
Novel Effects of Statins on Cancer via Autophagy


 ,
,
Abstract
:1. Introduction
2. Cancer
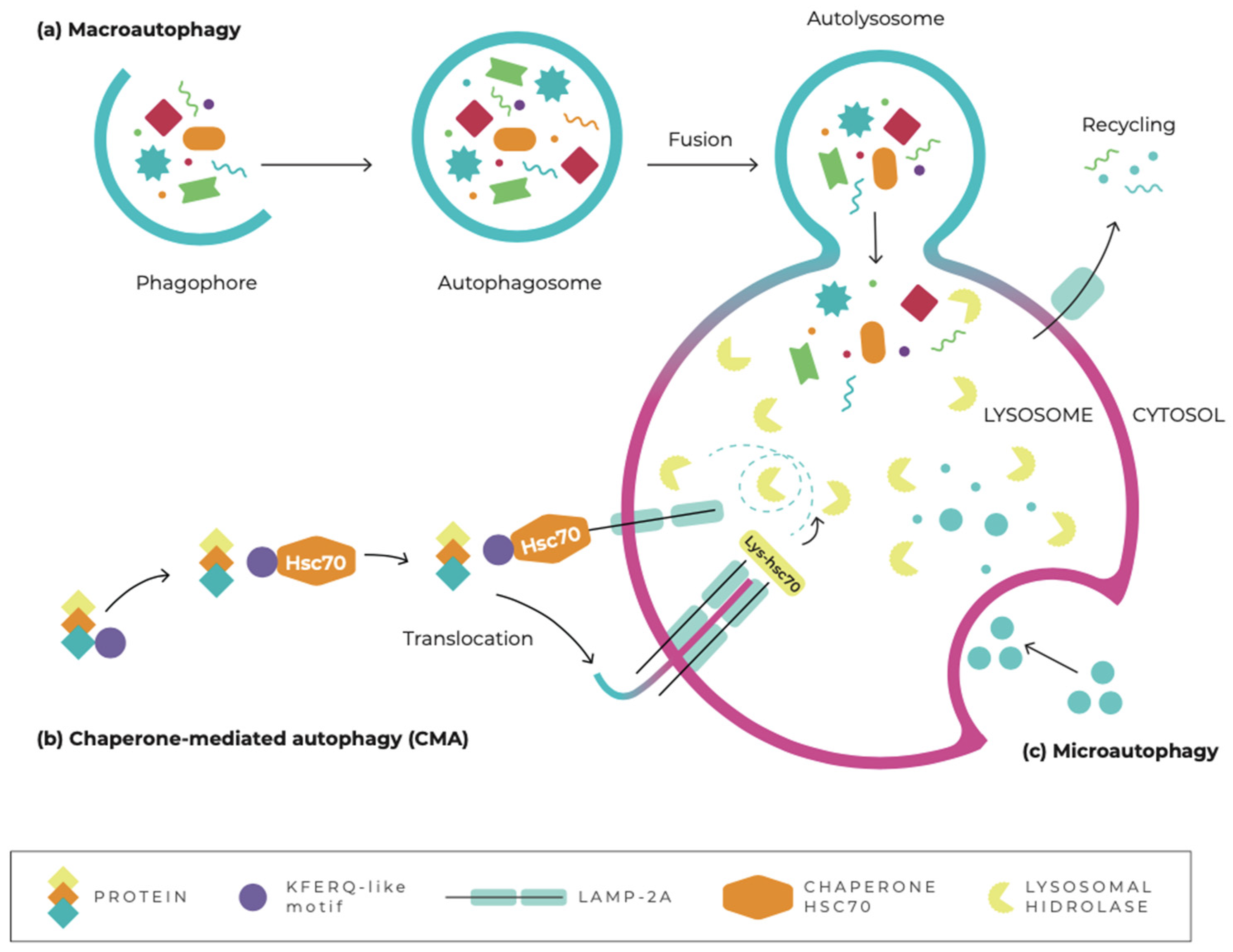
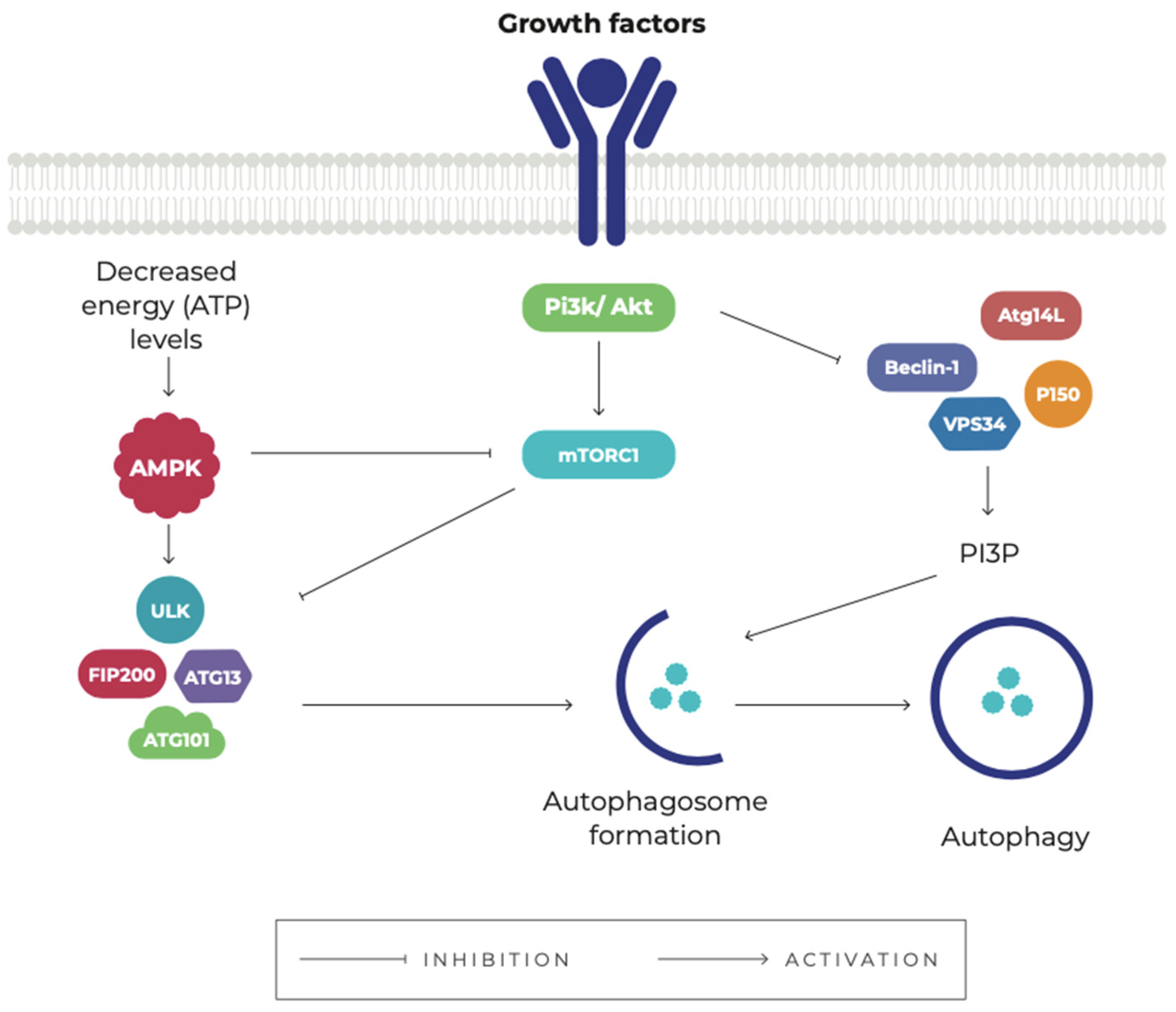
3. Autophagy
4. The Role of Autophagy in Cancer
4.1. The Role of Autophagy in Tumor Suppression
4.2. The Role of Autoaphagy as a Tumor Promoter
4.3. Autophagy as a Regulator of Cancer Metastasis
4.4. Autophagy as a Target for Cancer Therapy
5. Statins
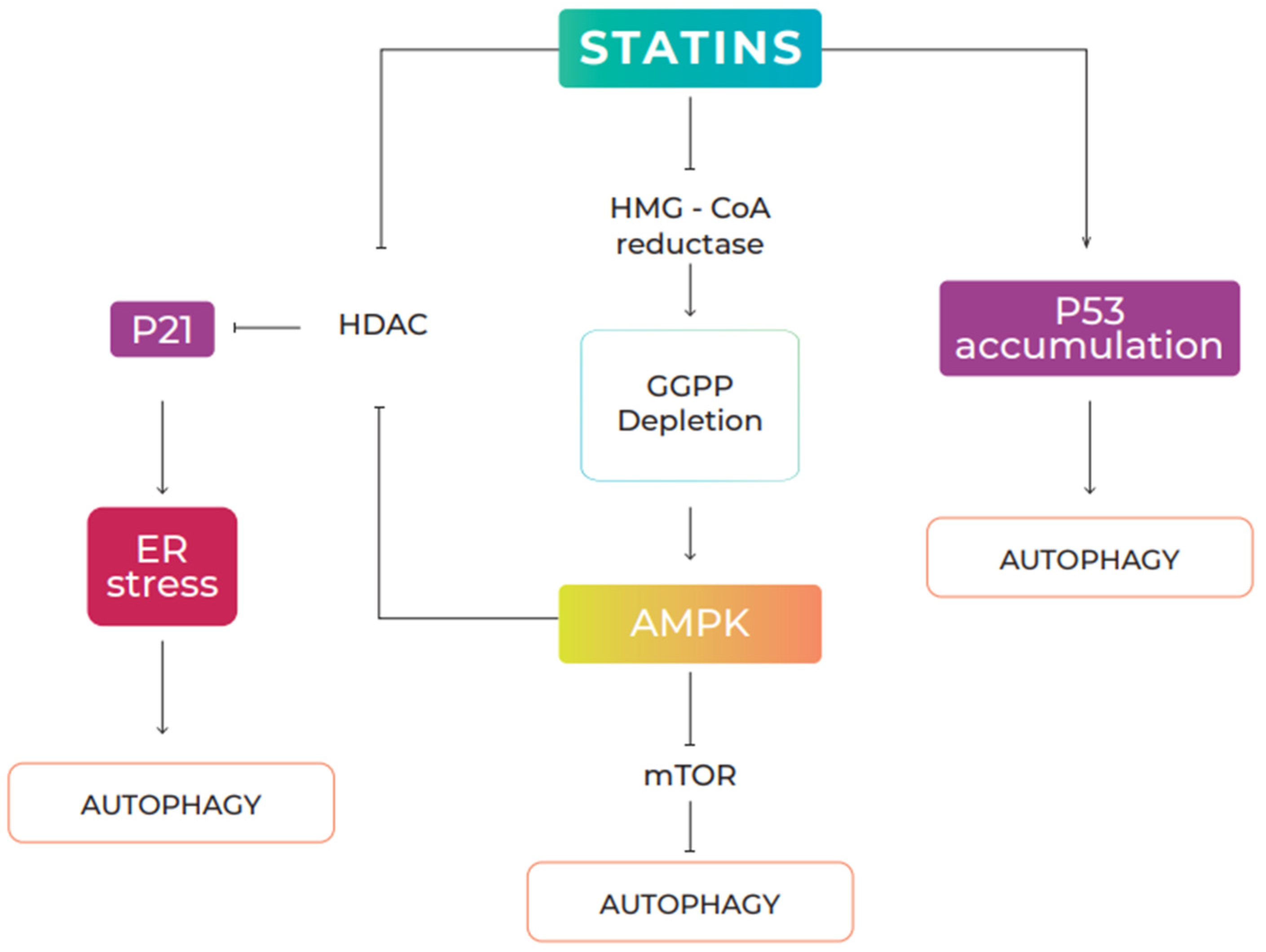
5.1. Statins and Autophagy
5.2. Effects of Statins on Cancer via Autophagy
6. Clinical Trials

{kind=link}
{kind=link}
{kind=link}
| Trial Name (Identifier) | Patient Population (Condition) | Treatment Groups | Enrollment (Participants) | Sponsor |
|---|---|---|---|---|
| Donor Atorvastatin Treatment in Preventing Severe Acute GVHD After Nonmyeloablative Peripheral Blood Stem Cell Transplant in Patients With Hematological Malignancies (NCT01527045) [199] | Hematological malignancies | Drug: Atorvastatin calcium Drug: Cyclosporine Drug: Fludarabine phosphate Drug: Mycophenolate mofetil Procedures: Nonmyeloablative allogeneic hematopoietic stem-cell transplantation and peripheral blood stem-cell transplantation. Radiation: Total-body irradiation | 47 | Fred Hutchinson Cancer Research Center |
| Safety & Efficacy of Atorvastatin for Prophylaxis of Acute Graft Versus Host Disease in Patients With Hematological Malignancies HLA- Donor Hematopoietic Stem Cell Transplantation (NCT01491958) [200] | Acute myelogenous leukemia Acute lymphocytic leukemia Myelodysplastic syndrome | Drug: Atorvastatin Drug: Tacrolimus Drug: Methotrexate | 40 | Ohio State University Comprehensive Cancer Center |
| Pilot Study of Statin Therapy in Young Adult Survivors of Childhood Cancer [201] (NCT01733953) | Cardiovascular disease Childhood ALL Childhood NHL | Drug: Atorvastatin Drug: Sugar pill (placebo) | 27 | University of Minnesota |
| Atorvastatin Calcium and Celecoxib in Treating Patients With Rising PSA Levels After Local Therapy for Prostate Cancer (NCT01220973) [202] | Prostate cancer | Drug: Atorvastatin calcium Drug: Celecoxib Other: Laboratory Biomarker analysis | 27 | Rutgers, The State University of New Jersey |
| Trial Name (Identifier) | Phase | Patient Population (Condition) | Treatment Groups | Enrollment (Participants) | Sponsor |
|---|---|---|---|---|---|
| Study to Assess the Effect of AZD9291 on the Blood Levels of Simvastatin in Patients With EGFRm+ NSCLC (NCT02197234) [203] | 1 | Non-small-cell lung cancer | Procedure: Pharmacokinetic sampling—AZD9291. Drug: Simvastatin Drug: AZD9291 tablet dosing. | 52 | AstraZeneca |
