
Targeting Nuclear Receptors in Lung Cancer—Novel Therapeutic Prospects

 ,
,  and
and
Abstract
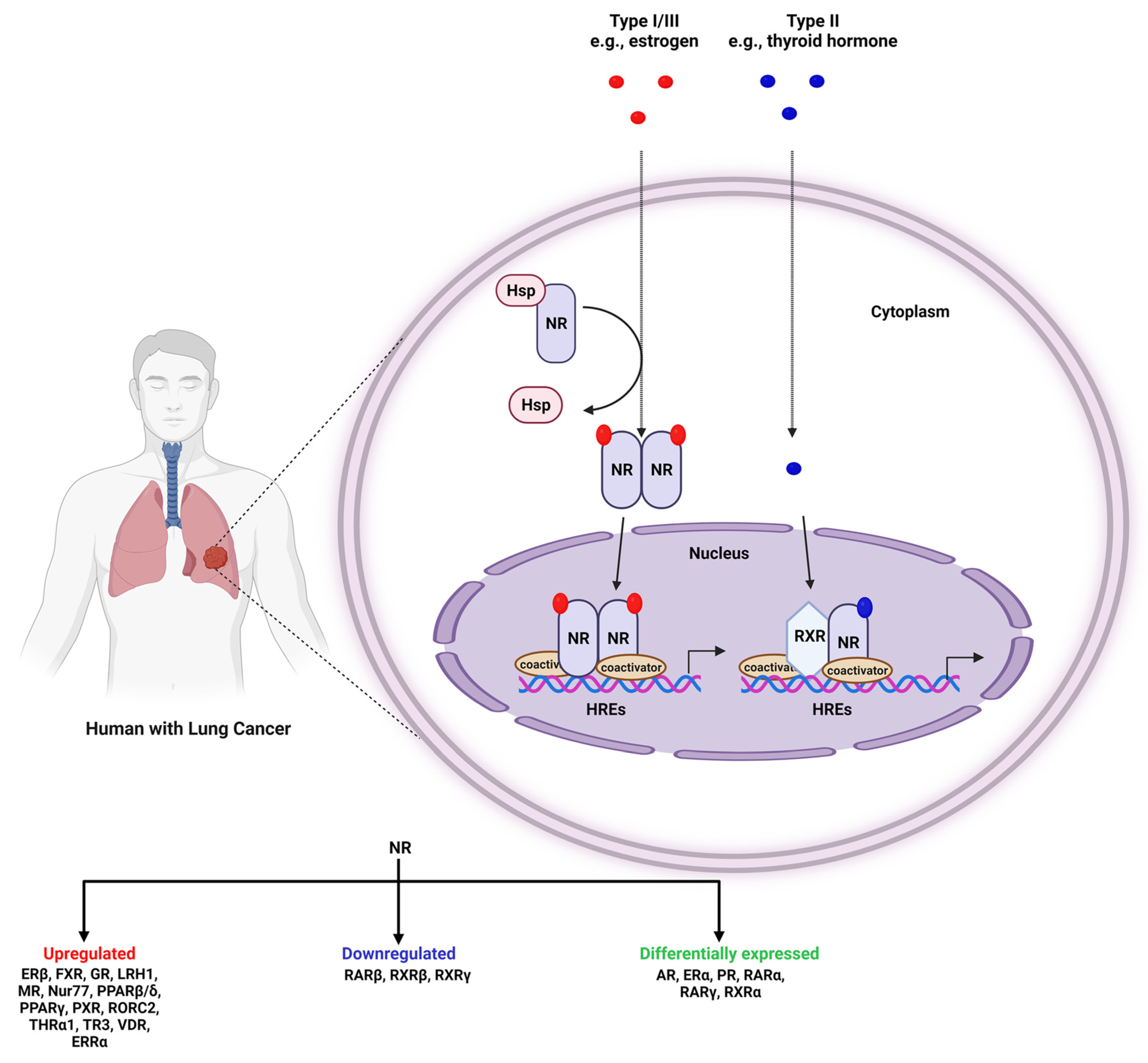
:1. Introduction
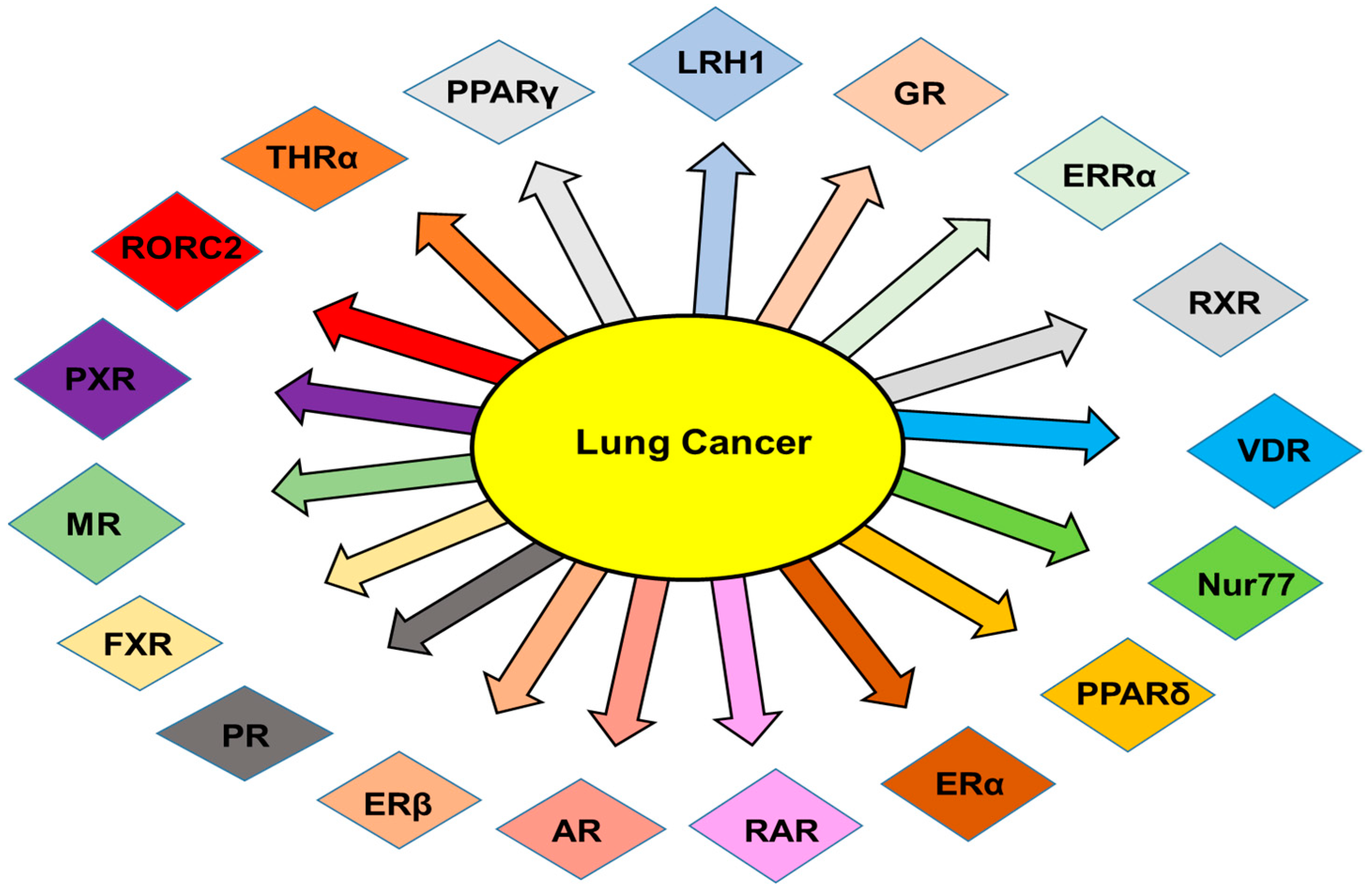
2. NRs in Lung Cancer
2.1. Androgen Receptors (ARs)

2.2. Estrogen Receptors (ERs)
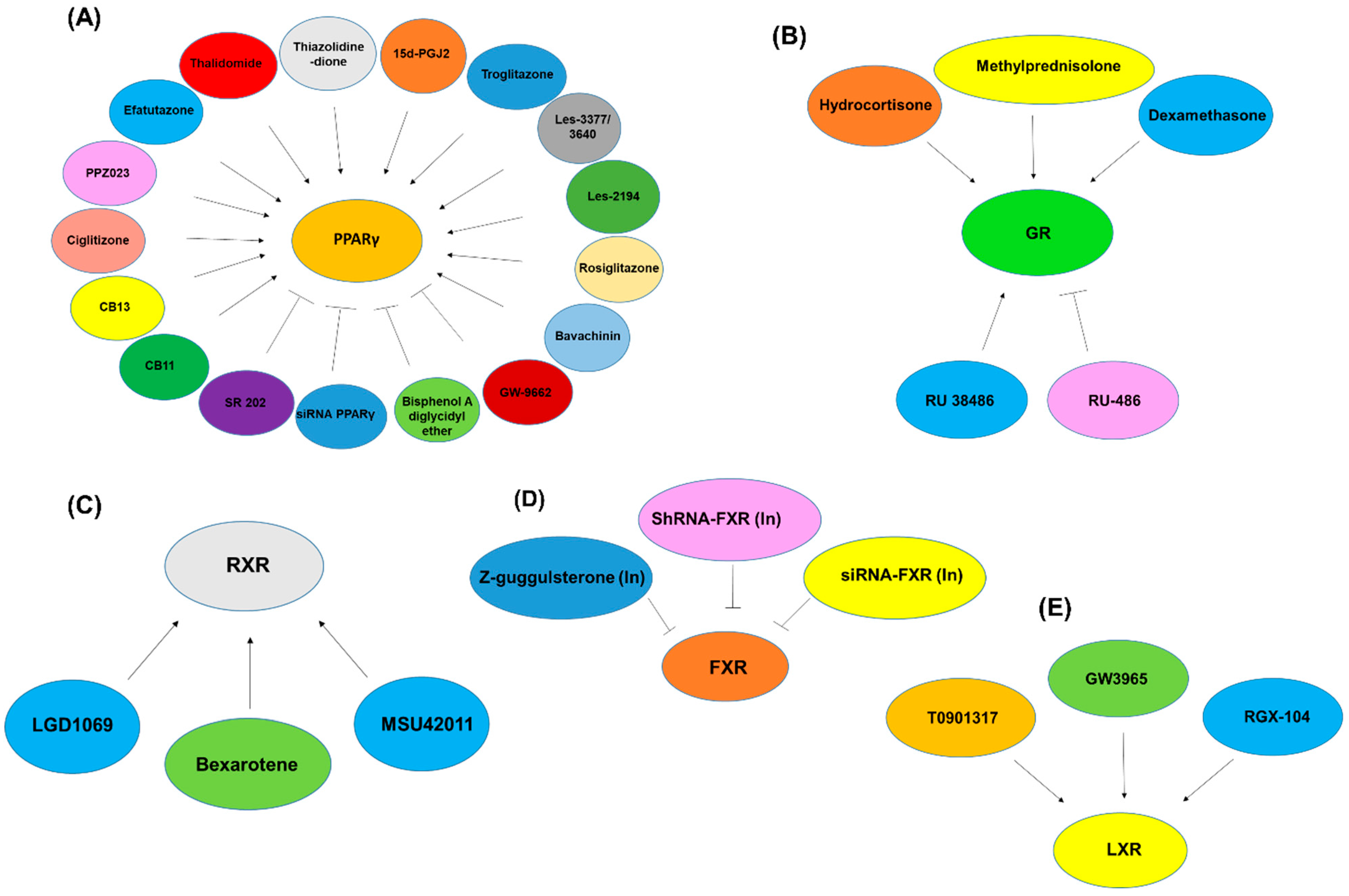
2.3. Peroxisome-Proliferator-Activated Receptors (PPARs)
2.4. Retinoic Acid Receptors (RARs)
2.5. Retinoic X Receptors (RXRs)
2.6. Vitamin D Receptors (VDRs)
2.7. Liver X Receptors (LXRs)
2.8. Estrogen-Related Receptors (ERRs)
2.9. Farnesoid X Receptor (FXR)
2.10. Pregnane X Receptor (PXR) or NR1I2
2.11. Liver Receptor Homologue 1 (LRH-1)
2.12. Glucocorticoid Receptor (GR)
2.13. Progesterone Receptor (PR)
2.14. Thyroid Hormone Receptors (TRs)
3. Biomarkers Using NRs in Lung Cancer
4. Epigenetic Changes in NRs in Lung Cancer
5. Clinical Trials in NRs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nuclear Receptors (NRs) | In Vitro/In Vivo/Clinical | Models/Cell Lines/Tissue | Expression (Down-/Upregulation) | Reference |
|---|---|---|---|---|
| AR | In vivo | Athymic nude mice | Down | [74] |
| In vitro | NSCLC cell lines | Down | [75] | |
| In vivo | Lewis’s lung cancer C57BL/6 mice | Up | [76] | |
| COUP-TF | In vitro | Calu-6, H460, H596, SK-MES-1, H661 | Up | [395] |
| ER | In vivo | Athymic nude mice | Down | [74] |
| Clinical | Lung cancer tissues | Down | [101] | |
| In vitro | NSCLC cell lines | Down | [75] | |
| Clinical | NSCLC tissues | Up | [95] | |
| Clinical | Lung carcinoma tissues | Up | [102] | |
| Clinical | NSCLC tissues | Up | [103] | |
| ERRα | Clinical | NSCLC tissues | Up | [298] |
| In vitro | A549, H1793, H1395, H358 | Up | [298] | |
| ERα | Clinical | NSCLC tissues | Up | [104] |
| In vitro | 91T, 784T, 54T | Up | [106] | |
| In vitro | 128-88T, H23, A549, Calu-6 | Down | [106] | |
| Clinical | NSCLC tissues | Up | [107] | |
| Clinical | NSCLC specimens | Up | [105] | |
| Clinical | NSCLC specimens | Up | [108] | |
| In vivo | Xenograft SCID mice | Up | [106] | |
| Clinical | NSCLC tissues | Down | [109] | |
| Clinical | Lung cancer tissues | Down | [110] | |
| Clinical | NSCLC tissues | Down | [112] | |
| In vivo | Lewis’s lung cancer C57BL/6 mice | Up | [76] | |
| ERβ | Clinical | NSCLC tissues | Up | [104] |
| Clinical | NSCLC tissues | Up | [107] | |
| Clinical | NSCLC specimens | Down | [105] | |
| In vivo | Xenograft SCID mice | Up | [106] | |
| Clinical | NSCLC tissues | Up | [109] | |
| Clinical | Lung cancer tissues | Up | [110] | |
| Clinical | NSCLC tissues | Up | [112] | |
| Clinical | NSCLC specimens | Up | [111] | |
| Clinical | Lung cancer specimens | Up | [100] | |
| In vitro | ERF-LC-OK, PC-3, DMS114, PC-6, | Up | [100] | |
| Clinical | Lung cancer tissues | Up | [113] | |
| Clinical | Primary lung tumor tissues | Up | [115] | |
| Clinical | NSCLC specimens | Up | [116] | |
| Clinical | NSCLC tissues | Up | [117] | |
| In vitro | A549 | Up | [117] | |
| In vivo | Lewis’s lung cancer C57BL/6 mice | Up | [76] | |
| Clinical | Primary NSCLC samples | Up | [118] | |
| Clinical | NSCLC tissues | Up | [114] | |
| FXR | Clinical | NSCLC tissues | Up | [308] |
| In vitro | H1975 and H1299 | Up | [308] | |
| GR | In vivo | Athymic nude mice | Up | [74] |
| Clinical | Lung cancer tissues | Up | [101] | |
| Clinical | Lung neoplastic tissues | Up | [396] | |
| In vitro | NSCLC cell lines | Up | [397] | |
| In vitro | EPLC-32M1, H157, EPLC-272H, U-1752, A-549, H596, LCLC-97TM1 | Up | [336] | |
| Clinical | NSCLC specimens | Up | [398] | |
| LRH1 | Clinical | NSCLC tissues | Up | [326] |
| In vitro | A549, NCI-H157, H1299, SK-MES-1 | Up | [327] | |
| LRH-1 (Nr5a2) | Clinical | Lung cancer tissues | Up | [325] |
