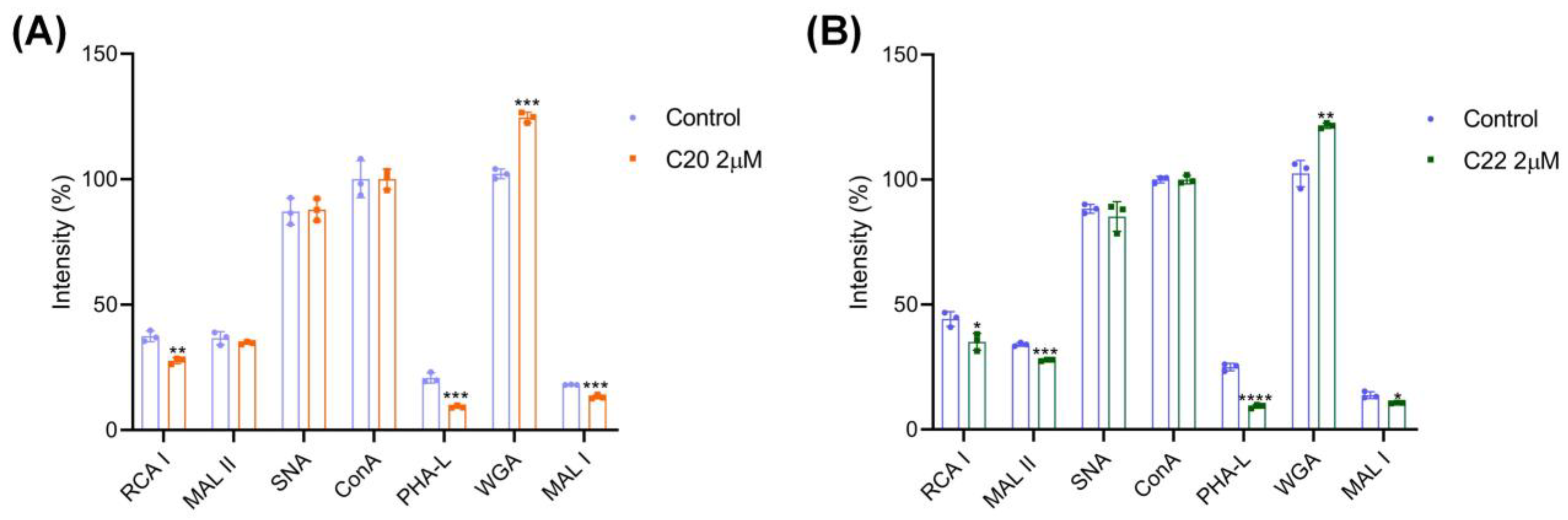
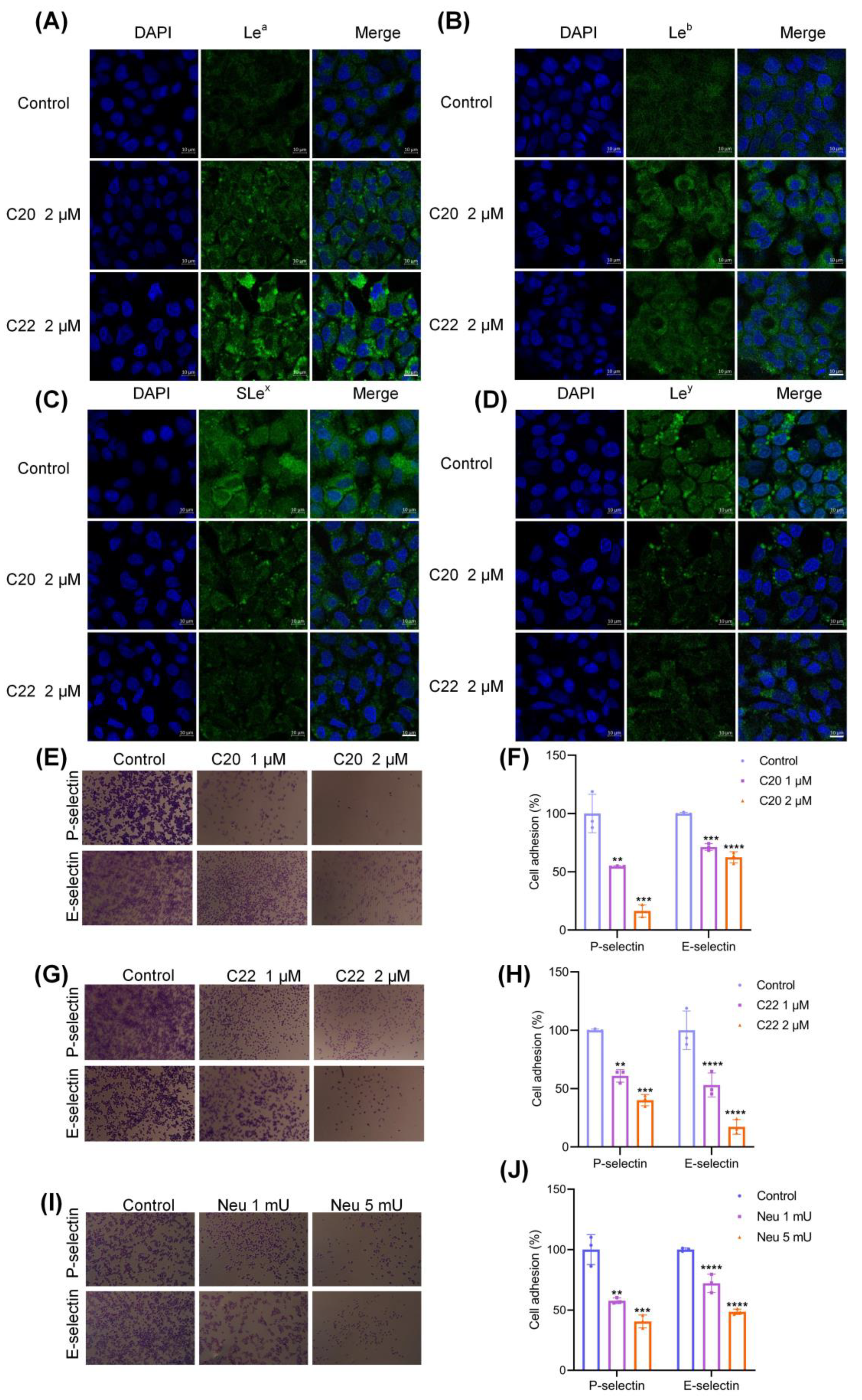
2.2. C20/C22 Modified Cell Surface Glycosylation and Inhibited Binding to Selectins
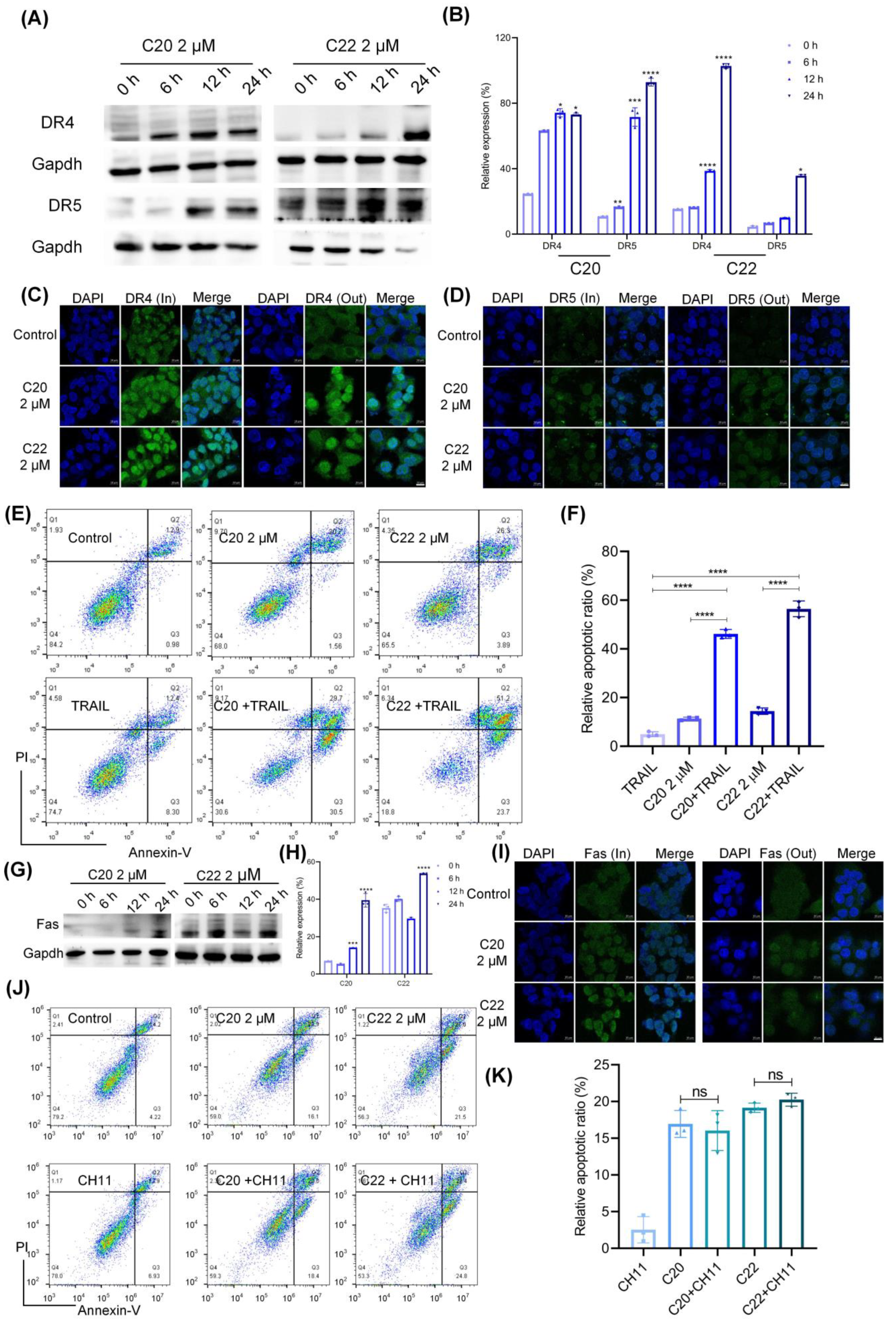
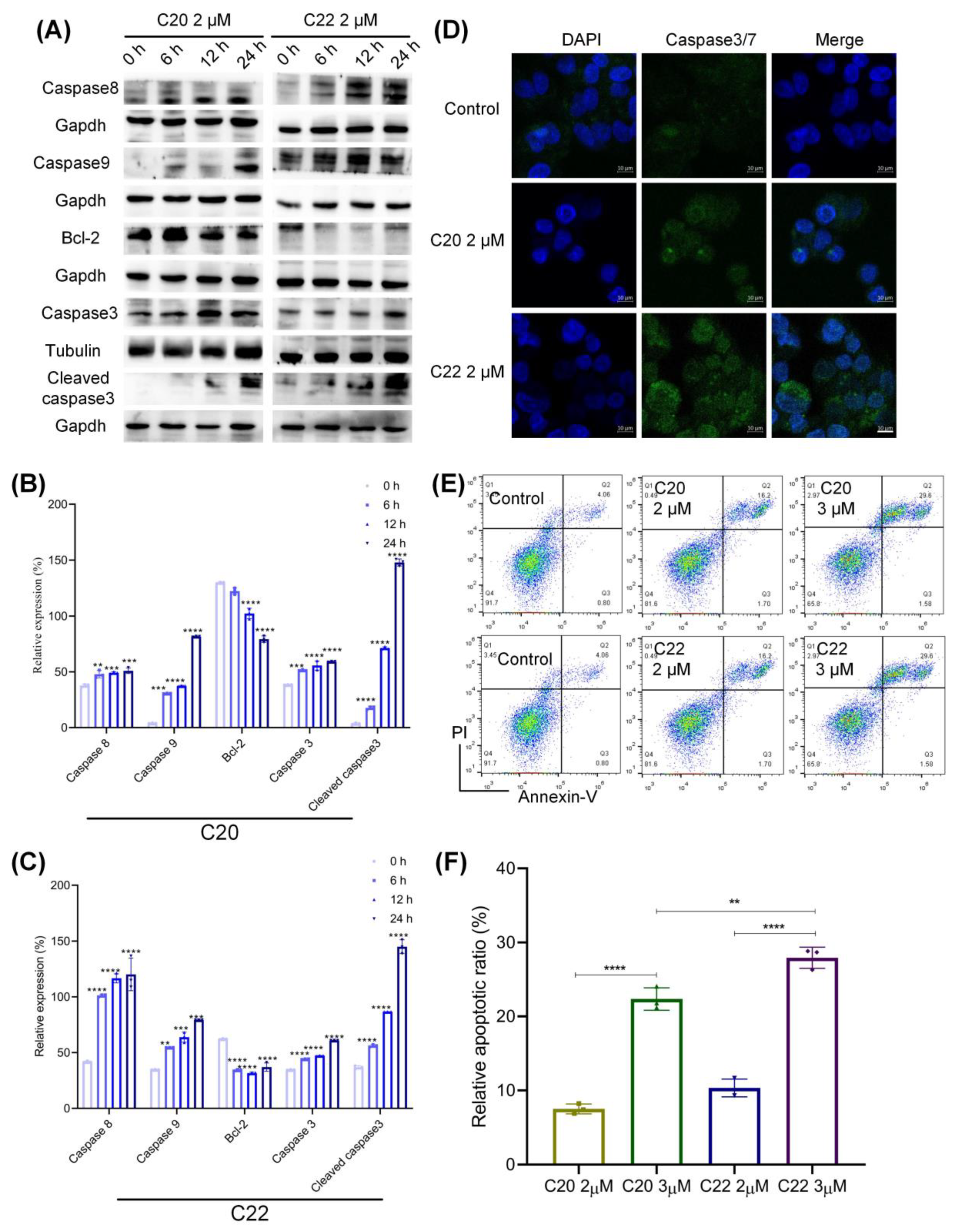
2.3. C20/C22 Increased the Susceptibility of HepG2 to TRAIL-Induced Apoptosis
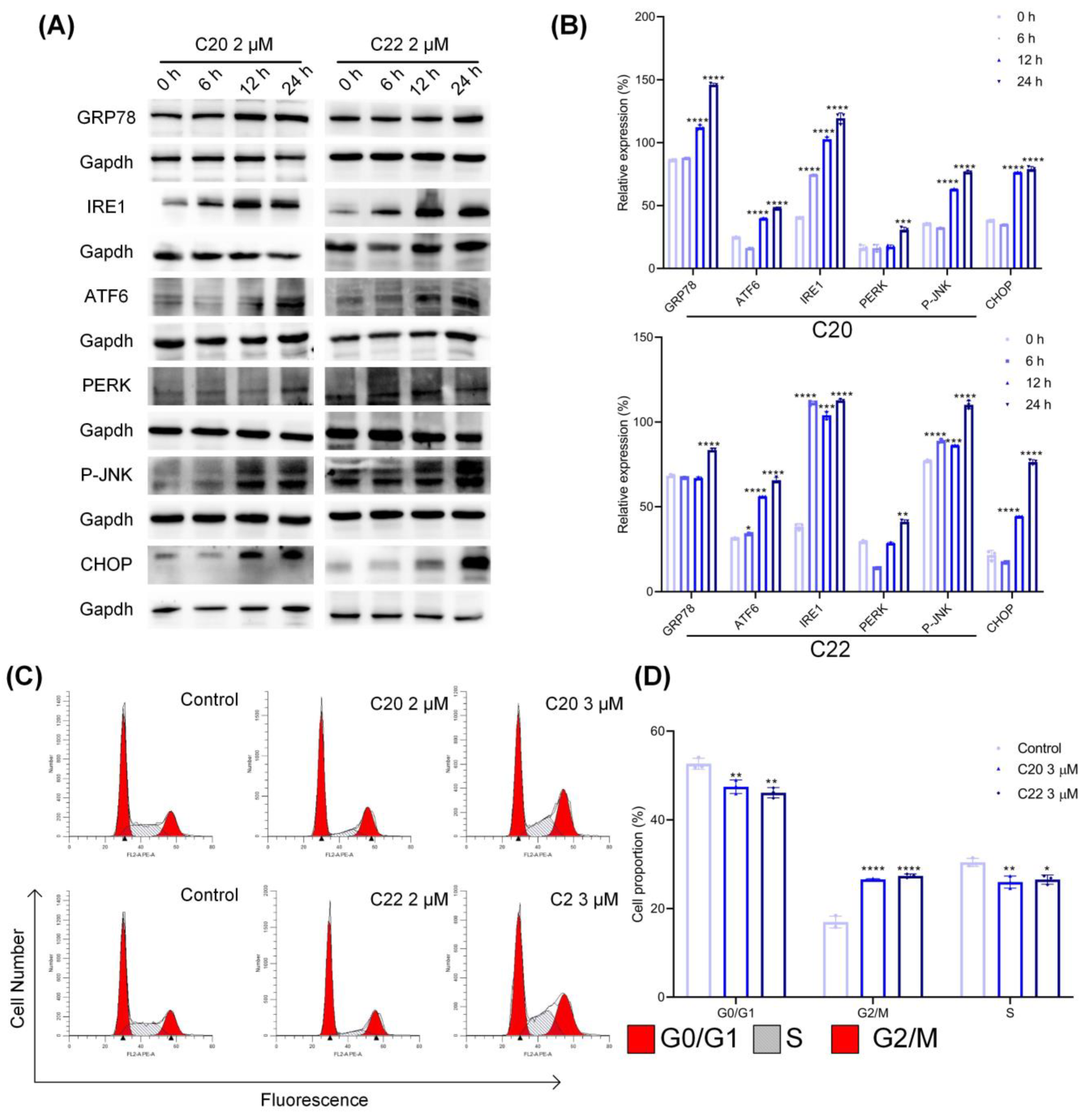
2.4. Treatment with C20/C22 Induced Endoplasmic Reticulum (ER) Stress and Cell-Cycle Arrest
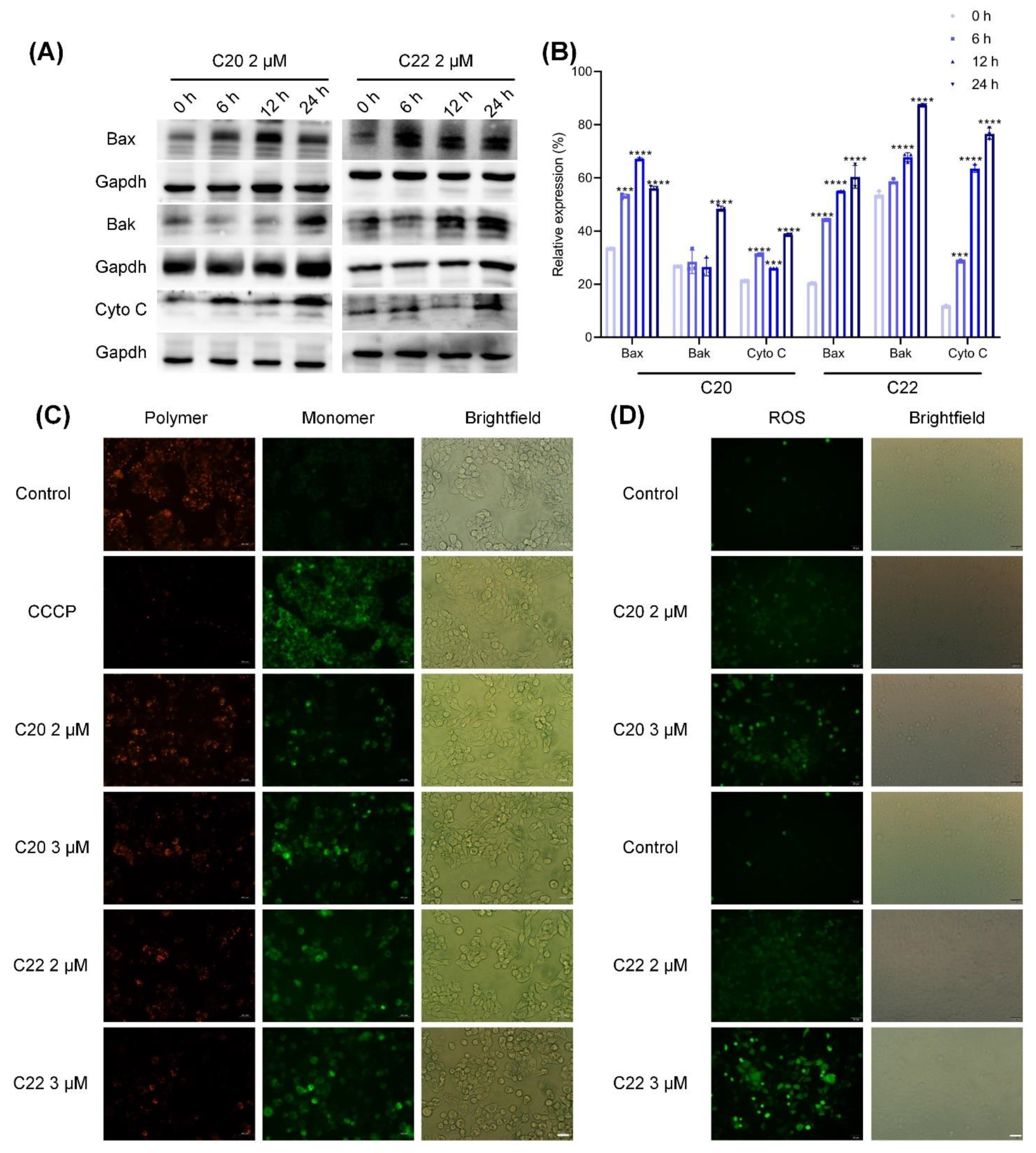
2.5. C20/C22 Induced Mitochondrial Dysfunction and Intrinsic Apoptosis of HepG2 Cells through the ER Stress Pathway
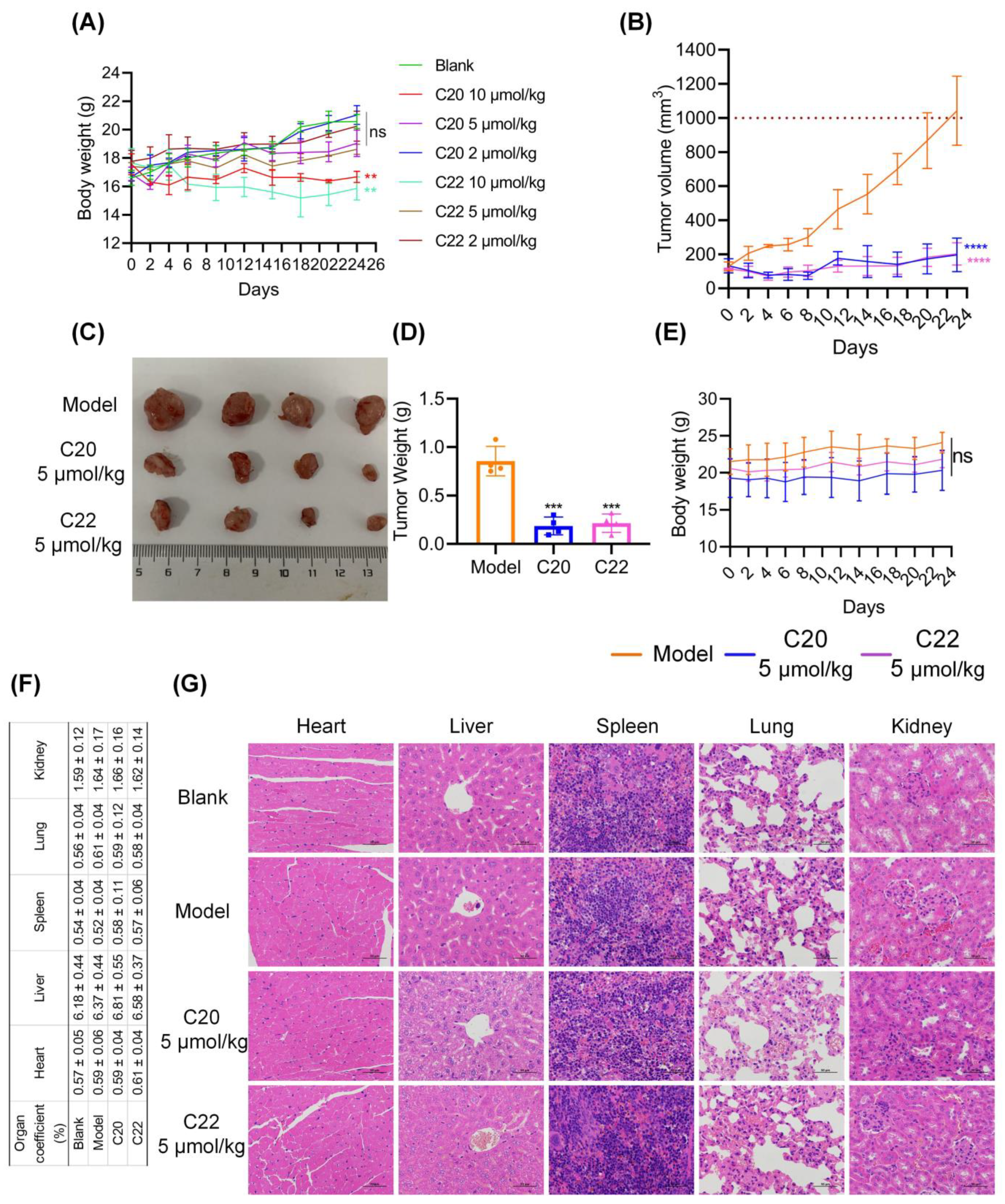
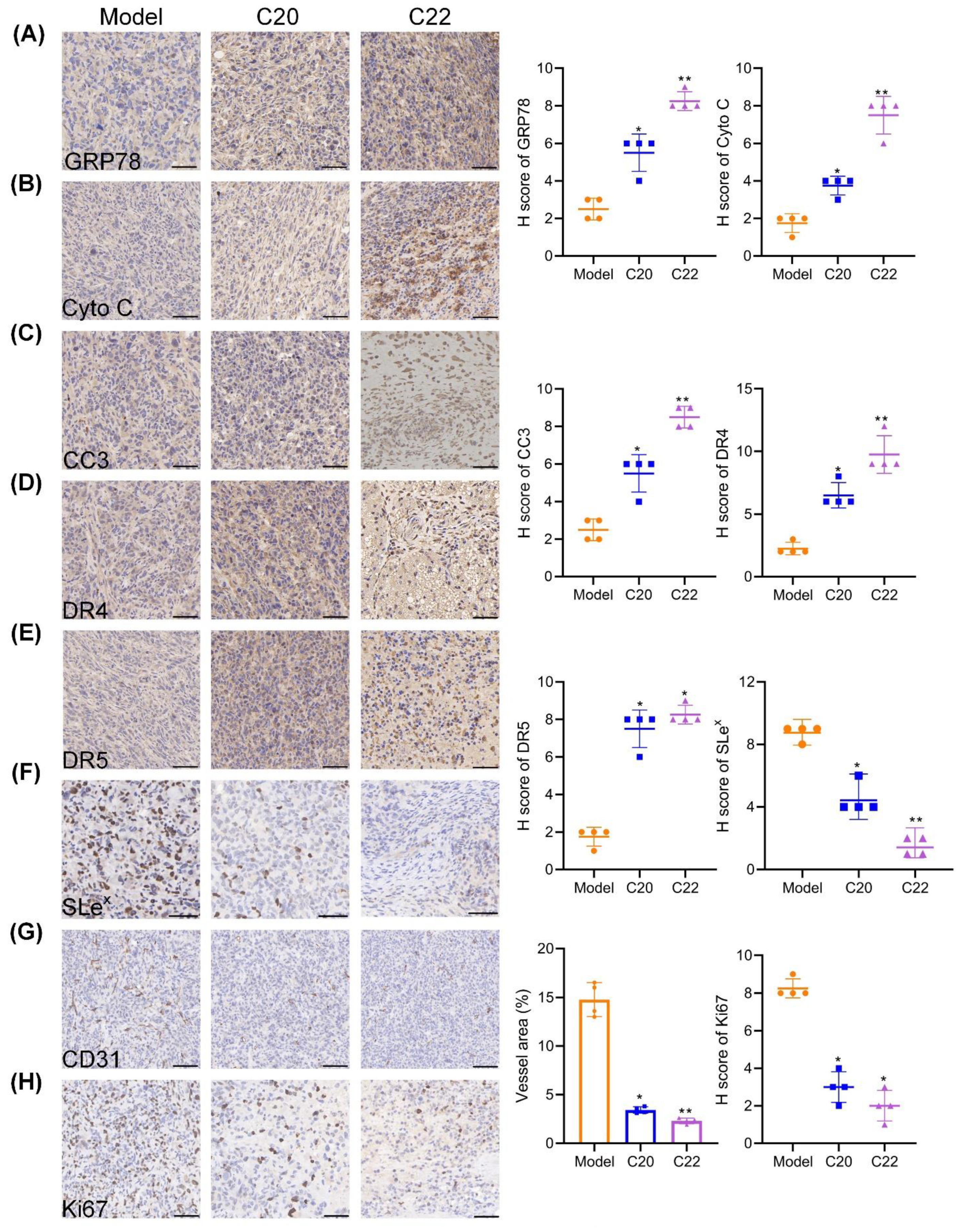
2.6. C20 and C22 Exhibit Antitumor Effects In Vivo without Causing the Histopathological Changes in Major Organs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Cultures
4.3. Cell Viability
4.4. Adhesion to HUVEC Cells
4.5. Cell Invasion Assays
4.6. Wound Healing
4.7. Immunofluorescence
4.8. Selectin Recognition Analysis
4.9. Lectin Staining Assays
4.10. Western Blot Analysis
4.11. Mitochondrial Membrane Potential Assay
4.12. Active Oxygen Species (ROS) Detection
4.13. Cell-Cycle Analysis
4.14. Cell Apoptosis Tests
4.15. In Vivo Toxicity and the Antitumor Effect of C20/C22
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartke, J.; Johnson, M.; Ghabril, M. The diagnosis and treatment of hepatocellular carcinoma. Semin. Diagn. Pathol. 2017, 34, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharm. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Wei-Qi, J.; Jin, D. New insights on sorafenib resistance in liver cancer with correlation of individualized therapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188382. [Google Scholar] [CrossRef] [PubMed]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in liver cancer and possible treatment approaches. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188314. [Google Scholar] [CrossRef]
- Islami, F.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Ward, E.M.; Jemal, A. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA Cancer J. Clin. 2017, 67, 273–289. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Herrera, H.; Block, T. Glycosylation and liver cancer. Adv. Cancer Res. 2015, 126, 257–279. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Chick, J.M.; Kolarich, D.; Haynes, P.A.; Robertson, G.R.; Tsoli, M.; Jankova, L.; Clarke, S.J.; Packer, N.H.; Baker, M.S. Liver membrane proteome glycosylation changes in mice bearing an extra-hepatic tumor. Mol. Cell. Proteomics 2011, 10, M900538mcp200. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Gao, Y.; Luan, X.; Melamed, J.; Brockhausen, I. Role of Glycans on Key Cell Surface Receptors That Regulate Cell Proliferation and Cell Death. Cells 2021, 10, 1252. [Google Scholar] [CrossRef]
