
Nano-Silica Carriers Coated by Chloramphenicol: Synthesis, Characterization, and Grinding Trial as a Way to Improve the Release Profile

 ,
,  ,
,  ,
,  , , , and
, , , and
Abstract
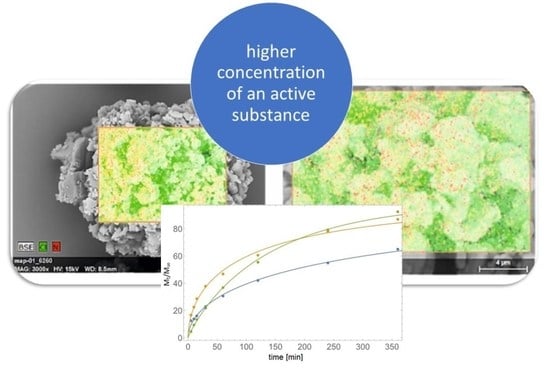
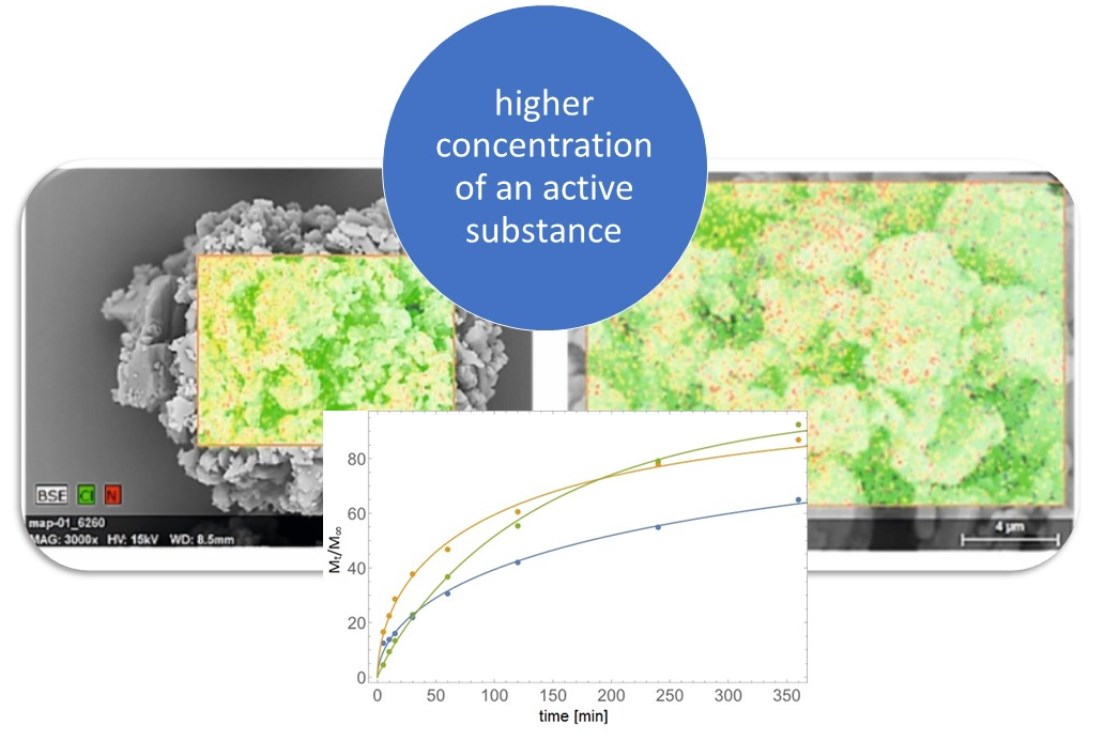
:
1. Introduction
2. Results and Discussion
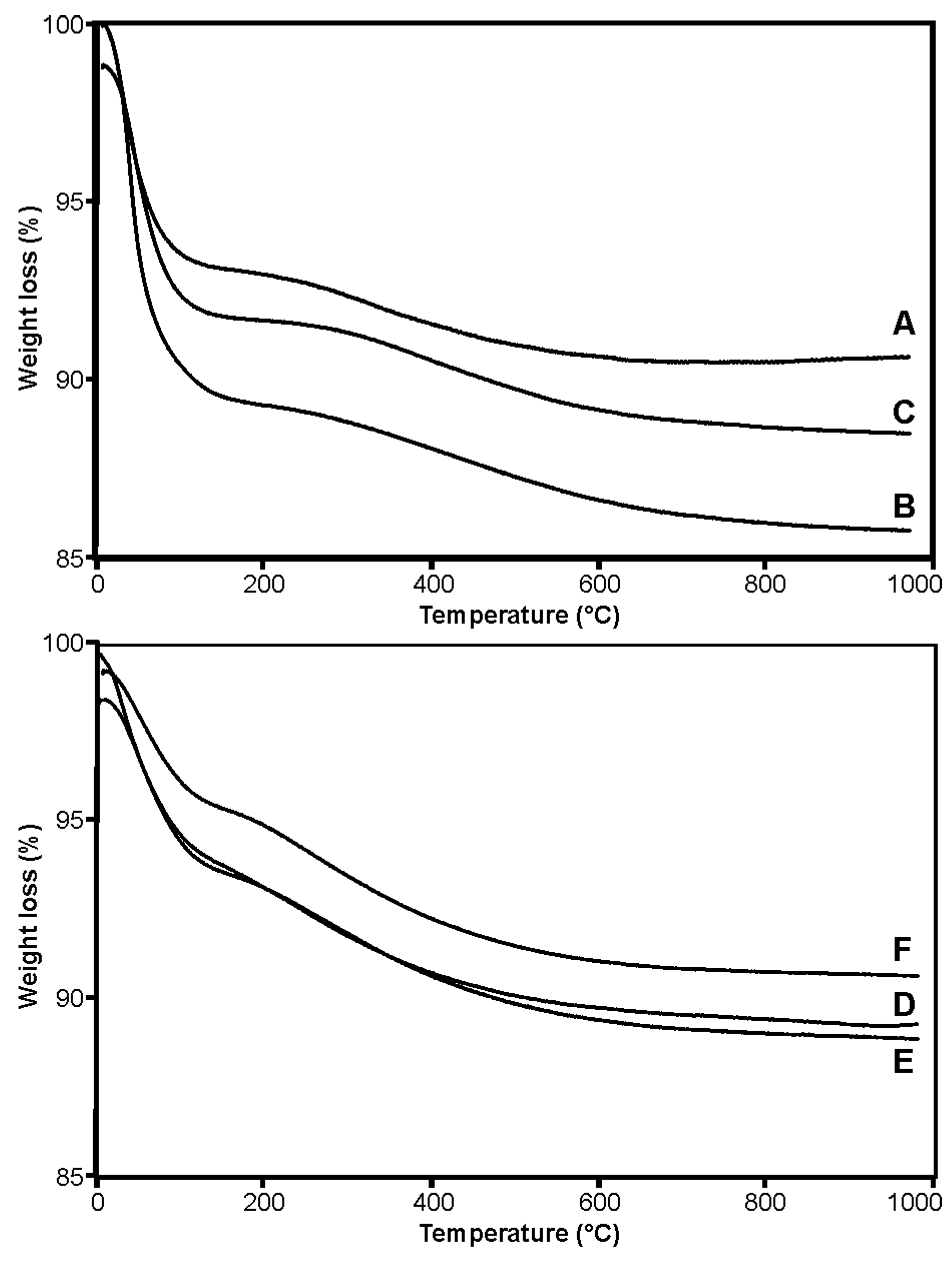
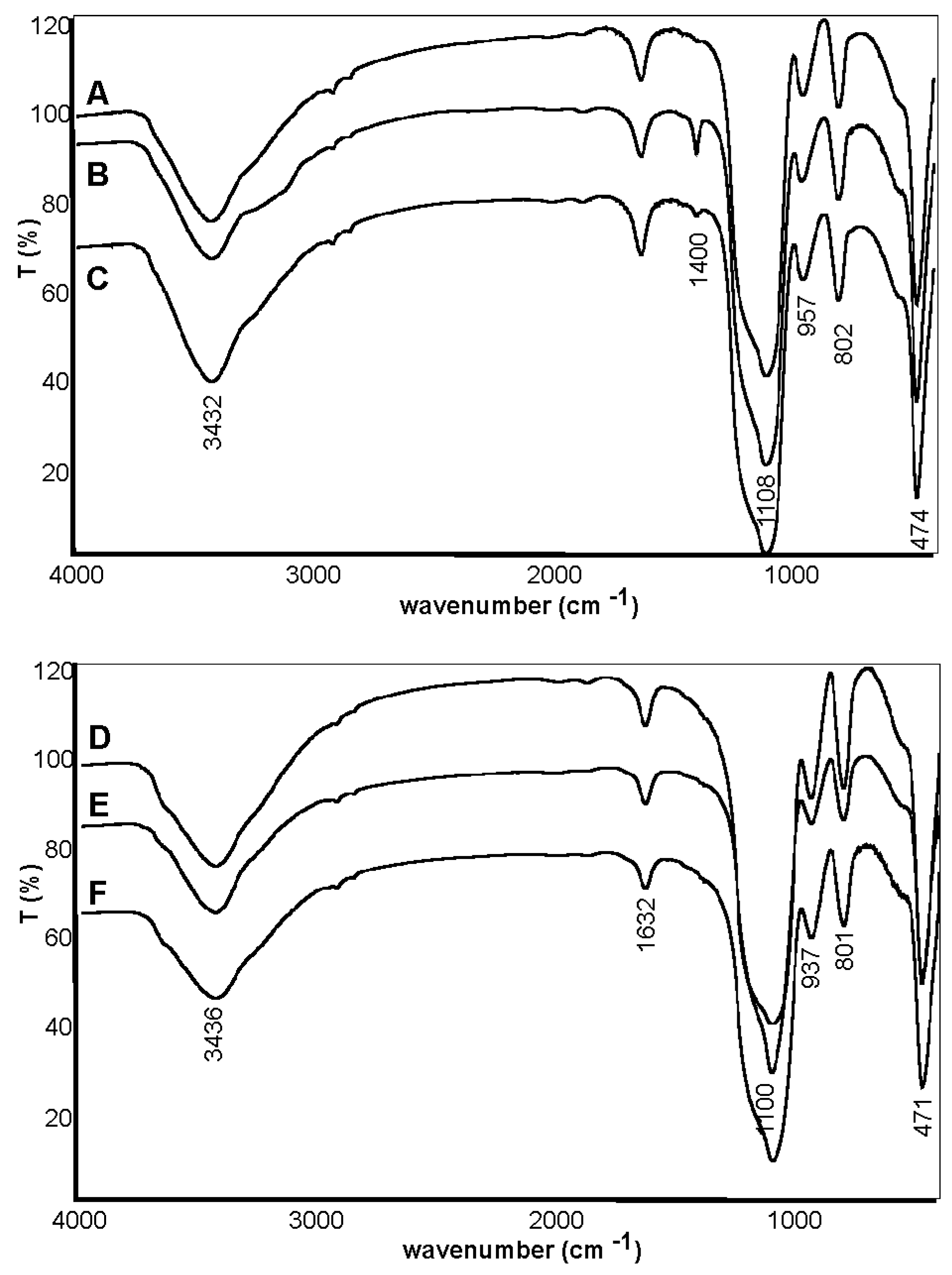
2.1. Synthesis and Characterization of Silica Carriers
2.2. Synthesis and Characterization of Silicon Nanoparticles Coated with Chloramphenicol
2.3. Study of the Release of Chloramphenicol from the Carbopol-Based Gels
2.3.1. The Weilbull Model
2.3.2. The Mahalonobis Distance
2.3.3. Model-Independent Coefficients of Similarity and Difference (F1, F2)
3. Materials and Methods
3.1. Materials
3.2. Characterization Techniques
3.3. The Analysis of Release Profiles and Statistical Analysis
- -
- model-dependent based on Weibull function;
- -
- model-independent based on Mahalanobis distance;
- -
- model-independent similarity and difference coefficients (F1, F2).
3.4. Synthesis Procedures
Silica Carrier Synthesis
3.5. Silica Carrier Grinding
3.6. Coating of Silica Carrier with Chloramphenicol
3.7. Coating of Ground Silica Carrier with Chloramphenicol
3.8. Preparation of the Carbopol Gels
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ge, K.; Ren, H.; Sun, W.; Zhao, Q.; Jia, G.; Zang, A.; Zhang, C.; Zhang, J. Walnut Kernel-like Mesoporous Silica Nanoparticles as Effective Drug Carrier for Cancer Therapy In Vitro. J. Nanopart. Res. 2016, 18, 81. [Google Scholar] [CrossRef]
- Baumgartner, A.; Planinšek, O. Application of Commercially Available Mesoporous Silica for Drug Dissolution Enhancement in Oral Drug Delivery. Eur. J. Pharm. Sci. 2021, 167, 106015. [Google Scholar] [CrossRef]
- Nagarajan, R.; Alan Hatton, T. Nanoparticles: Synthesis, Stabilization, Passivation, and Functionalization. In ACS Symposium Series 996; American Chemical Society: Washington, DC, USA, 2008; Volume 1, pp. 2–14. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Kim, L.U.; Kim, C.K. Size Control of Silica Nanoparticles and Their Surface Treatment for Fabrication of Dental Nanocomposites. Biomacromolecules 2007, 8, 215–222. [Google Scholar] [CrossRef]