| MR | Clinical | Lung cancer tissues | Up | [399] |
| Nur77 | In vitro | H520, H292 | Up | [395] |
| PPARβ/δ | Clinical | Lung cancer tissues | Up | [155] |
| PPARγ | In vitro | A549, LTEP-P | Up | [147] |
| Clinical | Lung cancer tissues | Down | [148] | |
| Clinical | NSCLC specimens | Up | [149] | |
| Clinical | NSCLC tissues | Down | [150] | |
| In vitro | H441, A549, H322, H1944 | Up | [400] | |
| Clinical | NSCLC tissue | Up | [400] | |
| In vitro | H841, A549, PC14 | Up | [151] | |
| Clinical | NSCLC specimens | Up | [152] | |
| In vivo | NNK-induced A/J mouse tumor | Up | [153] | |
| In vitro | Precancerous human bronchial epithelial cells (HBECs) | Up | [154] | |
| PR | In vivo | Athymic nude mice | Down | [74] |
| Clinical | Lung cancer tissues | Down | [101] | |
| Clinical | NSCLC tissues | Down | [95] | |
| Clinical | NSCLC tissues | Up | [104] | |
| Clinical | NSCLC tissues | Up | [107] | |
| In vitro | A549, LCSC#2, 1–87 | Up | [107] | |
| Clinical | Primary lung tumor tissues | Down | [115] | |
| Clinical | NSCLC tissues | Down | [401] | |
| PRα | Clinical | NSCLC specimens | Up | [402] |
| PXR | In vitro | A549, NCI-H358, HCC827, H1650, H1299 | Up | [316] |
| RARα | Clinical | Lung cancerous tissue | Up | [207] |
| In vitro | Calu-1, H647, Al188 | Up | [222] | |
| In vitro | EBC-1 | Up | [208] | |
| In vitro | Calu-1 | Up | [209] | |
| In vitro | Rat tracheobronchial epithelial cell line SPOC-1 | Up | [210] | |
| Clinical | NSCLC tissues | Down | [211] | |
| Clinical | NSCLC tissues | Down | [212] | |
| Clinical | NSCLC specimens | Down | [213] | |
| Clinical | NSCLC specimens | Down | [214] | |
| RARβ | In vitro | H125, SK-MES-1, A1188, H596 | Down | [222] |
| In vitro | Calu-1 | Down | [209] | |
| Clinical | NSCLC tissues | Down | [211] | |
| Clinical | Lung cancer tissues | Down | [216] | |
| In vitro | BEAS-2B-R1 | Down | [216] | |
| Clinical | NSCLC tissues | Down | [212] | |
| Clinical | NSCLC specimens | Up | [217] | |
| Clinical | NSCLC specimens | Down | [213] | |
| In vitro | Calu-6, H146, H661, SK-MES-1 | Down | [205] | |
| In vitro | Calu-3, H292 | Up | [205] | |
| Clinical | Lung cancerous tissue | Down | [207] | |
| Clinical | Lung cancerous tissue | Down | [206] | |
| Clinical | Bronchial biopsy specimens | Down | [218] | |
| RARγ | In vitro | H125, H647, SK-LU-1, H292 | Up | [222] |
| In vitro | Calu-1 | Up | [209] | |
| In vitro | SPOC-1 | Up | [210] | |
| Clinical | NSCLC tissues | Down | [212] | |
| Clinical | Lung cancerous tissue | Down | [207] | |
| Clinical | Lung cancerous tissue | Down | [206] | |
| In vitro | SK-MES-1, Calu-1, H157, H226, H460, H1792, H1648, H1944 | Up | [221] | |
| RORC2 | Clinical | Lung cancer tissues | Up | [403] |
| RXR | Clinical | NSCLC specimens | Down | [214] |
| RXRα | Clinical | Lung cancerous tissue | Up | [207] |
| Clinical | NSCLC tissues | Down | [212] | |
| Clinical | NSCLC tissues | Down | [245] | |
| RXRβ | In vitro | SPOC-1 | Up | [210] |
| Clinical | NSCLC tissues | Down | [212] | |
| Clinical | NSCLC tissues | Down | [245] | |
| Clinical | Lung cancerous tissue | Down | [207] | |
| Clinical | Lung cancerous tissue | Down | [206] | |
| RXRγ | Clinical | NSCLC tissues | Down | [212] |
| Clinical | NSCLC tissues | Down | [245] | |
| Clinical | NSCLC specimens | Down | [214] | |
| THRα1 | Clinical | NSCLC tissues | Up | [355] |
| TR3 (NR41A and Nur77) | In vitro | H460, Calu-6 | Up | [404] |
| Clinical | NSCLC tissues | Up | [405] | |
| VDR | In vitro | NSCLC cell lines | Up | [257] |
| In vitro | SCLC cell lines | Up | [257] | |
| In vitro | EBC-1 | Up | [208] | |
| Clinical | NSCLC tissues | Up | [258] | |
| Clinical | NSCLC tissues | Up | [262] | |