- Piñero, F.; Dirchwolf, M.; Pessôa, M.G. Biomarkers in Hepatocellular Carcinoma: Diagnosis, Prognosis and Treatment Response Assessment. Cells 2020, 9, 1370. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silsirivanit, A. Glycosylation markers in cancer. Adv. Clin. Chem. 2019, 89, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Vajaria, B.N.; Patel, P.S. Glycosylation: A hallmark of cancer? Glycoconj. J. 2017, 34, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wang, K.; Gao, Y. The critical role of B4GALT4 in promoting microtubule spindle assembly in HCC through the regulation of PLK1 and RHAMM expression. J. Cell. Physiol. 2022, 237, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Fuster, M.M.; Esko, J.D. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer. 2005, 5, 526–542. [Google Scholar] [CrossRef]
- Hennet, T. The galactosyltransferase family. Cell. Mol. Life Sci. 2002, 59, 1081–1095. [Google Scholar] [CrossRef]
- Indellicato, R.; Zulueta, A.; Caretti, A.; Trinchera, M. Complementary Use of Carbohydrate Antigens Lewis a, Lewis b, and Sialyl-Lewis a (CA19.9 Epitope) in Gastrointestinal Cancers: Biological Rationale Towards A Personalized Clinical Application. Cancers 2020, 12, 1509. [Google Scholar] [CrossRef]
- Mare, L.; Trinchera, M. Suppression of beta 1,3galactosyltransferase beta 3Gal-T5 in cancer cells reduces sialyl-Lewis a and enhances poly N-acetyllactosamines and sialyl-Lewis x on O-glycans. Eur. J. Biochem. 2004, 27, 186–194. [Google Scholar] [CrossRef]
- Brockhausen, I. The role of galactosyltransferases in cell surface functions and in the immune system. Drug News Perspect. 2006, 19, 401–409. [Google Scholar] [CrossRef]
- Chen, Y.; Su, L.; Huang, C.; Wu, S.; Qiu, X.; Zhao, X.; Meng, Q.; Meng, Y.M.; Kong, X.; Wang, M.; et al. Galactosyltransferase B4GALT1 confers chemoresistance in pancreatic ductal adenocarcinomas by upregulating N-linked glycosylation of CDK11(p110). Cancer Lett. 2021, 500, 228–243. [Google Scholar] [CrossRef]