- Liberman, A.; Mendez, N.; Trogler, W.C.; Kummel, A.C. Synthesis and Surface Functionalization of Silica Nanoparticles for Nanomedicine. Surf. Sci. Rep. 2014, 69, 132–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasmin, R.; Rao, S.; Bremmell, K.; Prestidge, C. Synergistic Role of Solid Lipid and Porous Silica in Improving the Oral Delivery of Weakly Basic Poorly Water Soluble Drugs. Eur. J. Pharm. Sci. 2017, 96, 508–514. [Google Scholar] [CrossRef]
- Borawake, D.D.; Pande, V.V.; Giri, M.A. Mesoporous Silica Nanoparticles as Theranostic Platform for Smart Drug De-Livery: A Review. J. Nanomed Nanosci 2017, 5, 125. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled Growth of Monodisperse Silica Spheres in the Micron Size Range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Tabatabaei, S.; Shukohfar, A.; Aghababazadeh, R.; Mirhabibi, A. Experimental Study of the Synthesis and Characterisation of Silica Nanoparticles via the Sol-Gel Method. J. Phys. Conf. Ser. 2006, 26, 371. [Google Scholar] [CrossRef]
- Finnie, K.S.; Bartlett, J.R.; Barbé, C.J.A.; Kong, L. Formation of Silica Nanoparticles in Microemulsions. Langmuir 2007, 23, 3017–3024. [Google Scholar] [CrossRef]
- Singh, L.P.; Bhattacharyya, S.K.; Kumar, R.; Mishra, G.; Sharma, U.; Singh, G.; Ahalawat, S. Sol-Gel Processing of Silica Nanoparticles and Their Applications. Adv. Colloid Interface Sci. 2014, 214, 17–37. [Google Scholar] [CrossRef]
- Jafari, S.; Derakhshankhah, H.; Alaei, L.; Fattahi, A.; Varnamkhasti, B.S.; Saboury, A.A. Mesoporous Silica Nanoparticles for Therapeutic/Diagnostic Applications. Biomed. Pharmacother. 2019, 109, 1100–1111. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhi, Z.; Jiang, T.; Zhang, J.; Wang, Z.; Wang, S. Spherical Mesoporous Silica Nanoparticles for Loading and Release of the Poorly Water-Soluble Drug Telmisartan. J. Control. Release. 2010, 145, 257–263. [Google Scholar] [CrossRef]
- Ghaferi, M.; Koohi Moftakhari Esfahani, M.; Raza, A.; al Harthi, S.; Ebrahimi Shahmabadi, H.; Alavi, S.E. Mesoporous Silica Nanoparticles: Synthesis Methods and Their Therapeutic Use-Recent Advances. J. Drug Target. 2020, 29, 131–154. [Google Scholar] [CrossRef]