| Simvastatin in Preventing a New Breast Cancer in Women at High Risk for a New Breast Cancer (NCT00334542) [204] | 2 | Breast cancer | Drug: Simvastatin | 50 | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins |
| Pre-Operative Statin Therapy Versus Placebo in Human Prostate Cancer (NCT00572468) [205] | N/A | Prostate cancer | Drug: 40 mg of Simvastatin Other: Placebo | 42 | VA Office of Research and Development |
| Detection and Prevention of Anthracycline-Related Cardiac Toxicity With Concurrent Simvastatin (NCT02096588) [206] | 2 | Breast cancer Stages I, II, and III of breast cancer | Drug: Simvastatin Drug: Doxorubicin/ cyclophosphamide | 34 | Avon Foundation |
| Trial Name (Identifier) | Phase | Patient Population (Condition) | Treatment Groups | Enrollment (Participants) | Sponsor |
|---|---|---|---|---|---|
| Study of Effectiveness of Lovastatin to Prevent Radiation-Induced Rectal Injury (NCT00580970) [207] | 2 | Prostate cancer | Drug: Lovastatin | 73 | Virginia Commonwealth University |
| Phase 2 Study of Lovastatin as Breast Cancer Chemoprevention (NCT00285857) [208] | 2 | Breast cancer | Drug: Lovastatin | 30 | Stanford University |
| Study to Assess the Effect of AZD9291 on the Blood Levels of Rosuvastatin, in Patients with EGFRm+ Non-small Cell Lung Cancer (NCT02317016) [209] | 1 | Non-small-cell lung cancer | Procedure: Pharmacokinetic sampling—AZD9291 Drug: AZD9291 tablet dosing Drug: Rosuvastatin | 44 | AstraZeneca |
| Rosuvastatin to Lower Circulating Tissue Factor Bearing Microparticles in Metastatic Breast Cancer (NCT01299038) [210] | 2 | Breast cancer | Drug: Rosuvastatin | 20 | Beth Israel Deaconess Medical Center |
| Idarubicin, Cytarabine and Pravastatin (IAP) for Induction of Newly Diagnosed Acute Myeloid Leukemia (NCT01831232) [211] | N/A | Acute myeloid leukemia | Drug: Pravastatin sodium Drug: Idarubicin Drug: Cytarabine Other: Laboratory biomarker analysis | 24 | Fred Hutchinson Cancer Research Center |
7. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roy, P.S.; Saikia, B. Cancer and cure: A critycal analysis. Indian J. Cancer 2016, 53, 441–442. [Google Scholar] [PubMed]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Bray, F. Observatorio Global del Cáncer: Cancer Today; Agencia Internacional de Investigación Sobre el Cáncer: Lyon, France, 2021; Available online: https://gco.iarc.fr/today/home (accessed on 10 December 2021).