- Zhou, H.; Ma, H.; Wei, W.; Ji, D.; Song, X.; Sun, J.; Zhang, J.; Jia, L. B4GALT family mediates the multidrug resistance of human leukemia cells by regulating the hedgehog pathway and the expression of p-glycoprotein and multidrug resistance-associated protein 1. Cell Death Dis. 2013, 4, e654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, P.K.; Hsiao, M.; Hsu, T.L.; Chang, C.F.; Wu, C.Y.; Chen, B.R.; Huang, H.W.; Liao, K.S.; Chen, C.C.; Chen, C.L.; et al. Signaling pathway of globo-series glycosphingolipids and β1,3-galactosyltransferase V (β3GalT5) in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3518–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revoredo, L.; Wang, S.; Bennett, E.P.; Clausen, H.; Moremen, K.W.; Jarvis, D.L.; Ten Hagen, K.G.; Tabak, L.A.; Gerken, T.A. Mucin-type O-glycosylation is controlled by short- and long-range glycopeptide substrate recognition that varies among members of the polypeptide GalNAc transferase family. Glycobiology 2016, 26, 360–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berois, N.; Mazal, D.; Ubillos, L.; Trajtenberg, F.; Nicolas, A.; Sastre-Garau, X.; Magdelenat, H.; Osinaga, E. UDP-N-acetyl-D-galactosamine: Polypeptide N-acetylgalactosaminyltransferase-6 as a new immunohistochemical breast cancer marker. J. Histochem. Cytochem. 2006, 54, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, T.; Suzuki, A.; Asaka, R.; Ishikawa, K.; Yamada, Y.; Kobara, H.; Nakayama, J.; Shiozawa, T. Immunohistochemical expression of core 2 β1,6-N-acetylglucosaminyl transferase 1 (C2GnT1) in endometrioid-type endometrial carcinoma: A novel potential prognostic factor. Histopathology 2013, 62, 986–993. [Google Scholar] [CrossRef]
- Du, J.; Hong, S.; Dong, L.; Cheng, B.; Lin, L.; Zhao, B.; Chen, Y.G.; Chen, X. Dynamic Sialylation in Transforming Growth Factor-β (TGF-β)-induced Epithelial to Mesenchymal Transition. J. Biol. Chem. 2015, 290, 12000–12013. [Google Scholar] [CrossRef] [Green Version]
- Borsig, L.; Wong, R.; Hynes, R.O.; Varki, N.M.; Varki, A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc. Natl. Acad. Sci. USA 2002, 99, 2193–2198. [Google Scholar] [CrossRef] [Green Version]
- St Hill, C.A.; Baharo-Hassan, D.; Farooqui, M. C2-O-sLeX glycoproteins are E-selectin ligands that regulate invasion of human colon and hepatic carcinoma cells. PLoS ONE 2011, 6, e16281. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.G.; Balmaña, M.; Macedo, J.A.; Poças, J.; Fernandes, Â.; de-Freitas-Junior, J.C.M.; Pinho, S.S.; Gomes, J.; Magalhães, A.; Gomes, C.; et al. Glycosylation in cancer: Selected roles in tumour progression, immune modulation and metastasis. Cell Immunol. 2018, 333, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Kannagi, R.; Izawa, M.; Koike, T.; Miyazaki, K.; Kimura, N. Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 2004, 95, 377–384. [Google Scholar] [CrossRef]