- An, J.; Yang, X.Q.; Cheng, K.; Song, X.L.; Zhang, L.; Li, C.; Zhang, X.S.; Xuan, Y.; Song, Y.Y.; Fang, B.Y.; et al. In Vivo Computed Tomography/Photoacoustic Imaging and NIR-Triggered Chemo-Photothermal Combined Therapy Based on a Gold Nanostar-, Mesoporous Silica-, and Thermosensitive Liposome-Composited Nanoprobe. ACS Appl. Mater. Interfaces 2017, 9, 41748–41759. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Q.; Han, N.; Bai, L.; Li, J.; Liu, J.; Che, E.; Hu, L.; Zhang, Q.; Jiang, T.; et al. Mesoporous Silica Nanoparticles in Drug Delivery and Biomedical Applications. Nanomedicine 2015, 11, 313–327. [Google Scholar] [CrossRef]
- Huh, S.; Wiench, J.W.; Yoo, J.C.; Pruski, M.; Lin, V.S.Y. Organic Functionalization and Morphology Control of Mesoporous Silicas via a Co-Condensation Synthesis Method. Chem. Mater. 2003, 15, 4247–4256. [Google Scholar] [CrossRef]
- Slowing, I.I.; Vivero-Escoto, J.L.; Wu, C.W.; Lin, V.S.Y. Mesoporous Silica Nanoparticles as Controlled Release Drug Delivery and Gene Transfection Carriers. Adv. Drug Deliv. Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Burleigh, M.C.; Dai, S.; Hagaman, E.W.; Barnes, C.E.; Xue, Z.L. Stepwise Assembly of Surface Imprint Sites on MCM-41 for Selective Metal Ion Separations. ACS Symp. Ser. 2001, 778, 146–158. [Google Scholar] [CrossRef]
- Gounani, Z.; Asadollahi, M.A.; Pedersen, J.N.; Lyngsø, J.; Skov Pedersen, J.; Arpanaei, A.; Meyer, R.L. Mesoporous Silica Nanoparticles Carrying Multiple Antibiotics Provide Enhanced Synergistic Effect and Improved Biocompatibility. Colloids Surf. B. 2019, 175, 498–508. [Google Scholar] [CrossRef]
- Subramaniam, S.; Thomas, N.; Gustafsson, H.; Jambhrunkar, M.; Kidd, S.P.; Prestidge, C.A. Rifampicin-Loaded Mesoporous Silica Nanoparticles for the Treatment of Intracellular Infections. Antibiotics 2019, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, H.; Sakai, T.; Teshima, R.; Nemoto, S.; Akiyama, H. Total Determination of Chloramphenicol Residues in Foods by Liquid Chromatography-Tandem Mass Spectrometry. Food Chem. 2017, 230, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Stratton, C.W. Chloramphenicol. Antimicrob. Infect. Dis. News. 2002, 18, 89–91. [Google Scholar] [CrossRef]