- Barbalata, C.I.; Tefas, L.R.; Achim, M.; Tomuta, I.; Porfire, A.S. Statins in risk-reduction and treatment of cancer. World J. Clin. Oncol. 2020, 11, 573–588. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cancer. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 10 December 2021).
- Ho, C.J.; Gorski, S.M. Molecular mechanisms underlying autophagy-mediated treatment resistance in cancer. Cancers 2019, 11, 1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polivka, J.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef]
- Dey, N.; De, P.; Leyland-Jones, B. Pi3k-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, L.; Constantin, S.; Constatin, S.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef] [PubMed]
- Afify, S.M.; Oo, A.K.K.; Hassan, G.; Seno, A.; Seno, M. How can we turn the PI3K/AKT/mTOR pathway down? Insights into inhibition and treatment of cáncer. Expert Rev. Anticancer Ther. 2021, 21, 605–619. [Google Scholar] [CrossRef]
- Xu, J.L.; Yuan, L.; Tang, Y.C.; Xu, Z.Y.; Xu, H.D.; Cheng, X.D.; Qin, J.J. The Role of Autophagy in Gastric Cancer Chemoresistance: Friend or Foe? Front. Cell Dev. Biol. 2020, 8, 621428. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cáncer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef]
- Botti, J.; Djavaheri-Mergny, M.; Pilatte, Y.; Codogno, P. Autophagy signaling and the cogwheels of cancer. Autophagy 2006, 2, 67–73. [Google Scholar] [CrossRef]
- Chen, N.; Debnath, J. Autophagy and Tumorigenesis. FEBS Lett. 2010, 584, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Z.; Xie, L.; Xu, L.; Xu, D.; Liu, X. Statins, autophagy and cancer metastasis. Int. J. Biochem. Cell Biol. 2013, 45, 745–752. [Google Scholar] [CrossRef]
- Hassanabad, A.F. Current perspectives on statins as potential anti-cancer therapeutics: Clinical outcomes and underlying molecular mechanisms. Transl. Lung Cancer Res. 2019, 8, 692–699. [Google Scholar] [CrossRef]
- Beckwitt, C.H.; Clark, A.M.; Ma, B.; Whaley, D.; Oltvai, Z.N.; Wells, A. Statins attenuate outgrowth of breast cancer metastases. Br. J. Cancer 2018, 119, 1094–1105. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.M.; Fang, Z.; Sun, W.; Clark, L.H.; Stine, J.E.; Tran, A.Q.; Bae-Jump, V.L. Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am. J. Cancer Res. 2017, 7, 2478–2490. [Google Scholar]
- Chou, C.W.; Lin, C.H.; Hsiao, T.H.; Lo, C.C.; Hsieh, C.Y.; Huang, C.C.; Sher, Y.-P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Liu, L.; Zhao, Z.; Yin, L.; Bauer, N.; Nwaeburu, C.C.; Gladkich, J.; Gross, W.; Hackert, T.; Sticht, C.; et al. Simvastatin inhibits sonic hedgehog signaling and stemness features of pancreatic cancer. Cancer Lett. 2018, 426, 14–24. [Google Scholar] [CrossRef]
- Alipour Talesh, G.; Trezeguet, V.; Merched, A. Hepatocellular Carcinoma and Statins. Biochemistry 2020, 59, 3393–3400. [Google Scholar] [CrossRef]
- Oliveira, K.A.; Dal-Cim, T.; Lopes, F.G.; Ludka, F.K.; Nedel, C.B.; Tasca, C.I. Atorvastatin Promotes Cytotoxicity and Reduces Migration and Proliferation of Human A172 Glioma Cells. Mol. Neurobiol. 2018, 55, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Song, Y.; Zhang, J.; Li, R.; Wang, Z.; Zhu, X. Atorvastatin suppresses the progression of cervical cancer via regulation of autophagy. Am. J. Transl. Res. 2020, 12, 5252–5268. [Google Scholar]
- Hu, M.-B.; Zhang, J.-W.; Gao, J.-B.; Qi, Y.-W.; Gao, Y.; Xu, L.; Ma, Y.; Wei, Z.-Z. Atorvastatin induces autophagy in MDA-MB-231 breast cancer cells. Ultrastruct. Pathol. 2018, 42, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Maeda, M.; Motojima, K. Hydrophobic statins induce autophagy and cell death in human rhabdomyosarcoma cells by depleting geranylgeranyl diphosphate. Eur. J. Pharmacol. 2012, 674, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Vilimanovich, U.; Bosnjak, M.; Bogdanovic, A.; Markovic, I.; Isakovic, A.; Kravic-Stevovic, T.; Mircic, A.; Trajkovic, V.; Bumbasirevic, V. Statin-mediated inhibition of cholesterol synthesis induces cytoprotective autophagy in human leukemic cells. Eur. J. Pharmacol. 2015, 765, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Garner, H.; de Visser, K.E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 2020, 20, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Lewandowska, A.M.; Rudzki, M.; Rudzki, S.; Lewandowski, T.; Laskowska, B. Environmental risk factors for cancer—review paper. Ann. Agric. Environ. Med. 2019, 26, 1–7. [Google Scholar] [CrossRef]
- Devouassoux-Shisheboran, M.; Genestie, C.; Ray-Coquard, I. Dualistic classification of epithelial ovarian cancer: Is it clinically relevant? Bull. Cancer 2016, 103, 252–258. [Google Scholar] [CrossRef]
- Katz, D.; Palmerini, E.; Pollack, S.M. More Than 50 Subtypes of Soft Tissue Sarcoma: Paving the Path for Histology-Driven Treatments. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 925–938. [Google Scholar] [CrossRef]
- Juliusson, G.; Hough, R. Leukemia. Prog. Tumor Res. 2016, 43, 87–100. [Google Scholar]
- Mugnaini, E.N.; Ghosh, N. Lymphoma. Prim. Care—Clin. Off. Pract. 2016, 43, 661–675. [Google Scholar] [CrossRef]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef]
- Panda, M.; Biswal, B.K. Cell signaling and cancer: A mechanistic insight into drug resistance. Mol. Biol. Rep. 2019, 46, 5645–5659. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- da Silva Santos, E.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.D.B.V.; Eloy, J.O. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int. J. Pharm. 2021, 592, 120082. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 1–19. [Google Scholar] [CrossRef]
- Xiu, M.X.; Liu, Y.M.; Kuang, B.H. The oncogenic role of Jagged1/Notch signaling in cancer. Biomed. Pharmacother. 2020, 129, 110416. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Asghariazar, V.; Sakhinia, E.; Mansoori, B.; Mohammadi, A.; Baradaran, B. Tumor suppressor microRNAs in lung cancer: An insight to signaling pathways and drug resistance. J. Cell. Biochem. 2019, 120, 19274–19289. [Google Scholar] [CrossRef]
- Cao, X.; Hou, J.; An, Q.; Assaraf, Y.G.; Wang, X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist. Updates 2020, 49, 100671. [Google Scholar] [CrossRef] [PubMed]
- NaghiZadeh, S.; Mohammadi, A.; Duijf, P.H.G.; Baradaran, B.; Safarzadeh, E.; Cho, W.C.-S.; Mansoori, B. The role of miR-34 in cancer drug resistance. J. Cell. Physiol. 2020, 235, 6424–6440. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Deng, R.; Cai, R.; Lu, X.; Luo, Y.; Wang, Z.; Zhu, Y.; Yin, M.; Ding, Y.; Lin, J. TUSC3 induces drug resistance and cellular stemness via Hedgehog signaling pathway in colorectal cancer. Carcinogenesis 2020, 41, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Hawsawi, Y.M.; Al-Numair, N.S.; Sobahy, T.M.; Al-Ajmi, A.M.; Al-Harbi, R.M.; Baghdadi, M.A.; Alamer, O.M. The role of BRCA1/2 in hereditary and familial breast and ovarian cancers. Mol. Genet. Genom. Med. 2019, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Lundqvist, E.Å.; Fujiwara, K.; Seoud, M. Principles of chemotherapy. Int. J. Gynaecol. Obstet. 2015, 131, S146–S149. [Google Scholar] [CrossRef] [Green Version]