- Laubli, H.; Borsig, L. Altered Cell Adhesion and Glycosylation Promote Cancer Immune Suppression and Metastasis. Front. Immunol. 2019, 10, 2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.C.; Chen, L.H.; Gillespie, S.; Kiejda, K.A.; Mhaidat, N.; Wang, Y.F.; Thorne, R.; Zhang, X.D.; Hersey, P. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res. 2007, 67, 5880–5888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, T.; Yoshida, T.; Nakata, S.; Horinaka, M.; Wakada, M.; Mizutani, Y.; Miki, T.; Sakai, T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005, 65, 6364–6370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xiong, W.; Tang, Y. Tunicamycin suppresses breast cancer cell growth and metastasis via regulation of the protein kinase B/nuclear factor-κB signaling pathway. Oncol. Lett. 2018, 15, 4137–4142. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dai, L.; Wang, M.; Feng, F.; Xiao, Y. Tunicamycin Induces Hepatic Stellate Cell Apoptosis Through Calpain-2/Ca(2 +)-Dependent Endoplasmic Reticulum Stress Pathway. Front. Cell Dev. Biol. 2021, 9, 684857. [Google Scholar] [CrossRef]
- Patsos, G.; Hebbe-Viton, V.; Robbe-Masselot, C.; Masselot, D.; San Martin, R.; Greenwood, R.; Paraskeva, C.; Klein, A.; Graessmann, M.; Michalski, J.C.; et al. O-glycan inhibitors generate aryl-glycans, induce apoptosis and lead to growth inhibition in colorectal cancer cell lines. Glycobiology 2009, 19, 382–398. [Google Scholar] [CrossRef]
- Tian, E.; Ten Hagen, K.G.; Shum, L.; Hang, H.C.; Imbert, Y.; Young, W.W., Jr.; Bertozzi, C.R.; Tabak, L.A. An inhibitor of O-glycosylation induces apoptosis in NIH3T3 cells and developing mouse embryonic mandibular tissues. J. Biol. Chem. 2008, 283, 4460. [Google Scholar] [CrossRef]
- Song, L.; Linstedt, A.D. Inhibitor of ppGalNAc-T3-mediated O-glycosylation blocks cancer cell invasiveness and lowers FGF23 levels. Elife 2017, 6, e24051. [Google Scholar] [CrossRef] [Green Version]
- Kosuge, Y.; Koen, Y.; Ishige, K.; Minami, K.; Urasawa, H.; Saito, H.; Ito, Y. S-allyl-L-cysteine selectively protects cultured rat hippocampal neurons from amyloid beta-protein- and tunicamycin-induced neuronal death. Neuroscience 2003, 122, 885–895. [Google Scholar] [CrossRef]