- Mehlhorn, A.; Rahimi, P.; Joseph, Y. Aptamer-Based Biosensors for Antibiotic Detection: A Review. Biosensors 2018, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Duan, N.; Wu, S.; Xu, B.; Wang, Z. Chemiluminescent Aptasensor for Chloramphenicol Based on N-(4-Aminobutyl)-N-Ethylisoluminol-Functionalized Flower-like Gold Nanostructures and Magnetic Nanoparticles. Anal. Bioanal. Chem. 2015, 407, 7907–7915. [Google Scholar] [CrossRef]
- Ingebrigtsen, S.G.; Didriksen, A.; Johannessen, M.; Škalko-Basnet, N.; Holsæter, A.M. Old Drug, New Wrapping−A Possible Comeback for Chloramphenicol? Int. J. Pharm. 2017, 526, 538–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzel Kaya, G.; Yilmaz, E.; Deveci, H. A Novel Silica Xerogel Synthesized from Volcanic Tuff as an Adsorbent for High-Efficient Removal of Methylene Blue: Parameter Optimization Using Taguchi Experimental Design. J. Chem. Technol. Biotechnol. 2019, 94, 2729–2737. [Google Scholar] [CrossRef]
- Ştefǎnescu, M.; Stoia, M.; Ştefǎnescu, O.; Popa, A.; Simon, M.; Ionescu, C. The Interaction between TEOS and Some Polyols. J. Therm. Anal. Calorim. 2007, 88, 19–26. [Google Scholar] [CrossRef]
- Shao, Z.; Luo, F.; Cheng, X.; Zhang, Y. Superhydrophobic Sodium Silicate Based Silica Aerogel Prepared by Ambient Pressure Drying. Mater. Chem. Phys. 2013, 141, 570–575. [Google Scholar] [CrossRef]
- Vecchio Ciprioti, S.; Tuffi, R.; Dell’Era, A.; Dal Poggetto, F.; Bollino, F. Thermal Behavior and Structural Study of SiO2/Poly(ε-Caprolactone) Hybrids Synthesized via Sol-Gel Method. Materials 2018, 11, 275. [Google Scholar] [CrossRef] [Green Version]
- Rubio, F.; Rubio, J.; Oteo, J.L. A FT-IR Study of the Hydrolysis of Tetraethylorthosilicate (TEOS). Spectrosc. Lett. 2006, 31, 199–219. [Google Scholar] [CrossRef]
- Iijima, M.; Omori, S.; Hirano, K.; Kamiya, H. Free-Standing, Roll-Able, and Transparent Silicone Polymer Film Prepared by Using Nanoparticles as Cross-Linking Agents. Adv. Powder Technol. 2013, 24, 625–631. [Google Scholar] [CrossRef]