| Clinical | Biopsy specimens | Up | [263] | |
| Clinical | Lung cancer tissues | Down | [264] |
| Nuclear Receptors (NRs) | In Vitro/In Vivo/Clinical | Models/Cell Lines | Agonist/Antagonist | Results | Reference |
|---|---|---|---|---|---|
| AR | In vitro | 86M1, 21H, 16HV, 24H | Testosterone (Ac) | ↑ cell growth | [77] |
| In vitro | H526, 86M1, 21H | Flutamide or Cyproterone acetate (In) | ↓ cell growth | [77] | |
| In vitro | H1355 | 5α-Dihydrotestosterone (DHT) (Ac) | ↑ cell growth | [78] | |
| In vitro | A549 | DHT (Ac) | ↑ cell growth, ↑ CD1, | [79] | |
| ER | In vitro | H460 | Tamoxifen + paclitaxel (In) | ↓ cell growth, ↑ apoptosis | [406] |
| In vitro | H23 | β-Estradiol (Ac) | ↑ cell growth | [106] | |
| In vitro | H23 | Estradiol-17β (E2β) (Ac) | ↑ pMAPK, ↑ cell growth, ↑ VEGF | [120] | |
| In vitro | H23 | Faslodex (In) | ↓ pMAPK, ↓ cell growth, ↓ VEGF | [120] | |
| In vitro | A549 | 17β-estradiol (E2) (Ac) | Restore ER mRNA, ↑ acetylated histone 3 and histone 4 | [121] | |
| In vitro | 201T, 273T, A549 | E2 (Ac) | ↑ cell proliferation, ↑ pMAPK, ↑ pEGFR, | [122] | |
| In vitro | 201T, 273T, A549 | Fulvestrant + gefitinib | ↓ cell growth, ↑ apoptosis | [122] | |
| In vitro | NSCLC cells | Estrogen (Ac) | ↑ ERβ, ↑ metastasis, ↑ MMP-2, ↑ TLR4 | [123] | |
| In vitro | PC9, Hcc827 | β-estradiol (Ac) | ↑ ERβ1 | [118] | |
| In vitro | H23, A549 | β-estradiol (Ac) | ↑ cell proliferation | [106] | |
| In vivo | Xenograft SCID mice | β-estradiol (Ac) | ↑ cell proliferation | [106] | |
| In vivo | Xenograft SCID mice | ICI 182,780 (In) or β-estradiol + ICI 182,780 | ↓ cell proliferation | [106] | |
| In vitro | H23 | siRNA-ERα or siRNA-ERβ | ↓ ERα/ERβ, ↓ cell growth | [124] | |
| In vitro | A549, H1650 | Tamoxifen (In- ER) + gefitinib | ↓ cell growth, ↑ apoptosis, ↑ ERβ | [125] | |
| ERRα | In vitro | A549, H1793 | siRNA-ERRα (In) or XCT-790 (In) | ↓ proliferation, ↓ migration, ↓ invasion, ↓ fibronectin (FN), ↓ vimentin, ↓ MMP-2, ↓ IL-6 | [298] |
| ERβ | In vitro | Calu-6, 201T | ShRNA- ERβ | ↓ cell growth, ↑ apoptosis | [119] |
| In vitro | A549 | ShRNA- ERβ (In) | ↓ pERK, ↓ MMP-2, ↓ MMP-9, cell ↓ proliferation, ↓ invasion | [117] | |
| In vivo | Lung metastatic mouse model | ShRNA- ERβ (In) | ↓ tumor growth, ↓ metastasis | [117] | |
| In vitro | A549, H1793 | Estrogen (E2) (Ac) | ↑ cell growth, ↑ ERβ, ↑ IL6, ↑ migration | [114] | |
| In vitro | A549, H1793 | Fulvestrant (Ful) (In) | ↓ cell growth, ↓ IL6, | [114] | |
| In vivo | Urethane-induced mouse model | Estrogen (E2) (Ac) | ↑ tumor, ↑ ERβ, ↑ IL6, ↑ p-↑ p38MAPK, ↑ p-AKT, ↑ p-Stat3 | [114] | |
| FXR | In vitro | H1975, H1299 | Z-guggulsterone (In) | ↓ proliferation, ↓ CD1, ↓ CDK2, ↓ CDK4, ↓ CDK6, ↓ p-Rb | [308] |
| In vitro | H1975, H1299 | siRNA-FXR (In) | ↓ proliferation, ↓ CD1, ↓ pRb | [308] | |
| In vivo | NSCLC xenograft | ShRNA-FXR (In) | ↓ tumor growth, | [308] | |
| GR | In vivo | Athymic nude mice | Hydrocortisone (Ac) or RU 38,486 (In) | ↓ tumor size | [74] |
| In vitro | C10 | Dexamethasone (Dex) (Ac) | ↓ cell proliferation, ↓ GR, ↓ K-RAS | [335] | |
| In vitro | A5, LM2 cells | Dex (Ac) | ↓ GR | [335] | |
| In vitro | 32M1, H157, A549, 97TM1 | Dex (Ac) | ↓ cell growth | [336] | |
| In vitro | 32M1, H157, A549, 97TM1 | RU-486 (In) | ↑ cell growth | [336] | |
| In vitro | H82, H345, H510A, N592, H2081 | Methylprednisolone (MP) (Ac) | ↓ cell growth, ↑ apoptosis | [342] | |
| In vitro | A549, Calu-1 cells | Dex (Ac) | ↓ pRB, ↓ CDK2, CDK4, ↓ cyclin D, ↓ E2F, ↓ Myc, ↑ p21(Cip1), ↓ ERK/MAPK | [337] | |
| In vitro | A549 | Dex (Ac) | ↑ 15-PGDH | [338] | |
| In vivo | SCLC xenograft mice | Infection with GR-expressing adenovirus | ↓ tumor growth, ↓ Bcl-2, ↓ Bcl-xL, ↑ apoptosis, | [343] | |