- Fairchild, A.; Tirumani, S.H.; Rosenthal, M.H.; Howard, S.A.; Krajewski, K.M.; Nishino, M.; Shinagare, A.B.; Jagannathan, J.P.; Ramaiya, N.H. Hormonal therapy in oncology: A primer for the radiologist. Am. J. Roentgenol. 2015, 204, W620–W630. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Tooze, S.A.; Dikic, I. Autophagy Captures the Nobel Prize. Cell 2016, 167, 1433–1435. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M.K. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83. [Google Scholar] [PubMed]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2019, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S. Microautophagy—Distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef]
- Kim, S.; Choi, S.; Kang, D. Quantitative and qualitative analysis of autophagy flux using imaging. BMB Rep. 2020, 53, 241–247. [Google Scholar] [CrossRef]
- Tamargo-Gómez, I.; Fernández, Á.F.; Mariño, G. Pathogenic single nucleotide polymorphisms on autophagy-related genes. Int. J. Mol. Sci. 2020, 21, 8196. [Google Scholar] [CrossRef]
- Wirawan, E.; Vanden Berghe, T.; Lippens, S.; Agostinis, P.; Vandenabeele, P. Autophagy: For better or for worse. Cell Res. 2012, 22, 43–61. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Ramos Batara, D.C.; Choi, M.-C.; Shin, H.-U.; Kim, H.; Kim, S.-H. Friend or Foe: Paradoxical Roles of Autophagy in Gliomagenesis. Cells 2021, 10, 1411. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. MTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [PubMed] [Green Version]
- Li, Y.; Chen, Y. AMPK and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar] [PubMed]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective. Oxid. Med. Cell Longev. 2019, 2019, 8730816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Bari, M.A.A.; Xu, P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 2020, 1467, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.-K.; Song, J.; Lu, J.; et al. Balancing mTOR signaling and autophagy in the treatment of Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy Akpedje. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhou, X. Research progress of mTOR inhibitors. Eur. J. Med. Chem. 2020, 208, 112820. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Kang, J.H.; Lee, S. Autophagy in neurodegenerative diseases: A hunter for aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Cretoiu, D.; Li, G.; Xiao, J. Exercise-mediated regulation of autophagy in the cardiovascular system. J. Sport Health Sci. 2020, 9, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.; Ljubojevic-Holzer, S.; Madeo, F.; Sedej, S. Autophagy in cardiovascular health and disease. Prog. Mol. Biol. Transl. Sci. 2020, 172, 87–106. [Google Scholar] [PubMed]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020, 36, 101679. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Lapaquette, P.; Nguyen, H.T.T.; Faure, M. L’autophagie garante de l’immunité et de l’inflammation. Med. Sci. 2017, 33, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.C.; Livingston, M.J.; Liang, X.L.; Dong, Z. Cell Apoptosis and Autophagy in Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 557–584. [Google Scholar]
- Wong, S.Q.; Kumar, A.; Mills, J.; Lapierre, L.R. Autophagy in aging and longevity. Hum. Genet. 2020, 139, 277–290. [Google Scholar] [CrossRef]
- Tao, T.; Xu, H. Autophagy and Obesity and Diabetes. Adv. Exp. Med. Biol. 2019, 1206, 445–461. [Google Scholar]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14, 1297. [Google Scholar] [CrossRef]
- Mani, S.; Swargiary, G.; Chadha, R. Mitophagy impairment in neurodegenerative diseases: Pathogenesis and therapeutic interventions. Mitochondrion 2021, 57, 270–293. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Murthy, A. Targeting Autophagy to Treat Cancer: Challenges and Opportunities. Front. Pharmacol. 2020, 11, 590344. [Google Scholar] [CrossRef]
- Jogalekar, M.P.; Veerabathini, A.; Gangadaran, P. Recent developments in autophagy-targeted therapies in cancer. Exp. Biol. Med. 2021, 246, 207–212. [Google Scholar] [CrossRef]
- Zappavigna, S.; Luce, A.; Vitale, G.; Merola, N.; Facchini, S.; Caraglia, M. Autophagic cell death: A new frontier in cancer research. Adv. Biosci. Biotechnol. 2013, 04, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Smaili, S.S. Autophagy and intermittent fasting: The connection for cancer therapy? Clinics 2018, 73, e814s. [Google Scholar] [CrossRef]
- Kung, C.P.; Budina, A.; Balaburski, G.; Bergenstock, M.K.; Murphy, M.E. Autophagy in tumor suppression and cancer therapy. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 71–100. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Maheswari, U.; Sadras, S.R. Mechanism and regulation of autophagy in cancer. Crit. Rev. Oncog. 2018, 23, 269–280. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Ávalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F.G. Tumor Suppression and Promotion by Autophagy. BioMed Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin1. Sci. Exch. 1999, 402, 672–676. [Google Scholar]
- Qu, X.; Yu, J.; Bhagat, G.; Faruya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; Pledger, W.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef]
- An, C.H.; Kim, M.S.; Yoo, N.J.; Park, S.W.; Lee, S.H. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol. Res. Pract. 2011, 207, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Imamura, Y.; Jenkins, R.W.; Cañadas, I.; Aref, A.; Brannon, A.; Oki, E.; Castoreno, A.; Zhu, Z.; Barbie, D.A.; et al. Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol. Res. 2017, 4, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Jiang, L.; Fu, X.; Wang, W.; Ma, J.; Tian, T.; Nan, K. Cytoplasmic liver kinase B1 promotes the growth of human lung adenocarcinoma by enhancing autophagy. Cancer Sci. 2018, 109, 3055–3067. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.-Y. Autophagy and hallmarks of cancer. Crit. Rev. Oncog. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; White, E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy 2013, 9, 1636–1638. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Wang, B.R.; Wang, Y.G. Role of autophagy in tumorigenesis, metastasis, targeted therapy and drug resistance of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 4643–4651. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.; Wang, P.; Zhang, J.; Fu, L.; Ouyang, L.; Wang, J. Deconvoluting the relationships between autophagy and metastasis for potential cancer therapy. Apoptosis 2016, 21, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar]