- Yen, J.H.; Wu, P.S.; Chen, S.F.; Wu, M.J. Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells. Int. J. Mol. Sci. 2017, 18, 852. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Vlahakis, J.Z.; Szarek, W.A.; Brockhausen, I. Selective inhibition of glycosyltransferases by bivalent imidazolium salts. Bioorg. Med. Chem. 2013, 21, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.J.; Hu, R.H.; Chou, C.H.; Hsu, C.L.; Liu, Y.W.; Huang, J.; Hung, J.S.; Lai, I.R.; Juan, H.F.; Yu, S.L.; et al. Knockdown of GALNT1 suppresses malignant phenotype of hepatocellular carcinoma by suppressing EGFR signaling. Oncotarget 2015, 6, 5650–5665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.S.; Chang, H.Y.; Li, C.P.; Liu, J.M.; Huang, T.S. Tumor beta-1,4-galactosyltransferase IV overexpression is closely associated with colorectal cancer metastasis and poor prognosis. Clin. Cancer Res. 2005, 11, 8615–8622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.M.; Wang, Y.H.; Hung, J.T.; Lin, Y.J.; Huang, Y.L.; Liao, G.S.; Hsu, Y.L.; Wu, J.C.; Yu, A.L. High B3GALT5 expression confers poor clinical outcome and contributes to tumor progression and metastasis in breast cancer. Breast Cancer Res. 2021, 23, 5. [Google Scholar] [CrossRef]
- Beaman, E.M.; Brooks, S.A. The extended ppGalNAc-T family and their functional involvement in the metastatic cascade. Histol. Histopathol. 2014, 29, 293–304. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Kurita, T.; Koi, C.; Murakami, M.; Kagami, S.; Hachisuga, T.; Masanori, H.; Morimoto, Y.; Izumi, H. GalNAc-T6 in the relationship with invasion ability of endometrial carcinomas and prognostic significance. Am. J. Cancer Res. 2017, 7, 1188–1197. [Google Scholar]
- St Hill, C.A.; Farooqui, M.; Mitcheltree, G.; Gulbahce, H.E.; Jessurun, J.; Cao, Q.; Walcheck, B. The high affinity selectin glycan ligand C2-O-sLex and mRNA transcripts of the core 2 beta-1,6-N-acetylglucosaminyltransferase (C2GnT1) gene are highly expressed in human colorectal adenocarcinomas. BMC Cancer 2009, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Furukawa, K. Sequential action of Ets-1 and Sp1 in the activation of the human beta-1,4-galactosyltransferase V gene involved in abnormal glycosylation characteristic of cancer cells. J. Biol. Chem. 2007, 282, 27702–27712. [Google Scholar] [CrossRef] [Green Version]