| LXR | In vitro | A549, HCC827-8-1 | T0901317 (Ac) + gefitinib | ↓ migration, ↓ invasion, ↓ MMP9, ↑ E-cadherin | [277] |
| In vivo | BALB/c nude mice | T0901317 (Ac) + gefitinib | ↓ migration, ↓ MMP9, | [277] | |
| In vivo | Homograft murine model | GW3965 or RGX-104 (Ac) | ↓ myeloid-derived suppressor cell (MDSC) | [279] | |
| In vitro | A549 | T0901317 (Ac) | ↓ migration, ↓ invasion, ↓ MMP-9, ↓ NF-κB/MMP-9 | [278] | |
| In vitro | HCC827/GR-8-2 | GW3965 + gefitinib | ↓ cell proliferation, ↑ apoptosis, ↓ NF-κB | [281] | |
| In vitro | H827-7-2, H827-7-4 | GW3965 + gefitinib | ↓ cell proliferation, ↑ apoptosis, ↓ pAKT, ↓ pNF-κB | [287] | |
| LXRα | In vitro | HCC827-GR, PC9-GR | T0901317 | ↓ proliferation, ↑ LXRα, ↑ ABCA1 | [288] |
| In vitro | HCC827-GR, PC9-GR | siRNA- LXRα (In) | ↓ LXRα, ↓ ABCA1, ↑ proliferation | [288] | |
| NR0B1 | In vitro | A549 | siRNA- NR0B1 (In) | ↓ Bcl-2, ↓ MMP-2 | [407] |
| PPARα | In vivo | TC-1 lung tumor-mice model | AVE8134, Wyeth-14,643, Bezafibrate (Ac) | ↓ EET, ↑ 11-HETE, ↑ proliferation, ↑ angiogenesis, ↑ AKT/ERK, ↑ | [408] |
| PPARβ/δ | In vitro | A549, H23, H157 | GW501516 (Ac) | ↑ cell growth, ↓ apoptosis, ↑ pAkt, ↑ PDK1, ↓ PTEN, ↑ Bcl-xL, and COX-2 | [155] |
| In vitro | A549, H1838 | GW0742 or GW501516 (Ac) | ↑ Angptl4 | [156] | |
| In vitro | H157, H1838 | GW501516 (Ac) | ↑ EP4, ↑ cell proliferation | [157] | |
| PPARγ | In vitro | H441, H358 | Ciglitizone or 15d-PGJ2 (Ac) | ↓ cell growth, ↑ cell death, ↑ HTI56 | [400] |
| In vitro | H157, H322, H520, H522, H1299, H1334, H1944, A549 | Ciglitizone or 15d-PGJ2 (Ac) | ↓ cell growth, ↑ cell death | [400] | |
| In vitro | H441, H358, H322 | Ciglitizone or 15d-PGJ2 (Ac) | ↑ gelsolin, ↑ Mad, ↑ p21, ↑ PPARγ, ↑ HTI56 ↓ MUC1, ↓ SP-A, ↓ CC10 | [400] | |
| In vitro | H157, H1299 | Ciglitizone (Ac) | ↓ MMP-2, | [400] | |
| In vitro | H441, H358 | Ciglitizone(Ac) | ↓ cyclin D1, ↓ pRb | [400] | |
| In vitro | H358 | 15d-PGJ2 (Ac) | ↓ cyclin D1, ↓ pRb | [400] | |
| In vitro | H841, A549, PC14 | Troglitazone (Tro) or 15d-PGJ2 (Ac) | ↓ cell growth, ↑ cell death | [151] | |
| In vitro | A549, H345, N417 | 15d-PGJ2 (Ac) | ↓ cell growth, ↑ apoptosis | [409] | |
| In vitro | A549, N417 | Ciglitazone (Ac) | ↓ cell growth | [409] | |
| In vitro | A549, LTEP-P | 15d-PGJ(2) or ciglitazone (Ac) | ↓ cell growth, ↑ apoptosis, ↑ Caspase-3, ↑ bax, ↑ bcl-2 | [158] | |
| In vivo/In vitro | Nude mice-A549 cells | Ciglitazone (Ac) | ↓ cell growth, ↑ PPARγ, ↓ cyclin D1, ↑ P21 | [410] | |
| In vitro | A549, LTEP-P | 15d-PGJ2 or ciglitazone (Ac) | ↓ cell viability, ↑ apoptosis | [147] | |
| In vitro | A549 | Tro (Ac) | ↑ PPARγ activity, ↓ cell growth, ↓ cyclins D and E, ↓ Erk1/2 | [149] | |
| In vivo | A549-tumor-bearing SCID mice | Tro or Pio (Ac) | ↓ tumor | [149] | |
| In vitro | H1838, H2106 | 15d-PGJ2, rosiglitazone (BRL49653) or Tro (Ac) | ↓ fibronectin (Fn), ↓ pCREB, ↓ Sp1 | [164] | |
| In vitro | H1838, H2106 | GW-9662 (In) | ↑ fibronectin (Fn) | [164] | |
| In vitro | H1838, H2106 | siRNA- PPARγ (In) | ↑ fibronectin (Fn) | [164] | |
| In vitro | A549, H2122 | ciglitazone, PGA1, or 15-deoxy-12,14-PGJ2 (Ac) | ↓ cell growth | [165] | |
| In vitro | H2122 | ciglitazone | ↑ E-cadherin, ↓ cell growth | [165] | |
| In vivo/In vitro | Nude rats-H2122-PPAR(gamma) cells | Implantation of H2122- PPARγ cells into the lungs of nude rats | ↓ cell growth, ↓ metastasis, | [166] | |
| In vitro | SQ-5, EBC-1, ABC-1, RERF-LC-OK | Tro (Ac) | ↓ cell growth, ↑ apoptosis, ↑ GADD153 | [167] | |
| In vitro | H460, H1299, H661 | Thalidomide (Ac) | ↑ PPARγ, ↓ NFκB, ↑ apoptosis, ↓ growth-related oncogene (GRO), ↓ epithelial cell derived-neutrophil activating peptide-78 (ENA-78), ↓ angiotensin, ↓ IL-8, ↓ COX-2 | [168] | |