- Koutsoulas, C.; Pippa, N.; Demetzos, C.; Zabka, M. Preparation of Liposomal Nanoparticles Incorporating Terbinafine in Vitro Drug Release Studies. Int. J. Nanosci. Nanotechnol. 2014, 14, 4529–4533. [Google Scholar] [CrossRef]
- Xua, Y.L.; Gong, X.L.; Peng, C.; Sun, Y.Q.; Jiang, W.Q.; Zhang, Z. Shear Thickening Fluids Based on Additives with Different Concentrations and Molecular Chain Lengths. Chin. J. Chem. Phys. 2013, 23, 342. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and Comparison of Dissolution Profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Jans, H.; Liu, X.; Austin, L.; Maes, G.; Huo, Q. Dynamic Light Scattering as a Powerful Tool for Gold Nanoparticle Bioconjugation and Biomolecular Binding Studies. Anal. Chem. 2009, 81, 9425–9432. [Google Scholar] [CrossRef]
- Carvalho, P.M.; Felício, M.R.; Santos, N.C.; Gonçalves, S.; Domingues, M.M. Application of Light Scattering Techniques to Nanoparticle Characterization and Development. Front. Chem. 2018, 6, 237. [Google Scholar] [CrossRef]
- Sadana, G.S.; Ghogare, A.B. Simultaneous Determination of Chloramphenicol and Benzocaine in Topical Formulations by High-Performance Liquid Chromatography. J. Chromatogr. A. 1991, 542, 515–520. [Google Scholar] [CrossRef]
- Al-Rimawi, F.; Kharoaf, M. Analysis of Chloramphenicol and Its Related Compound 2-Amino-1-(4-Nitrophenyl)Propane-1,3-Diol by Reversed-Phase High-Performance Liquid Chromatography with U.V. Detection. Chromatogr. Res. Int. 2011, 2011, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Moffat, A.C.; Osselton, M.D.; Widdop, B.; Watts, J. Clarke’s Analysis of Drugs and Poisons . In Pharmaceuticals, Body Fluids and Postmortem Material; Pharmaceutical Press: London, UK, 2011. [Google Scholar]
- EMA. Guideline on the Investigation of Bioequivalence; Committee for Medicinal Products for Human Use; EMEA: London, UK, 2008; Doc. Ref. CPMP/EWP/QWP/1401/98; Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003011.pdf (accessed on 1 April 2022).
- FDA. Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms; FDA: Rockville, MD, USA, 1997. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/dissolution-testing-immediate-release-solid-oral-dosage-forms (accessed on 1 April 2022).