- Schjoldager, K.T.; Vakhrushev, S.Y.; Kong, Y.; Steentoft, C.; Nudelman, A.S.; Pedersen, N.B.; Wandall, H.H.; Mandel, U.; Bennett, E.P.; Levery, S.B.; et al. Probing isoform-specific functions of polypeptide GalNAc-transferases using zinc finger nuclease glycoengineered SimpleCells. Proc. Natl. Acad. Sci. USA 2012, 109, 9893–9898. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lin, D.; Xu, L.; Jiang, Z.; Zhou, Y.; Wu, S. An anti-human ppGalNAcT-2 monoclonal antibody. Hybridoma 2011, 30, 549–554. [Google Scholar] [CrossRef]
- Kim, Y.S.; Yoo, H.S.; Ko, J.H. Implication of aberrant glycosylation in cancer and use of lectin for cancer biomarker discovery. Protein Pept. Lett. 2009, 16, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.J.; Gallagher, E.S. Overview of Characterizing Cancer Glycans with Lectin-Based Analytical Methods. Methods Mol. Biol. 2019, 1928, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Wang, F. The physiological and pathological roles and applications of sialyl Lewis x, a common carbohydrate ligand of the three selectins. Glycoconj. J. 2020, 37, 277–291. [Google Scholar] [CrossRef] [PubMed]
- St Hill, C.A.; Bullard, K.M.; Walcheck, B. Expression of the high-affinity selectin glycan ligand C2-O-sLeX by colon carcinoma cells. Cancer Lett. 2005, 217, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Chen, H.; Yao, X.P.; Cong, W.M.; Wu, M.C. E-selectin and its ligand-sLeX in the metastasis of hepatocellular carcinoma. Hepatobiliary Pancreat Dis. Int. 2002, 1, 80–82. [Google Scholar] [PubMed]
- Ferreira, I.G.; Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Glycosylation as a Main Regulator of Growth and Death Factor Receptors Signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Wen, T.; Yan, R.; Kim, S.R.; Stowell, S.R.; Wang, W.; Wang, Y.; An, G.; Cummings, R.D.; Ju, T. O-glycans on death receptors in cells modulate their sensitivity to TRAIL-induced apoptosis through affecting on their stability and oligomerization. FASEB J. 2020, 34, 11786–11801. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Costa, A.F.; Campos, D.; Reis, C.A.; Gomes, C. Targeting Glycosylation: A New Road for Cancer Drug Discovery. Trends Cancer 2020, 6, 757–766. [Google Scholar] [CrossRef]
- Chen, C.; Khismatullin, D.B. Lipopolysaccharide induces the interactions of breast cancer and endothelial cells via activated monocytes. Cancer Lett. 2014, 345, 75–84. [Google Scholar] [CrossRef]
- Deschepper, F.M.; Zoppi, R.; Pirro, M.; Hensbergen, P.J.; Dall’Olio, F.; Kotsias, M.; Gardner, R.A.; Spencer, D.I.R.; Videira, P.A. L1CAM as an E-selectin Ligand in Colon Cancer. Int. J. Mol. Sci. 2020, 21, 8286. [Google Scholar] [CrossRef] [PubMed]