- Hashimoto, I.; Koizumi, K.; Tatematsu, M.; Minami, T.; Cho, S.; Takeno, N.; Nakashima, A.; Sakurai, H.; Saito, S.; Tsukada, K.; et al. Blocking on the CXCR4/mTOR signalling pathway induces the anti-metastatic properties and autophagic cell death in peritoneal disseminated gastric cancer cells. Eur. J. Cancer 2008, 44, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Marsh, T.; Kenific, C.M.; Suresh, D.; Gonzalez, H.; Eliah, R.S.; Mei, W.; Tankka, A.; Leidal, A.M.; Kalavacherla, S.; Woo, K.; et al. Autophagic Degradation of NBR1 Restricts Metastatic Outgrowth During Mammary Tumor Progression. Dev. Cell 2020, 52, 591–604. [Google Scholar] [CrossRef]
- Marsh, T.; Debnath, J. Autophagy suppresses breast cancer metastasis by degrading NBR1. Autophagy 2020, 16, 1164–1165. [Google Scholar] [CrossRef]
- Kim, Y.N.; Koo, K.H.; Sung, J.Y.; Yun, U.J.; Kim, H. Anoikis resistance: An essential prerequisite for tumor metastasis. Int. J. Cell Biol. 2012, 2012, 306879. [Google Scholar] [CrossRef] [Green Version]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Wittmann, T.; Debnath, J. Autophagy in adhesion and migration. J. Cell Sci. 2016, 129, 3685–3693. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-F.; Shi, Y.-H.; Ding, Z.-B.; Ke, A.-W.; Gu, C.-Y.; Hui, B.; Zhou, J.; Qiu, S.-J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, A.; Hait, N.C. Autophagy-mediated tumor cell survival and progression of breast cancer metastasis to the brain. J. Cancer 2021, 12, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.Y.; Zhang, X.Y.; Wang, X.F.; Sun, B.C. Autophagy enhances the aggressiveness of human colorectal cancer cells and their ability to adapt to apoptotic stimulus. Cancer Biol. Med. 2012, 9, 105–110. [Google Scholar] [PubMed]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Ding, X.F.; Bouamar, H.; Pressley, K.; Sun, L.Z. Everolimus induces G1 cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells. Am. J. Physiol.—Cell Physiol. 2019, 317, C244–C252. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef]
- Dong, Y.; Wu, Y.; Zhao, G.; Ye, Z.; Xing, C.; Yang, X. Inhibition of autophagy by 3-MA promotes hypoxia-induced apoptosis in human colorectal cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1047–1054. [Google Scholar]
- Schott, C.R.; Ludwig, L.; Mutsaers, A.J.; Foster, R.A.; Wood, G.A. The autophagy inhibitor spautin-1, either alone or combined with doxorubicin, decreases cell survival and colony formation in canine appendicular osteosarcoma cells. PLoS ONE 2018, 13, e0206427. [Google Scholar] [CrossRef]
- Buzun, K.; Gornowicz, A.; Lesyk, R.; Bielawski, K.; Bielawska, A. Autophagy Modulators in Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 5804. [Google Scholar] [CrossRef]
- Wang, H.; Li, D.; Li, X.; Ou, X.; Liu, S.; Zhang, Y.; Ding, J.; Xie, B. Mammalian target of rapamycin inhibitor RAD001 sensitizes endometrial cancer cells to paclitaxel-induced apoptosis via the induction of autophagy. Oncol. Lett. 2016, 12, 5029–5035. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.E.G.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schettini, F.; Sobhani, N.; Ianza, A.; Triulzi, T.; Molteni, A.; Lazzari, M.C.; Strina, C.; Milani, M.; Corona, S.P.; Sirico, M.; et al. Immune system and angiogenesis-related potential surrogate biomarkers of response to everolimus-based treatment in hormone receptor-positive breast cancer: An exploratory study. Breast Cancer Res. Treat. 2020, 184, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Ciruelos, E.; Jerusalem, G.; Martin, M.; Tjan-Heijnen, V.C.G.; Neven, P.; Gavila, J.; Montemurro, F.; Generali, D.; Lang, I.; Martínez-Serrano, M.J.; et al. Everolimus plus exemestane in hormone-receptor-positive, HER2-negative locally advanced or metastatic breast cancer: Incidence and time course of adverse events in the phase IIIb BALLET population. Clin. Transl. Oncol. 2020, 22, 1857–1866. [Google Scholar] [CrossRef]
- Tannir, N.M.; Msaouel, P.; Ross, J.A.; Devine, C.E.; Chandramohan, A.; Gonzalez, G.M.N.; Wang, X.; Wang, J.; Corn, P.G.; Lim, Z.D.; et al. Temsirolimus versus Pazopanib (TemPa) in Patients with Advanced Clear-cell Renal Cell Carcinoma and Poor-risk Features: A Randomized Phase II Trial. Eur. Urol. Oncol. 2019, 3, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Haas, N.B.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J.; et al. Autophagy Inhibition to Augment mTOR Inhibition: A Phase I/II Trial of Everolimus and Hydroxychloroquine in Patients with Previously Treated Renal Cell Carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.; Jabbour, S.; Orlick, M.; Riedlinger, G.; Guo, Y.; White, E.; Aisner, J. Phase Ib/II study of hydroxychloroquine in combination with chemotherapy in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Treat. Res. Commun. 2019, 21, 100158. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Lin, J.-F.; Wen, S.-I.; Yang, S.-C.; Tsai, T.-F.; Chen, H.-E.; Chou, K.-Y.; Hwang, T.I.-S. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J. Med Sci. 2017, 33, 215–223. [Google Scholar] [CrossRef]
- Bigelsen, S. Evidence-based complementary treatment of pancreatic cancer: A review of adjunct therapies including paricalcitol, hydroxychloroquine, intravenous vitamin C, statins, metformin, curcumin, and aspirin. Cancer Manag. Res. 2018, 10, 2003–2018. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cao, F.; Li, M.; Li, P.; Yu, Y.; Xiang, L.; Xu, T.; Lei, J.; Tai, Y.Y.; Zhu, J.; et al. Hydroxychloroquine induced lung cancer suppression by enhancing chemo-sensitization and promoting the transition of M2-TAMs to M1-like macrophages. J. Exp. Clin. Cancer Res. 2018, 37, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, S.; Xie, R.; Qin, L.; Yu, M.; Xiao, W.; Hu, C.; Yu, W.; Qian, Z.; Ouyang, L.; He, Q.; et al. Aggregable Nanoparticles-Enabled Chemotherapy and Autophagy Inhibition Combined with Anti-PD-L1 Antibody for Improved Glioma Treatment. Nano Lett. 2019, 19, 8318–8332. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Tang, J.; Liang, Y.; Jin, R.; Cai, X. Suppression of autophagy by chloroquine sensitizes 5-fluorouracil-mediated cell death in gallbladder carcinoma cells. Cell Biosci. 2014, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.G.; Sun, R.J.; Yang, X.Y.; Liu, D.Y.; Lei, D.P.; Jin, T.; Pan, X.-L. Chloroquine-enhanced efficacy of cisplatin in the treatment of hypopharyngeal carcinoma in xenograft mice. PLoS ONE 2015, 10, e0126147. [Google Scholar] [CrossRef]
- Chen, Y.S.; Song, H.X.; Lu, Y.; Li, X.; Chen, T.; Zhang, Y.; Xue, J.X.; Liu, H.; Kan, B.; Yang, G.; et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis. Esophagus 2010, 24, 437–443. [Google Scholar] [CrossRef]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of p53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Guo, Z.; Xia, X.; Liu, Y.; Huang, C.; Jiang, L.; Wang, X.; Liu, J.; Huang, H. Inhibition of EGFR signaling with Spautin-1 represents a novel therapeutics for prostate cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Xiao, J.; Feng, X.; Huang, X.-Y.; Huang, Z.; Huang, Y.; Li, C.; Li, G.; Nong, S.; Wu, R.; Huang, Y.; et al. Spautin-1 Ameliorates Acute Pancreatitis via Inhibiting Impaired Autophagy and Alleviating Calcium Overload. Mol. Med. 2016, 22, 643–652. [Google Scholar] [CrossRef]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [Green Version]