- He, Y.; Liang, S.; Long, M.; Xu, H. Mesoporous Silica Nanoparticles as Potential Carriers for Enhanced Drug Solubility of Paclitaxel. Mater. Sci. Eng. C-Mater. Biol. Appl. 2017, 78, 12–17. [Google Scholar] [CrossRef]
- Dudhipala, N.; Gorre, T. Neuroprotective Effect of Ropinirole Lipid Nanoparticles Enriched Hydrogel for Parkinson’s Disease: In Vitro, Ex Vivo, Pharmacokinetic and Pharmacodynamic Evaluation. Pharmaceutics 2020, 12, 448. [Google Scholar] [CrossRef]
- Sareen, R.; Kumar, S.; D Gupta, G. Meloxicam Carbopol-Based Gels: Characterization and Evaluation. Curr. Drug Deliv. 2011, 8, 407–415. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
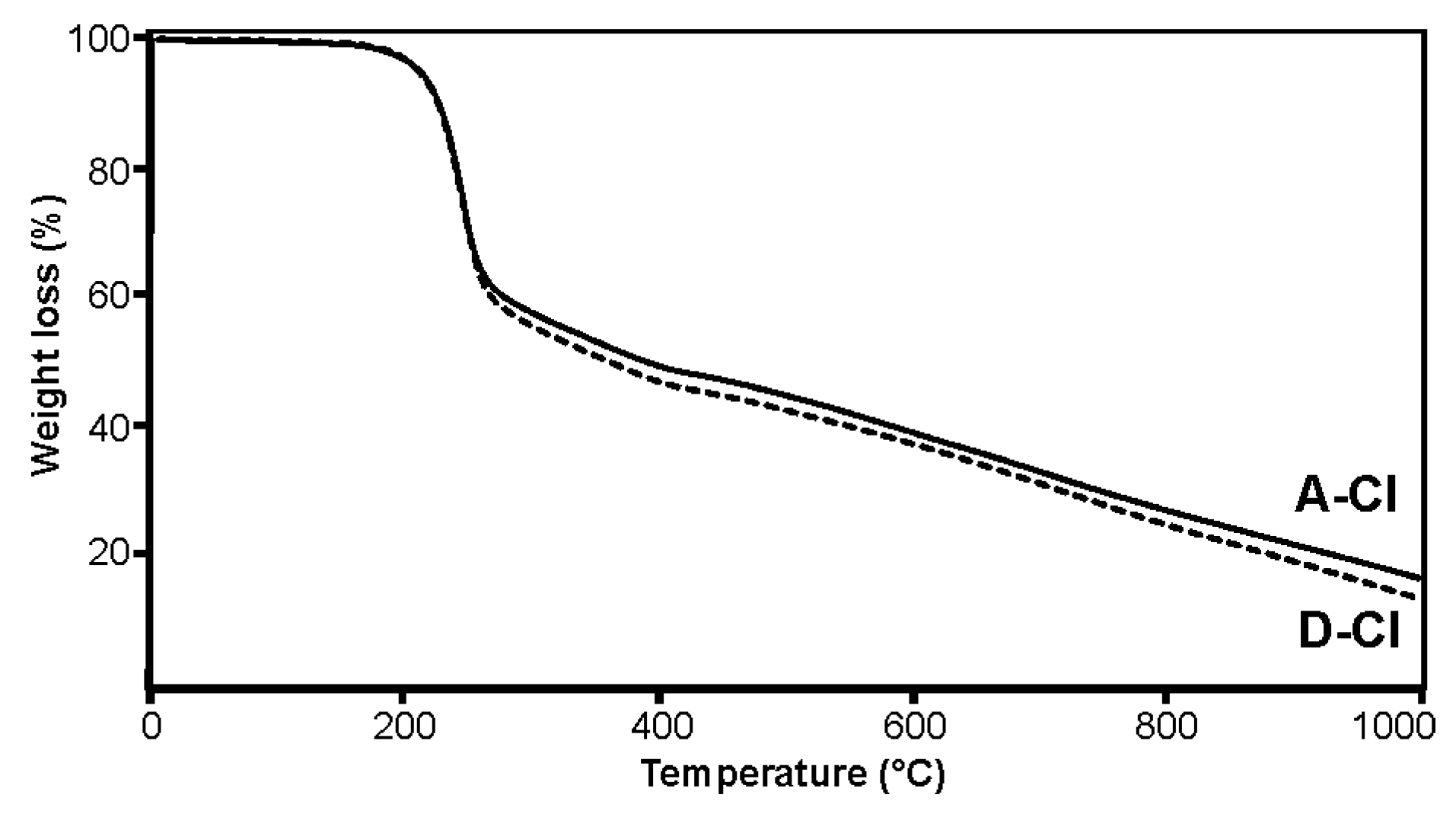{kind=link}
{kind=link}
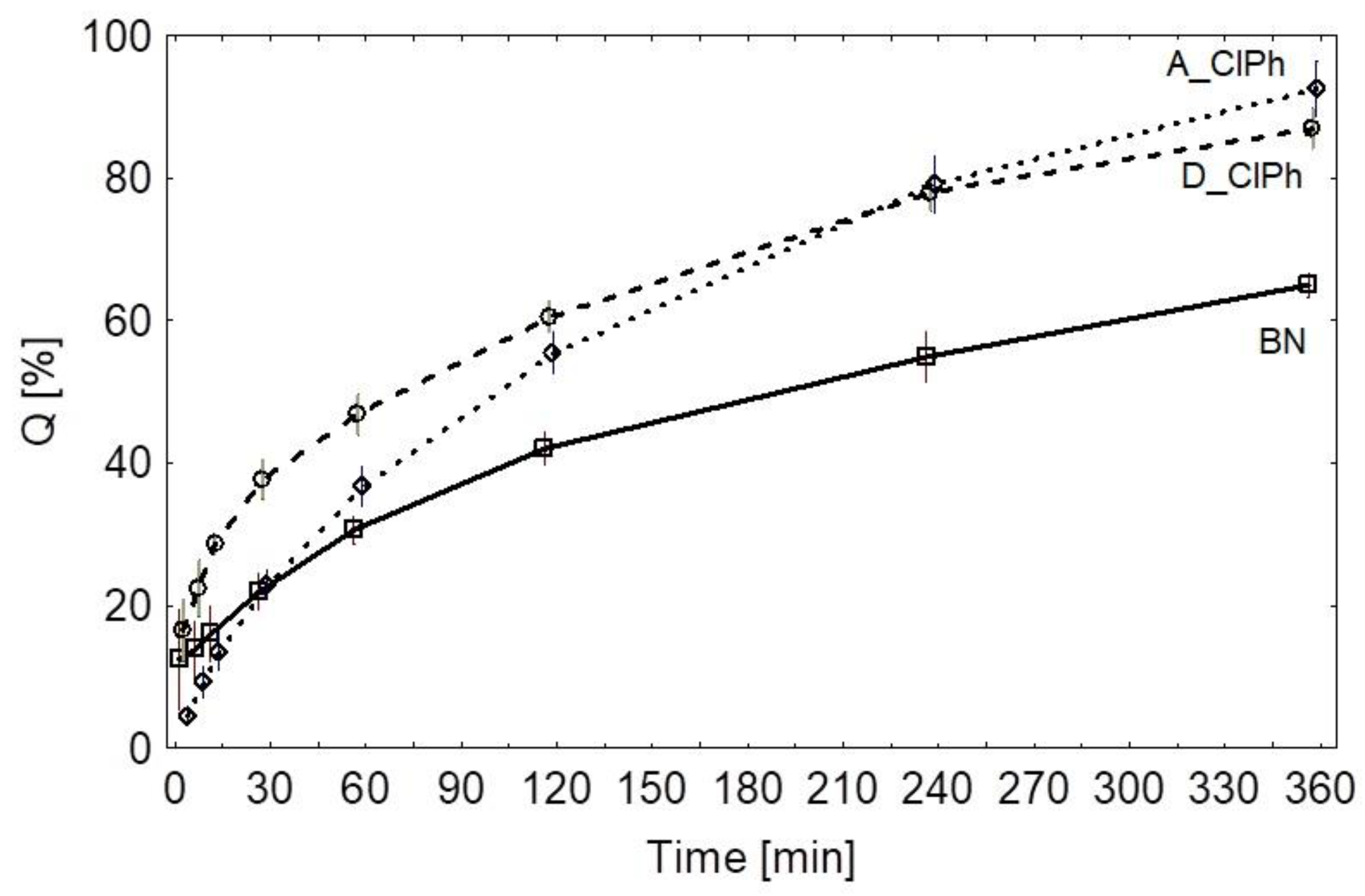{kind=link}
| Parameters | Synthesis 1 Carrier A | Synthesis 2 Carrier B | Synthesis 3 Carrier C | |
|---|---|---|---|---|
| Temperature [°C] | 65 | 25 | 35 | |
| Volume of added reagents [mL] | ammonium hydroxide 25% | 4.8 | 6.0 | 4.8 |
| water | 4.8 | 6.0 | 4.8 | |
| ethanol 96% | 72.0 | 90.0 | 72.0 | |
| TEOS | 6.4 | 8.0 | 6.4 | |
| TEOS insertion rate [mL/min] | 3.2 | 4.0 | 3.2 | |
| Volume of added ammonia solution [mL] | prior to the application of TEOS | 2.4 | 13.0 | 0 |
| after TEOS application | 14.5 | 15.0 | 13.0 | |
| Material | A | B | C | D | E | F | D_ClPh |
|---|---|---|---|---|---|---|---|
| Specific surface [m2/g] | 0.1871 | 13.8976 | 23.8035 | 35.0244 | 32.8918 | 32.7435 | 3.3736 |
| One-point analysis of specific surface area (p/p° = 0.25) | 0.1571 | 13.5829 | 22.9921 | 34.0508 | 32.2388 | 31.9001 | 3.1355 |
| Total pore volume in the range 1–300 nm (BJH adsorption) [cm3/g] | 0.000327 | 0.027005 | 0.050779 | 0.106719 | 0.105568 | 0.104813 | 0.010528 |
| Total pore volume in the range 1–300 nm (BJH desorption) [cm3/g] | 0.000292 | 0.029091 | 0.068072 | 0.134964 | 0.129747 | 0.121554 | 0.011746 |
| Average pore diameter (BJH adsorption) [nm] | 5.5172 | 6.8381 | 7.3497 | 10.5674 | 11.5499 | 11.2547 | 8.3944 |