- Natoni, A.; Macauley, M.S.; O’Dwyer, M.E. Targeting Selectins and Their Ligands in Cancer. Front. Oncol. 2016, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, S.; Hiraga, T.; Ikehara, Y.; Kudo, T.; Iwasaki, H.; Morozumi, K.; Akamatsu, S.; Tachikawa, T.; Narimatsu, H. Molecular mechanisms of expression of Lewis b antigen and other type I Lewis antigens in human colorectal cancer. Glycobiology 1999, 9, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Goelz, S.; Kumar, R.; Potvin, B.; Sundaram, S.; Brickelmaier, M.; Stanley, P. Differential expression of an E-selectin ligand (SLex) by two Chinese hamster ovary cell lines transfected with the same alpha (1,3)-fucosyltransferase gene (ELFT). J. Biol. Chem. 1994, 269, 1033–1040. [Google Scholar] [CrossRef]
- Kazaana, A.; Sano, E.; Yoshimura, S.; Makita, K.; Hara, H.; Yoshino, A.; Ueda, T. Promotion of TRAIL/Apo2L-induced apoptosis by low-dose interferon-β in human malignant melanoma cells. J. Cell. Physiol. 2019, 234, 13510–13524. [Google Scholar] [CrossRef]
- Guo, X.; Meng, Y.; Sheng, X.; Guan, Y.; Zhang, F.; Han, Z.; Kang, Y.; Tai, G.; Zhou, Y.; Cheng, H. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anticancer Drugs 2017, 28, 66–74. [Google Scholar] [CrossRef]
- Yoo, J.; Mashalidis, E.H.; Kuk, A.C.Y.; Yamamoto, K.; Kaeser, B.; Ichikawa, S.; Lee, S.Y. GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat. Struct. Mol. Biol. 2018, 25, 217–224. [Google Scholar] [CrossRef]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef]
- Lee, D.; Hokinson, D.; Park, S.; Elvira, R.; Kusuma, F.; Lee, J.M.; Yun, M.; Lee, S.G.; Han, J. ER Stress Induces Cell Cycle Arrest at the G2/M Phase Through eIF2α Phosphorylation and GADD45α. Int. J. Mol. Sci. 2019, 20, 6309. [Google Scholar] [CrossRef] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.H.; Tsai, F.J.; Hsu, Y.M.; Yin, M.C.; Chiu, H.Y.; Yang, J.S. Sensitivity of allyl isothiocyanate to induce apoptosis via ER stress and the mitochondrial pathway upon ROS production in colorectal adenocarcinoma cells. Oncol. Rep. 2020, 44, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.F.; Dong, Q.; Bai, Y.; Yuan, J.; Xu, Q.; Cao, C.; Liu, X. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic. Biol. Med. 2017, 103, 177–187. [Google Scholar] [CrossRef]