| In vivo | Nude mice-NCI-H1299 cells | Thalidomide (Ac) | ↓ tumor growth, ↑ PPARγ | [168] | |
| In vitro | H23, CRL-2066 | Tro (Ac) | ↓ cell growth, ↑ apoptosis, ↑ PPARγ, ↓ Bcl-w, ↓ Bcl-2, ↑ ERK1/2, ↑ p38, ↓ SAPK/JNK | [169] | |
| In vitro | H1838, H2106 | GW1929, PGJ2, ciglitazone, Tro or rosiglitazone | ↓ cell growth, ↑ pErk, ↓ EP2, | [170] | |
| In vitro | H345, H2081, H1838, H2106 | PGJ2 or ciglitazone | ↓ cell growth, ↑ apoptosis, ↑ p21, ↓ cyclin D1 | [171] | |
| In vitro | A549 | Rosiglitazone | ↓ cell growth, ↑ PPARγ, ↑ PTEN, | [173] | |
| In vitro | H23 | Troglitazone | ↑ ERK1/2, ↑ apoptosis, ↑ Akt | [172] | |
| In vitro | H23 | siRNA- PPARγ (In) | ↓ ERK1/2, ↓apoptosis, | [172] | |
| In vitro | H2122 | SR 202 (In) | ↓ PPARγ, ↑ cell growth, ↓ E-cadherin | [174] | |
| In vitro | A549, H157, H460, H1792 | Tro, cigolitazone and GW1929 | ↓ FLIPL, ↓ FLIPS, ↑ DR5, | [411] | |
| In vivo | A549, H460 | 15d-PGJ2 | ↓ cell growth, ↑ apoptosis, ↑ PPARγ, ↑ caspase3, ↓ Cyclin D1 | [175] | |
| In vivo | A549, H460 | Docetaxel | ↓ cell growth, ↑ apoptosis, ↑ caspase3, ↓ Cyclin D1 | [175] | |
| In vivo | A549, H460 | 15d-PGJ2 + docetaxel | ↓ cell growth, ↑ apoptosis, ↑ PPARγ, ↑ caspase3, ↓ Cyclin D11 | [175] | |
| In vivo | Athymic nu/nu mice- A549 and H460 cells | 15d-PGJ2 | ↓ tumor volume, ↓ PGE2, ↑ PPARγ, ↑ caspase3, ↑ caspase8, ↓ Cyclin D1, ↑ BAD, ↓ Bcl2 | [175] | |
| In vivo | Athymic nu/nu mice- A549 and H460 cells | Docetaxel | ↓ tumor volume, ↓ PGE2, ↑ PPARγ, ↑ caspase3, ↑ caspase8, ↑ caspase9, ↓ Cyclin D1, ↑ BAD, ↓ Bcl2, ↑ APAF1, ↑ BBC3, ↑ p53 | [175] | |
| In vivo | Athymic nu/nu mice- A549 and H460 cells | 15d-PGJ2 + docetaxel | ↓ tumor volume, ↓ PGE2, ↑ PPARγ, ↑ caspase3, ↑ caspase8, ↑ caspase9, ↓ Cyclin D1, ↑ BAD, ↓ Bcl2, ↑ APAF1, ↑ BBC3, ↑ p53 | [175] | |
| In vitro | CL1-0, A549 | Tro + Aspirin | ↓ cell growth, ↓ Cdk2, ↓ E2F-1, ↓ cyclin B1, ↓ cyclin D3, ↓ pRB, ↑ apoptosis, ↓ PI3K/Akt, ↑ p27, ↓ pRac1 | [412] | |
| In vitro | A549 | KR-62980 or rosiglitazone | ↓ cell growth, ↑ apoptosis, ↑ ROS, ↑ proline oxidase (POX), | [176] | |
| In vitro | A549 | Bisphenol A diglycidyl ether (In) | ↓ apoptosis | [176] | |
| In vitro | RERF-LC-AI, SK-MES-1, PC-14, A549 | Tro or ciglitazone | ↑ VEGF, ↑ neuropilin-1 | [177] | |
| In vitro | RERF-LC-AI, SK-MES-1, PC-14, A549 | GW9662 (In) | ↓ VEGF, ↓ neuropilin-1 | [177] | |
| In vitro | A549 | Tro and rosiglitazone | ↓ TGF-β-induced EMT, ↓ vimentin, ↓ N-cadherin, ↓ fibronectin, ↓ migration, ↓ invasion | [413] | |
| In vivo | Adenocarcinoma and SCC A/J mice | Pioglitazone | ↓ tumor, ↑ apoptosis | [414] | |
| In vitro | H1838, H2106, A549 | Rosiglitazone (Ac) | ↓ cell proliferation, ↓ alpha4 nicotinic acetylcholine receptor (nAChR), ↑ p38 MAPK, ↑ ERK 1/2, ↓ pAkt, ↑ p53 | [415] | |
| In vivo | A549-induced nude mice tumor | Ciglitazone (Ac) | ↓ cell proliferation, ↑ PPARγ, ↓ cyclin D1, ↑ P21 | [180] | |
| In vitro | A549R, H460R | CB13 (Ac) | ↓ cell viability, ↑ LDH, ↑ caspase-3, ↑ caspase-9, ↑ ROS, ↑ apoptosis | [181] | |
| In vitro | A549, H460 | CB11 (Ac) | ↓ cell viability, ↑ apoptosis, ↑ p-ATM, ↑ p-chk2, ↑ p-p53, ↑ GADD45α, ↑ LDH, ↑ caspase-3, ↑ PPARγ | [182] | |
| In vivo | A549 Xenograft mice | CB11 (Ac) | ↑ apoptosis, ↓ tumor volume, ↓ EMT, ↑ caspase-3, ↑ caspase-9, ↑ PPARγ | [182] | |
| In vitro | H1299, H460 | PIO (Ac) | ↓ cell proliferation, ↑ apoptosis, ↑ caspase-3, ↓ Myc, ↓ R-Ras, ↓ MAPK6, ↓ MAP3K8, ↓ Bcl-2, ↓ PCNA, ↓ laminin, ↑ CASP5, ↑ CASP4, ↑ CFLAR, ↑ PAWR, ↓ TGFβR1, ↓ SMAD3, ↓ pEGFR, ↓ pAKT, ↓ pMAPK | [179] | |
| In vitro | A549 | Bavachinin (BNN) (Ac) | ↓ cell viability, ↑ ROS, ↑ apoptosis | [416] | |
| In vitro | SCC-15 | Les-2194 (Ac) | ↑ ROS, ↓ Ki67 | [183] | |