- Semenkovich Clay, F.; Goldberg Ira, J. 41—Trastornos del metabolismo de los lípidos, Williams. In Tratado de Endocrinología, 14th ed.; Melmed, S., Auchus Richard, J., Goldfine Allison, B., Koenig Ronald, J., Rosen Clifford, J., Eds.; Elsevier: Amsterdam, Neither Land, 2021; pp. 1581–1619. ISBN 978-84-9113-851-8. [Google Scholar] [CrossRef]
- Robinson Jennifer, G. 195—Trastornos del metabolismo de los lípidos, Goldman-Cecil. In Tratado de Medicina Interna, 26th ed.; Goldman, L., Schafer Andrew, I., Eds.; Elsevier: Amsterdam, Neitherland, 2021; pp. 1357–1358. ISBN 978-84-9113-765-8. [Google Scholar] [CrossRef]
- Pedro-Botet Montoya, J.; Masana Marín, L.; Carmena Rodríguez, R. 227—Alteraciones del metabolismo de las lipoproteínas, Farreras Rozman. In Medicina Interna, 19th ed.; von Domarus, A., Farreras, P., Rozman, C., Cardellach, F., Nicolás, J.M., Cervera, R., Agust, A., Brugada, J., Campistol, J.M., et al., Eds.; Elsevier: Amsterdam, Netherlands, 2020; pp. 1813–1837. ISBN 978-84-9113-545-6. [Google Scholar] [CrossRef]
- Fleisher Lee, A.; Beckman Joshua, A. 11—Anestesia y cirugía no cardíaca en pacientes con enfermedades cardíacas, Braunwald. In Tratado de Cardiología, 11th ed.; Zipes Douglas, P., Libby, P., Bonow Robert, O., Mann Douglas, L., Tomaselli Gordon, F., Braunwald, E., Eds.; Elsevier: Amsterdam, Netherlands, 2019; pp. 102–116. ISBN 978-84-9113-398-8. [Google Scholar] [CrossRef]
- Genest, J.; Libby, P. 48—Trastornos de las lipoproteínas y enfermedad cardiovascular, Braunwald. In Tratado de Cardiología, 11th ed.; Zipes Douglas, P., Libby, P., Bonow Robert, O., Mann Douglas, L., Tomaselli Gordon, F., Braunwald, E., Eds.; Elsevier: Amsterdam, Netherlands, 2019; pp. 960–982. ISBN 978-84-9113-398-8. [Google Scholar]
- Reid Michael, A.; Sanderson Sydney, M.; Locasale Jason, W. 9—Metabolismo del cáncer, Abeloff. In Oncología Clínica, 6th ed.; Niederhuber, J.E., Armitage, J.O., Kastan, M.B., Doroshow, J.H., Tepper, J.E., Eds.; Elsevier: Amsterdam, Netherlands, 2020; pp. 127–138. ISBN 978-84-9113-520-3. Available online: https://www.clinicalkey.es/#!/content/3-s2.0-B9788491135203000096 (accessed on 10 December 2021). [CrossRef]
- Liao, Y.; Zhang, P.; Yuan, B.; Li, L.; Bao, S. Pravastatin protects against avascular necrosis of femoral head via autophagy. Front. Physiol. 2018, 9, 307. [Google Scholar] [CrossRef]
- Wei, Y.-M.; Li, X.; Xu, M.; Abais, J.M.; Chen, Y.; Riebling, C.R.; Boini, K.M.; Li, P.-L.; Zhang, Y. Enhancement of Autophagy by Simvastatin through Inhibition of Rac1-mTOR Signaling Pathway in Coronary Arterial Myocytes. Cell. Physiol. Biochem. 2013, 31, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Mutawe, M.M.; Sharma, P.; Yeganeh, B.; McNeill, K.D.; Klonisch, T.; Unruh, H.; Kashani, H.; Schaafsma, D.; Los, M.; et al. Mevalonate Cascade Regulation of Airway Mesenchymal Cell Autophagy and Apoptosis: A Dual Role for p53. PLoS ONE 2011, 6, e16523. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Björklund, M. The mevalonate pathway as a metabolic requirement for autophagy–implications for growth control, proteostasis, and disease. Mol. Cell Oncol. 2016, 3, e1143546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Yang, Y.-J.; Wang, H.; Dong, Q.-T.; Wang, T.-J.; Qian, H.-Y.; Xu, H. Autophagy Activation: A Novel Mechanism of Atorvastatin to Protect Mesenchymal Stem Cells from Hypoxia and Serum Deprivation via AMP-Activated Protein Kinase/Mammalian Target of Rapamycin Pathway. Stem Cells Dev. 2012, 21, 1321–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.-M.; Chen, C.-C. Life or death? Autophagy in anticancer therapies with statins and histone deacetylase inhibitors. Autophagy 2011, 7, 107–108. [Google Scholar] [CrossRef] [Green Version]
- Tricarico, P.M.; Crovella, S.; Celsi, F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. Int. J. Mol. Sci. 2015, 16, 16067–16084. [Google Scholar] [CrossRef] [Green Version]
- Longatti, A.; Tooze, S. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009, 16, 956–965. [Google Scholar] [CrossRef]
- Chua, C.E.L.; Gan, B.Q.; Tang, B.L. Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol. Life Sci. 2011, 68, 3349–3358. [Google Scholar] [CrossRef]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Popovic, D.; Akutsu, M.; Novak, I.; Harper, J.W.; Behrends, C.; Dikic, I. Rab GTPase-Activating Proteins in Autophagy: Regulation of Endocytic and Autophagy Pathways by Direct Binding to Human ATG8 Modifiers. Mol. Cell. Biol. 2012, 32, 1733–1744. [Google Scholar] [CrossRef] [Green Version]
- Bodemann, B.O.; Orvedahl, A.; Cheng, T.; Ram, R.R.; Ou, Y.H.; Formstecher, E.; Maiti, M.; .Hazelett, C.C.; Wauson, E.M.; Balakireva, M.; et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 2011, 144, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.-M.; Gao, S.; Zhang, Z.-M.; Shen, Z.-L.; Gao, K.; Chang, L.; Guo, Y.; Li, Z.; Wang, W. Atorvastatin activates autophagy and promotes neurological function recovery after spinal cord injury. Neural Regen. Res. 2016, 11, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Lee, S.-B.; Kim, H.J.; Yang, H.O.; Jang, W. Autophagic modulation by rosuvastatin prevents rotenone-induced neurotoxicity in an in vitro model of Parkinson’s disease. Neurosci. Lett. 2017, 642, 20–26. [Google Scholar] [CrossRef]
- Vosper, J.; Masuccio, A.; Kullmann, M.; Ploner, C.; Geley, S.; Hengst, L. Statin-induced depletion of geranylgeranyl pyrophosphate inhibits cell proliferation by a novel pathway of Skp2 degradation. Oncotarget 2014, 6, 2889–2902. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.T.; Ho, H.J.; Lin, J.T.; Shieh, J.J.; Wu, C.Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e2626. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Su, Z.; DeWitt, J.P.; Xie, L.; Chen, Y.; Li, X.; Han, L.; Li, D.; Xia, J.; Zhang, Y.; et al. Fluvastatin Prevents Lung Adenocarcinoma Bone Metastasis by Triggering Autophagy. EBioMedicine 2017, 19, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Yeganeh, B.; Stelmack, G.L.; Kashani, H.H.; Sharma, P.; Cunnington, R.; Freed, D.H. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012, 3, e330. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.S. Simvastatin Induces Mitochondrial Loss and Pten-Mediated Autophagy; San Diego State University: San Diego, CA, USA, 2011; Volume 1, Available online: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.991.4451&rep=rep1&type=pdf (accessed on 10 December 2021).