- Purohit, S.; Li, T.; Guan, W.; Song, X.; Song, J.; Tian, Y.; Li, L.; Sharma, A.; Dun, B.; Mysona, D.; et al. Multiplex glycan bead array for high throughput and high content analyses of glycan binding proteins. Nat. Commun. 2018, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Sun, H.; Wu, K.; Lyu, J.; Zhang, J.; Gu, F.; Ma, Y.; Shen, B.; Wang, C.; Chen, X.; et al. SLe(x) expression in invasive micropapillary breast carcinoma is associated with poor prognosis and can be combined with MUC1/EMA as a supplementary diagnostic indicator. Cancer Biol. Med. 2021, 18, 477–489. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, S.; Liu, Y.; Wang, Z.; Wang, X.; Yan, Q. Overexpression of fucosyltransferase VII (FUT7) promotes embryo adhesion and implantation. Fertil. Steril. 2009, 91, 908–914. [Google Scholar] [CrossRef]
- Gao, Y.; Chachadi, V.B.; Cheng, P.W.; Brockhausen, I. Glycosylation potential of human prostate cancer cell lines. Glycoconj. J. 2012, 29, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yip, J.; Harrison, M.; Brockhausen, I. Primary human osteoblasts and bone cancer cells as models to study glycodynamics in bone. Int. J. Biochem. Cell Biol. 2008, 40, 471–483. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, H.; Peng, L.; Kuhn, C.; Chelariu-Raicu, A.; Mahner, S.; Jeschke, U.; von Schönfeldt, V. Expression of the Carbohydrate Lewis Antigen, Sialyl Lewis A, Sialyl Lewis X, Lewis X, and Lewis Y in the Placental Villi of Patients With Unexplained Miscarriages. Front. Immunol. 2021, 12, 679424. [Google Scholar] [CrossRef]
- Zou, G.; Zhang, X.; Wang, L.; Li, X.; Xie, T.; Zhao, J.; Yan, J.; Wang, L.; Ye, H.; Jiao, S.; et al. Herb-sourced emodin inhibits angiogenesis of breast cancer by targeting VEGFA transcription. Theranostics 2020, 10, 6839–6853. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luan, X.; Sun, M.; Zhao, X.; Wang, J.; Han, Y.; Gao, Y. Bisimidazolium Salt Glycosyltransferase Inhibitors Suppress Hepatocellular Carcinoma Progression In Vitro and In Vivo. Pharmaceuticals 2022, 15, 716. https://doi.org/10.3390/ph15060716
Luan X, Sun M, Zhao X, Wang J, Han Y, Gao Y. Bisimidazolium Salt Glycosyltransferase Inhibitors Suppress Hepatocellular Carcinoma Progression In Vitro and In Vivo. Pharmaceuticals. 2022; 15(6):716. https://doi.org/10.3390/ph15060716
Chicago/Turabian StyleLuan, Xue, Ming Sun, Xue Zhao, Jingyi Wang, Ye Han, and Yin Gao. 2022. "Bisimidazolium Salt Glycosyltransferase Inhibitors Suppress Hepatocellular Carcinoma Progression In Vitro and In Vivo" Pharmaceuticals 15, no. 6: 716. https://doi.org/10.3390/ph15060716