| In vitro | SCC-15, A549 | Les-3377 (Ac) | ↑ ROS | [183] | |
| In vitro | SCC-15 | Les-3640 (Ac) | ↑ ROS, ↑ caspase-3, ↑ PPARγ | [183] | |
| In vitro | A549 | Rosiglitazone (Ac) | ↑ cell death, ↓ pAKT | [417] | |
| In vitro | H460, A549 | PPZ023 (Ac) | ↓ cell growth, ↑ LDH, ↑ apoptosis, ↑ PPARɣ, ↑ caspase-3, ↑ caspase-8, ↑ caspase-9 | [184] | |
| In vitro | H1993 | Thiazolidinedione (TZD) (Ac) | ↓ ALDH1A3 | [418] | |
| In vitro | HCC827-GR, PC9-GR | Efatutazone (Ac) | ↓ proliferation, ↑ PPARγ, ↑ LXRα, ↑ ABCA1 | [288] | |
| In vitro | Precancerous human bronchial epithelial cells (HBECs) | TZD (Ac) | ↓ COX2, ↓ cell growth, ↓ clonogenicity, ↓ cell migration | [154] | |
| PPARγ/RXR | In vitro | Calu-6 | ciglitazone (Ac- PPARγ) + SR11237 (Ac-RXR) | ↓ cell growth, ↑ RAR-β | [185] |
| In vitro | Calu-6 | ciglitazone (Ac- PPARγ) and SR11237 (Ac-RXR) + bisphenol A diglycidyl ether (In- PPARγ) | ↓ RAR-β | [185] | |
| PPARδ | In vitro | A549 | L-165041 (Ac) | ↓ cell growth, ↓ cyclin D, ↓ PCNA | [186] |
| In vitro | A549 | SR13904 (In) | ↓ cell proliferation, ↑ apoptosis | [187] | |
| PR | In vitro | A549, LCSC#2, 1-87 | Progesterone (Ac) | ↓ cell proliferation | [107] |
| In vitro | A549, LCSC#2, 1-87 | RU 38,486 (In) | ↑ cell proliferation | [107] | |
| In vivo/In vitro | Athymic nude mice cells (A549, 1-87, and LCSC#2) | Progesterone (Ac) | ↓ tumor volume, ↑ p21, ↑ p27, ↓ cyclin A, ↓ cyclin E, ↓ Ki67 | [107] | |
| PRα | In vitro | A549, PC-9, PC-9GR | P4/Org + gefitinib (Ac) | ↓ proliferation, ↓ invasion, ↓ migration, | [402] |
| PXR | In vitro | A549 | SR12813 (Ac) | ↑ PXR, ↑ CYP2C8, ↑ P-gp | [316] |
| In vitro | A549 | siRNA- PXR (In) | ↓ PXR, ↓ CYP2C8, ↓ P-gp | [316] | |
| RAR | Clinical | Bronchial biopsy specimens | 13-CRA (Ac) | ↑ RAR-β | [218] |
| Clinical | Bronchial brushing samples | 13-CRA (Ac) | ↑ RAR-β | [220] | |
| In vitro | H209-RAR-β | RA (Ac) | ↓ cell growth, ↑ p27Kip1and ↓ L-myc, ↑ apoptosis, ↓ cdk2 kinase activity | [419] | |
| In vitro | H82 | RA (Ac) | ↓ cell growth, ↑ p27Kip1, ↑ RAR-β, ↓ cdk2 kinase activity | [419] | |
| In vitro | H460, Calu-1, SK-LU-1, A549, H69 cells | RA (Ac) | ↑ RAR-β | [222] | |
| In vitro | EBC-1 | RA (Ac) | ↓ cell growth | [208] | |
| In vitro | H82 | RA (Ac) | ↓ cell growth | [215] | |
| In vivo | Male A/J mice | Isotretinoin (Ac) | ↓ tumor multiplicity, ↑ RAR-α, ↑ RAR-β, ↑ RAR-γ | [223] | |
| In vitro | Calu-1 | ATRA (Ac) | ↑ cell growth (serum-free medium) | [209] | |
| In vitro | H292G, H358, H157, H1792, H226a | ATRA (Ac) | ↓ cell growth | [209] | |
| In vitro | H345, H51 0 | 13-CRA (Ac) | ↑ RAR-β, ↓ cell growth | [219] | |
| In vitro | CH27 | RA (Ac) | ↓ cell growth, ↑ p27(Kip1), ↓ Cdk3, ↓ p21(CIP1/Waf1), ↑ RAR-β, ↓ c-Myc, ↓ cyclin A/Cdk2 activity | [224] | |
| In vitro | SCLC | RA (Ac) | ↑ gastrin-releasing peptide (GRP), ↑ cell growth | [225] | |
| In vitro | H460, SK-MES-1, H1792 | CD437 (Ac) | ↓ cell growth, ↑ apoptosis, ↑ c-Myc, ↑ ornithine decarboxylase (ODC), ↑ cdc25A | [226] | |
| In vitro | GLC82 | RA or 4-HPR (Ac) | ↓ cell growth, ↑ RAR-β2 | [228] | |
| In vitro | H460 | CD437 (Ac) | ↓ cell growth, ↑ apoptosis, ↑ p53, ↑ p21, ↑ Bax, ↑ Killer/DR5 | [227] | |
| In vitro | Rat tracheobronchial epithelial cell line SPOC-1 | SRI-6751-84 (Ac) RARα-selective retinoid (Ro40-6055) | ↑ transglutaminase (TGase II), ↑ apoptosis | [210] | |
| In vitro | Rat tracheobronchial epithelial cell line SPOC-1 | RARα-antagonist Ro41-5253 | ↓ TGase II induced by RAR-selective retinoid, ↑ apoptosis | [210] | |
| In vitro | BZR-T33 ras transformed human bronchial epithelial cell line | ATRA (Ac) | ↓ cell proliferation, ↑ RAR-γ, | [420] | |
| In vitro | H460 | RA (Ac) | ↑ EGFR, ↑ tumorigenicity | [229] | |