- Zhang, S.-X.; Qiu, L.; Xiao, C.-S.; Fang, Y.-Q. e0034 Inhibition of atorvastatin on the autophagy of vascular endothelial cells. Heart 2010, 96, A11–A12. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, T.P.; Björklund, M. Mevalonate Pathway Regulates Cell Size Homeostasis and Proteostasis through Autophagy. Cell Rep. 2015, 13, 2610–2620. [Google Scholar] [CrossRef] [Green Version]
- Szatmári, Z.; Kis, V.; Lippai, M.; Hegedus, K.; Faragó, T.; Lorincz, P.; Tanaka, T.; Juhász, G.; Sass, M. Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of Hook localization. Mol. Biol. Cell 2014, 25, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Modulatory effects of statins on the autophagy: A therapeutic perspective. J. Cell. Physiol. 2019, 235, 3157–3168. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.-M.; Liu, Y.-L.; Lin, Y.-C.; Shun, C.-T.; Wu, M.-S.; Chen, C.-C. Inhibition of Autophagy Enhances Anticancer Effects of Atorvastatin in Digestive Malignancies. Cancer Res. 2010, 70, 7699–7709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, A.; Childress, C.; Deitrick, K.; Lin, Q.; Rukstalis, D.; Yang, W. Statin-induced autophagy by inhibition of geranylgeranyl biosynthesis in prostate cancer PC3 cells. Prostate 2010, 70, 971–981. [Google Scholar] [CrossRef]
- Alarcon Martinez, T.; Zeybek, N.D.; Müftüoğlu, S. Evaluation of the Cytotoxic and Autophagic Effects of Atorvastatin on MCF-7 Breast Cancer Cells. Balkan Med. J. 2018, 35, 256–262. [Google Scholar] [CrossRef]
- Toepfer, N.; Childress, C.; Parikh, A.; Rukstalis, D.; Yang, W. Atorvastatin induces autophagy in prostate cancer PC3 cells through activation ofLC3transcription. Cancer Biol. Ther. 2011, 12, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Jeong, C.W.; Ku, J.H.; Kwak, C.; Kim, H.H. Inhibition of Autophagy Potentiates Atorvastatin-Induced Apoptotic Cell Death in Human Bladder Cancer Cells in Vitro. Int. J. Mol. Sci. 2014, 15, 8106–8121. [Google Scholar] [CrossRef] [Green Version]
- Elimam, H.; El-Say, K.M.; Cybulsky, A.V.; Khalil, H. Regulation of Autophagy Progress via Lysosomal Depletion by Fluvastatin Nanoparticle Treatment in Breast Cancer Cells. ACS Omega 2020, 5, 15476–15486. [Google Scholar] [CrossRef]
- Qi, X.-F.; Kim, D.-H.; Lee, K.-J.; Kim, C.-S.; Song, S.-B.; Cai, D.-Q.; Kim, S.-K. Autophagy contributes to apoptosis in A20 and EL4 lymphoma cells treated with fluvastatin. Cancer Cell Int. 2013, 13, 111. [Google Scholar] [CrossRef] [Green Version]
- Asakura, K.; Izumi, Y.; Yamamoto, M.; Yamauchi, Y.; Kawai, K.; Serizawa, A.; Mizushima, T.; Ohmura, M.; Kawamura, M.; Wakui, M.; et al. The Cytostatic Effects of Lovastatin on ACC-MESO-1 Cells. J. Surg. Res. 2011, 170, e197–e209. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Sane, K.M.; Kleinman, M.; Sloane, B.F.; Reiners, J.J.; Mattingly, R.R. Aborted Autophagy and Nonapoptotic Death Induced by Farnesyl Transferase Inhibitor and Lovastatin. J. Pharmacol. Exp. Ther. 2011, 337, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Felley-Bosco, E.; Marti, T.M.; Stahel, R.A. Differential effects of lovastatin on cisplatin responses in normal human mesothelial cells versus cancer cells: Implication for therapy. PLoS ONE 2012, 7, e45354. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; Pompili, C.; Gilardini Montani, M.S.; Romeo, M.A.; Gonnella, R.; D’Orazi, G.; Cirone, M. Lovastatin reduces PEL cell survival by phosphorylating ERK1/2 that blocks the autophagic flux and engages a cross-talk with p53 to activate p21. IUBMB Life 2021, 73, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, Y.; Yang, S.; Yen, H.; Tsai, H.; Hsieh, M.; Hsiao, Y. Pitavastatin and metformin synergistically activate apoptosis and autophagy in pancreatic cancer cells. Environ. Toxicol. 2021, 36, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Al-Qatati, A.; Aliwaini, S. Combined pitavastatin and dacarbazine treatment activates apoptosis and autophagy resulting in synergistic cytotoxicity in melanoma cells. Oncol. Lett. 2017, 14, 7993–7999. [Google Scholar] [CrossRef] [Green Version]
- Zeybek, N.D.; Gulcelik, N.E.; Kaymaz, F.F.; Sarisozen, C.; Vural, I.; Bodur, E.; Canpinar, H.; Usman, A.; Asan, E. Rosuvastatin induces apoptosis in cultured human papillary thyroid cancer cells. J. Endocrinol. 2011, 210, 105–115. [Google Scholar] [CrossRef]
- Castellanos-Esparza, Y.C.; Wu, S.; Huang, L.; Buquet, C.; Shen, R.; Sanchez-Gonzalez, B.; Latorre, E.A.G.; Boyer, O.; Varin, R.; Jim�Nez-Zamudio, L.; et al. Synergistic promoting effects of pentoxifylline and simvastatin on the apoptosis of triple-negative MDA-MB-231 breast cancer cells. Int. J. Oncol. 2018, 52, 1246–1254. [Google Scholar] [CrossRef] [Green Version]
- Misirkic, M.; Janjetovic, K.; Vucicevic, L.; Tovilovic, G.; Ristic, B.; Vilimanovich, U.; Harhaji-Trajkovic, L.; Sumarac-Dumanovic, M.; Micic, D.; Bumbasirevic, V.; et al. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol. Res. 2012, 65, 111–119. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Donor Atorvastatin Treatment in Preventing Severe Acute GVHD after Nonmyeloablative Peripheral Blood Stem Cell Transplant in Patients with Hematological Malignancies. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01217 307?term=NCT01217307&draw=2&rank=1 (accessed on 10 April 2022).
- Safety & Efficacy of Atorvastatin for Prophylaxis of Acute Graft Versus Host Disease in Patients with Hematological Malignancies HLA- Donor Hematopoietic Stem Cell Transplantation. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01491958 (accessed on 10 April 2022).
- Statin Therapy in Young Adult Survivors of Childhood Cancer. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01733953 (accessed on 10 April 2022).
- Atorvastatin Calcium and Celecoxib in Treating Patients with Rising PSA Levels after Local Therapy for Prostate Cancer. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01220973 (accessed on 10 April 2022).
- Study to Assess the Effect of AZD9291 on the Blood Levels of Simvastatin in Patients with EGFRm+ NSCLC. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02197234 (accessed on 10 April 2022).
- Simvastatin in Preventing a New Breast Cancer in Women at High Risk for a New Breast Cancer. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00334542 (accessed on 10 April 2022).
- Pre-Operative Statin Therapy Versus Placebo in Human Prostate Cancer. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00572468 (accessed on 10 April 2022).