| Average pore diameter (BJH desorption) [nm] | 12.9577 | 7.7064 | 10.1059 | 13.6115 | 13.1257 | 12.8671 | 11.7519 |
| Material | Average [nm] | Std. Dev. | RSD | Minimum [nm] | Median [nm] | Maximum [nm] |
|---|---|---|---|---|---|---|
| A | 107.7 | 22.0 | 20.4% | 62.8 | 112.8 | 131.0 |
| B | 181.4 | 16.0 | 8.8% | 143.8 | 186.6 | 196.9 |
| C | 125.1 | 16.0 | 12.8% | 84.2 | 131.9 | 137.3 |
| D | 389.7 | 139.3 | 35.7% | 112.2 | 438.0 | 556.5 |
| E | 215.3 | 23.3 | 10.8% | 188.8 | 212.9 | 289.2 |
| F | 260.0 | 77.8 | 29.9% | 168.7 | 257.3 | 440.4 |
| A_ClPh | 178.2 | 22.14 | 12.0% | 141.8 | 182.2 | 241.2 |
| D_ClPh | 156.3 | 32.3 | 20.7% | 100.3 | 151.8 | 229.1 |
| Sample | Weight [mg] | AUC Average | CAP [mg] | Content [mg/100 g] | Content Relative to Expected [%] |
|---|---|---|---|---|---|
| Only ClPh 0.5% (BN) | 100.36 | 566.0223 | 0.3955 | 492.6371 | 98.53% |
| A_ClPh (1:1) 0.5% | 103.24 | 624.4009 | 0.4368 | 528.8567 | 105.77% |
| D_ClPh (1:1) 0.5% | 101.35 | 582.7027 | 0.4073 | 502.3668 | 100.47% |
| Type of Gel | Average [Pa·s] | S.D. [Pa·s] | Coefficient of Variation [%] | pH |
|---|---|---|---|---|
| Without the silica and chloramphenicol | 272.71 | 20.45 | 7.50 | 7.02 |
| Without the silica and containing only chloramphenicol BN | 248.85 | 0.79 | 1.62 | 6.91 |
| Silica and chloramphenicol A_ClPh | 262.01 | 6.47 | 2.47 | 6.78 |
| Silica ground with chloramphenicol D_ClPh | 361.77 | 27.52 | 7.61 | 6.89 |
| Analysis Based on Mathematical Models [R2] | Model Independent Analysis | ||||||
|---|---|---|---|---|---|---|---|
| Formulation | First-Order Kinetics | 0-Order Kinetics | Higuchi | Hixson–Crowell | Pepas | Release Efficiency D.E. | Mean Dissolution Time MDT [min] |
| BN | 0.98274 | 0.93977 | 0.9968 | 0.97108 | 0.98975 | 0.45618 | 107.18720 |
| D_ClPh | 0.99026 | 0.89692 | 0.98464 | 0.96958 | 0.99338 | 0.65125 | 90.186017 |
| A_ClPh | 0.99600 | 0.93182 | 0.99622 | 0.99437 | 0.98206 | 0.62205 | 117.80174 |