| In vitro | H460a | RA (Ac) | ↑ EGFR, ↑ tumorigenicity | [230] | |
| In vitro | Calu-1, A549, H1792 | ATRA (Ac) | ↑ TIG3 | [232] | |
| In vitro | A549, H1792, H157, H460 | ATRA (Ac) | ↑ TIG3, ↑ RAR-β | [232] | |
| In vitro | A549 | beta-cryptoxanthin (Ac) | ↓ cell growth, ↓ cyclin D1, ↓ cyclin E, ↑ p21, ↑ RAR-β | [233] | |
| In vitro | A549 | ATRA (Ac) | ↑ VEGF-C, ↑ VEGF-D, ↑ VEGFR3, ↓ RXRα, | [234] | |
| In vitro | A549 | RA (Ac) | ↓ invasiveness, ↑ CRABP | [421] | |
| In vitro | Calu-6 | RA (Ac) | ↑ RAR-β | [422] | |
| In vitro | Calu-1 | RAR-α (Am80), RAR-β/γ (TTNN and SR3985) (Ac) | ↑ cell growth (serum-free medium) | [209] | |
| In vitro | Calu-1 | RAR-γ (CD2325 and SR11363) (Ac) | ↓ cell growth (serum-free medium) | [209] | |
| In vitro | NSCLC cell lines | 4HPR (Ac) | ↓ cell growth, ↑ apoptosis | [235] | |
| In vitro | A549, H226, H1648, SK-MES-l | 4-HPR (Ac) | ↓ cell growth, ↓ Bcl-2, ↑ apoptosis | [235] | |
| RAR/RAR-α | In vitro | Calu-1, H1792 | AGN193109 (In-RAR) or Ro 41-5253 (In- RAR-α) | ↓ TIG3 | [232] |
| RAR/RXR | In vitro | Calu-6, H460 | ATRA (Ac) | ↓ cell growth, ↑ RAR-β, ↑ apoptosis | [423] |
| In vivo | A/J mouse | 9Cra (Ac) | ↓ tumor multiplicity, ↑ RAR-β | [246] | |
| RARγ | In vitro | SK-MES-1, Calu-1, H157, H226, H460, H1792, H1648, H1944 | CD437 (Ac) | ↓ cell growth, ↑ apoptosis | [221] |
| RXR | In vivo | A549 xenograft model | LGD1069 (Ac) | ↓ cell growth, ↓ CD31, ↓ VEGF, ↓ JNK and ERK activation | [247] |
| In vivo | Vinyl-carbamate-induced A/J mouse model | MSU42011 (Ac) | ↓ tumor, ↑ CD8/CD4 ratio, ↑ CD25 T cells | [248] | |
| In vitro | A549 | Bexarotene (Ac) | ↑ PPARγ, ↑ PTEN, ↓ mTOR, | [249] | |
| TR3 (NR41A and Nur77) | In vitro | H460, Calu-6 | siRNA-TR3 (In) | ↓ cell growth | [404] |
| In vitro | A549, H460 | siRNA-TR3 (In) | ↓ cell growth, ↑ apoptosis, ↓ survivin, ↓ EGFR, ↓ bcl-2, ↓ c-myc, ↓ p70S6K, ↓ pS6RP, ↓ 4E-BP1, ↑ pAMPKα, ↑ sestrin | [405] | |
| VDR | In vitro | EBC-1 | calcitriol (Ac) | ↓ cell growth | [208] |
| In vivo/In vitro | C57BL/6 mice—green fluorescent protein-transfected Lewis lung carcinoma (LLC-GFP) cells | calcitriol (Ac) | ↓ MMP-2, ↓ MMP-9, ↓ VEGF, ↓ parathyroid-hormone-related protein (PTHrP) | [265] | |
| In vitro | H460 | Vitamin D | ↓ histidine-rich calcium-binding protein (HRC), ↓ cell migration, ↓ proliferation, ↑ apoptosis | [264] |
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Pass, H.I.; Carbone, D.P.; Johnson, D.H.; Minna, J.D.; Scagliotti, G.V.; Turrisi, A.T. Principles and Practice of Lung Cancer: The Official Reference Text of the International Association for the Study of Lung Cancer (IASLC); Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012. [Google Scholar]
- Wang, L.; Syn, N.L.; Subhash, V.V.; Any, Y.; Thuya, W.L.; Cheow, E.S.H.; Kong, L.; Yu, F.; Peethala, P.C.; Wong, A.L.; et al. Pan-HDAC inhibition by panobinostat mediates chemosensitization to carboplatin in non-small cell lung cancer via attenuation of EGFR signaling. Cancer Lett. 2018, 417, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Ong, P.S.; Wang, L.; Chia, D.M.; Seah, J.Y.; Kong, L.R.; Thuya, W.L.; Chinnathambi, A.; Lau, J.Y.; Wong, A.L.; Yong, W.P.; et al. A novel combinatorial strategy using Seliciclib((R)) and Belinostat((R)) for eradication of non-small cell lung cancer via apoptosis induction and BID activation. Cancer Lett. 2016, 381, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef] [Green Version]
- Sawhney, M.; Rohatgi, N.; Kaur, J.; Shishodia, S.; Sethi, G.; Gupta, S.D.; Deo, S.V.; Shukla, N.K.; Aggarwal, B.B.; Ralhan, R. Expression of NF-kappaB parallels COX-2 expression in oral precancer and cancer: Association with smokeless tobacco. Int. J. Cancer 2007, 120, 2545–2556. [Google Scholar] [CrossRef]