- Detection and Prevention of Anthracycline-Related Cardiac Toxicity with Concurrent Simvastatin. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02096588 (accessed on 10 April 2022).
- Study of Effectiveness of Lovastatin to Prevent Radiation-Induced Rectal Injury. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00580970 (accessed on 10 April 2022).
- Phase 2 Study of Lovastatin as Breast Cancer Chemoprevention. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00285857 (accessed on 10 April 2022).
- Study to Assess the Effect of AZD9291 on the Blood Levels of Rosuvastatin, in Patients With EGFRm+ Non-Small Cell Lung Cancer. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02317016 (accessed on 10 April 2022).
- Rosuvastatin to Lower Circulating Tissue Factor Bearing Microparticles in Metastatic Breast Cancer. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01299038 (accessed on 10 April 2022).
- Idarubicin, Cytarabine, and Pravastatin Sodium in Treating Patients with Acute Myeloid Leukemia or Myelodysplastic Syndromes. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01831232 (accessed on 10 April 2022).
- Combination Statin, Acetylsalicylic Acid and Dutasteride Use in Prostate Cancer. ClinicalTrials.gov. US National Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01428869 (accessed on 10 April 2022).
- Mira, E.; Manes, S. Immunomodulatory and anti-inflammatory activities of statins. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 237–247. [Google Scholar] [CrossRef]
- Tsakiri, A.; Tsiantoulas, D.; Frederiksen, J.; Svane, I.M. Increased immunopotency of monocyte derived dendritic cells from patients with optic neuritis is inhibited in vitro by simvastatin. Exp. Neurol. 2010, 221, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, C.C.; Zhang, M.; Li, X.L.; Zhang, P.; Yue, L.T.; Miao, S.; Wang, S.; Liu, Y.; Li, Y.B.; et al. Statin-modified dendritic cells regulate humoral immunity in experimental autoimmune myasthenia gravis. Mol. Cell. Neurosci. 2015, 68, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]



| Statin | Cancer Type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Atorvastatin | Breast cancer | MDA-MB-231 Cells | - | 0,5, 1, 2, 4, 8 µM | Reduced the viability of cancer cells by inducing autophagy [25]. |
| Atorvastatin | Breast cancer | MCF-7 | - | 5, 10, 20, 40 y 80 μM | Decreased the proliferation of breast cancer cells through the induction of both apoptosis and autophagy [184]. |
| Atorvastatin | Ovarian cancer | Hey and SKOV3 cells | - | 1–250 μM | Inhibited the growth of ovarian cancer cell lines associated with the induction of apoptosis, autophagy, cellular stress, and G1 cell-cycle arrest [19]. |
| Atorvastatin | Cervical Cancer | SiHa and Caski Cells | Female BALB/c nude mice | 0, 5, 10, y 20, 40, 80 μM (in vitro) 50 mg/kg (in vivo) | Reduced the viability of cervical cancer cells in vitro and in vivo by inducing apoptosis. ATO induced autophagy, and its inhibition was shown to enhance the anti-cancer effects of ATO on cervical cancer cells [24]. |
| Atorvastatin | Digestive malignancies | HCC cells (Hep3B, HepG2 and Huh7) CRC cells (HCT116 wt, HCT116 p21) | Female BALB nude mice | 50 μM (in vitro) 50 mg/kg (in vivo) | Inhibited cancer cell growth in vivo and in vitro by inducing apoptosis. ATO induced autophagy, and the pharmacological inhibition of autophagy was shown to enhance the anticancer effects of ATO in gastrointestinal malignancies [182]. |
| Atorvastatin | Bladder Cancer | T24 and J28 Cells | - | 0, 10, 20, 30, 40 y 50 μM | Enhanced ATP-induced apoptotic cell death in human bladder cancer cells in vitro through the pharmacological inhibition of autophagy [186]. |
| Statin | Cancer Type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Fluvastatin | Breast cancer | MCF-7 | - | 10 μM | Reduced cell viability through the depletion of lysosomal activities coupled with the accumulation of autophagosomes, leading to impaired autophagosome–lysosomal fusion in treated cells [187]. |
| Fluvastatin | Lung adenocarcinoma | A549 and SPC-A-1 cells | Female nude mice BALB/c | 10 μM (in vitro) 50 mg/kg (in vivo) | Suppressed bone metastasis from lung adenocarcinoma in vivo and in vitro by triggering autophagy through the p53–AMPK-mTOR pathway [175]. |
| Fluvastatin | Lymphoma | A20 and EL4 cells | - | 0–10 μM | Induced apoptosis in lymphoma cells by activating autophagy through increased LC3-II [188]. |
| Statin | Cancer Type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Lovastatin | Malignant pleural mesothelioma | ACC-MESO-1 Cells | Mice NOD/SCID/γnull (NOG) | 10 μM (in vitro) 12.5 mg/kg (in vivo) | Decreased viability and migration capacity of malignant pleural mesothelioma tumor cells by stimulating autophagy [189]. |
| Lovastatin | Malignant peripheral nerve sheath tumor | NF90-8 and ST88-14 Cells | - | 500 nM | Suppressed viability of cancer cells by inducing non-apoptotic cell death and altering autophagy flux [190]. |
| Lovastatin | Human mesothelioma | Cancer cells ZL55 | - | 2, 8 µM | Reduced the viability of tumor cells by inducing autophagy [191]. |
| Lovastatin | Primary effusion lymphoma (PEL) | BC3 and BCBL1 cells | - | 3, 10, 30 µM | Reduced the survival of PEL cells by triggering apoptotic cell death through the inhibition of autophagic flux [192]. |
| Statin | Cancer Type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Pitavastatin | Pancreatic cancer | ASPC-1 and PANC-1 cells | - | 10 µM | Decreased cell viability by triggering apoptosis, necrosis, and autophagy [193]. |
| Pitavastatin | Melanoma | Human melanoma cells A375 and WM115 | - | 0–5 µM | Induced autophagy and decreased viability of cancer cells [194]. |
| Statin | Cancer type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Rosuvastatin | Papillary thyroid carcinoma | B-CPAP and Nthy-ori 3-1 cells | - | 12,5, 18,5, 25, 50, 100 y 200 µM | Decreased the proliferation and induction of cell death in thyroid cells in a dose- and time-dependent manner [195]. |
| Statin | Cancer type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Simvastatin | Breast cancer | MDA-MB-231 cells | - | 0.50 µM | Reduced the viability of breast cancer cells by inhibiting autophagy [196]. |
| Simvastatin | Glioma | U251 and C6 cells | - | 0–50 µM | Increased the antiglioma effect through the inhibition of the AMPK-dependent autophagic response [197]. |
| Simvastatin | Brain cancer | GBM cells | - | 0–20 µM | Inhibited temozolomide-induced autophagy flux by blocking autophagolysosome formation [198]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mengual, D.; Medrano, L.E.; Villamizar-Villamizar, W.; Osorio-Llanes, E.; Mendoza-Torres, E.; Bolívar, S. Novel Effects of Statins on Cancer via Autophagy. Pharmaceuticals 2022, 15, 648. https://doi.org/10.3390/ph15060648
Mengual D, Medrano LE, Villamizar-Villamizar W, Osorio-Llanes E, Mendoza-Torres E, Bolívar S. Novel Effects of Statins on Cancer via Autophagy. Pharmaceuticals. 2022; 15(6):648. https://doi.org/10.3390/ph15060648
Chicago/Turabian StyleMengual, Daniela, Luz Elena Medrano, Wendy Villamizar-Villamizar, Estefanie Osorio-Llanes, Evelyn Mendoza-Torres, and Samir Bolívar. 2022. "Novel Effects of Statins on Cancer via Autophagy" Pharmaceuticals 15, no. 6: 648. https://doi.org/10.3390/ph15060648