| Sample | Parameter | Estimate | Standard Error | t-Statistic | p-Value | Confidence Interval (−) | Confidence Interval (+) |
|---|---|---|---|---|---|---|---|
| BN | k | 0.00282 | 0.00016 | 18.115 | 1.82121 × 10−6 | 0.00244 | 0.00320 |
| BN | b | 0.54563 | 0.02018 | 27.033 | 1.69271 × 10−7 | 0.49624 | 0.59501 |
| D_ClPh | k | 0.00834 | 0.00042 | 20.077 | 9.91565 × 10−7 | 0.00732 | 0.00935 |
| D_ClPh | b | 0.55761 | 0.02187 | 25.496 | 2.39898 × 10−7 | 0.50409 | 0.61112 |
| A_ClPh | k | 0.006992 | 0.000185 | 37.8444 | 2.27263 × 10−8 | 0.00654 | 0.00654 |
| A_ClPh | b | 0.896888 | 0.025522 | 35.1416 | 3.53872 × 10−8 | 0.834438 | 0.834438 |
| Comparison of the Profiles | Distance between the Profiles | Lower Limit of the Confidence Interval | Upper Limit of the Confidence Interval | Acceptable Distance between Profiles |
|---|---|---|---|---|
| BN vs. A_ClPh | 13.1292 | 6.29682 | 19.9616 | 8.06196 |
| BN vs. D_ClPh | 16.502 | 9.670 | 23.334 | 9.709 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balwierz, R.; Bursy, D.; Biernat, P.; Hudz, N.; Shanaida, M.; Krzemiński, Ł.; Skóra, P.; Biernat, M.; Siodłak, W.O. Nano-Silica Carriers Coated by Chloramphenicol: Synthesis, Characterization, and Grinding Trial as a Way to Improve the Release Profile. Pharmaceuticals 2022, 15, 703. https://doi.org/10.3390/ph15060703
Balwierz R, Bursy D, Biernat P, Hudz N, Shanaida M, Krzemiński Ł, Skóra P, Biernat M, Siodłak WO. Nano-Silica Carriers Coated by Chloramphenicol: Synthesis, Characterization, and Grinding Trial as a Way to Improve the Release Profile. Pharmaceuticals. 2022; 15(6):703. https://doi.org/10.3390/ph15060703
Chicago/Turabian StyleBalwierz, Radosław, Dawid Bursy, Paweł Biernat, Nataliia Hudz, Mariia Shanaida, Łukasz Krzemiński, Paweł Skóra, Monika Biernat, and Wioletta Ochędzan Siodłak. 2022. "Nano-Silica Carriers Coated by Chloramphenicol: Synthesis, Characterization, and Grinding Trial as a Way to Improve the Release Profile" Pharmaceuticals 15, no. 6: 703. https://doi.org/10.3390/ph15060703