- Sinha, R.; Kulldorff, M.; Curtin, J.; Brown, C.C.; Alavanja, M.C.; Swanson, C.A. Fried, well-done red meat and risk of lung cancer in women (United States). Cancer Causes Control 1998, 9, 621–630. [Google Scholar] [CrossRef]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [Green Version]
- Mayne, S.T.; Buenconsejo, J.; Janerich, D.T. Previous lung disease and risk of lung cancer among men and women nonsmokers. Am. J. Epidemiol. 1999, 149, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.T.; Blot, W.J.; Zheng, W.; Ershow, A.G.; Hsu, C.W.; Levin, L.I.; Zhang, R.; Fraumeni, J.F., Jr. Lung cancer among Chinese women. Int. J. Cancer 1987, 40, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K. Excess incidence of lung cancer among pulmonary tuberculosis patients. Jpn. J. Clin. Oncol. 1993, 23, 205–220. [Google Scholar] [PubMed]
- Wu, A.H.; Fontham, E.T.; Reynolds, P.; Greenberg, R.S.; Buffler, P.; Liff, J.; Boyd, P.; Henderson, B.E.; Correa, P. Previous lung disease and risk of lung cancer among lifetime nonsmoking women in the United States. Am. J. Epidemiol. 1995, 141, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Boice, J.D., Jr. 15 Ionizing Radiation. In Cancer Epidemiology and Prevention; Oxford University Press: Oxford, MS, USA, 2006; p. 259. [Google Scholar]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [CrossRef]
- McKenna, N.J.; O’Malley, B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Papacleovoulou, G.; Abu-Hayyeh, S.; Williamson, C. Nuclear receptor-driven alterations in bile acid and lipid metabolic pathways during gestation. Biochim. Biophys. Acta 2011, 1812, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.M.; Bothwell, A.L. The nuclear receptor PPARs as important regulators of T-cell functions and autoimmune diseases. Mol. Cells 2012, 33, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Z.S.; Czimmerer, Z.; Nagy, L. Nuclear receptor mediated mechanisms of macrophage cholesterol metabolism. Mol. Cell Endocrinol. 2013, 368, 85–98. [Google Scholar] [CrossRef]
- Gronemeyer, H.; Gustafsson, J.A.; Laudet, V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sethi, G.; Jain, A.K.; Jaiswal, A.K.; Aggarwal, B.B. Genetic deletion of NAD(P)H:quinone oxidoreductase 1 abrogates activation of nuclear factor-kappaB, IkappaBalpha kinase, c-Jun N-terminal kinase, Akt, p38, and p44/42 mitogen-activated protein kinases and potentiates apoptosis. J. Biol. Chem. 2006, 281, 19798–19808. [Google Scholar] [CrossRef] [Green Version]
- Green, K.A.; Carroll, J.S. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat. Rev. Cancer 2007, 7, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Manu, K.A.; Shanmugam, M.K.; Ramachandran, L.; Li, F.; Siveen, K.S.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Arfuso, F.; Kumar, A.P.; et al. Isorhamnetin augments the anti-tumor effect of capecitabine through the negative regulation of NF-kappaB signaling cascade in gastric cancer. Cancer Lett. 2015, 363, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2018, 128, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Poulsen, H.S. Immunohistochemical estrogen receptor determination in paraffin-embedded tissue. Prediction of response to hormonal treatment in advanced breast cancer. Cancer 1989, 64, 1901–1908. [Google Scholar] [CrossRef]
- Stierer, M.; Rosen, H.; Weber, R.; Hanak, H.; Spona, J.; Tuchler, H. Immunohistochemical and biochemical measurement of estrogen and progesterone receptors in primary breast cancer. Correlation of histopathology and prognostic factors. Ann. Surg. 1993, 218, 13–21. [Google Scholar] [CrossRef]
- Shang, Y. Molecular mechanisms of oestrogen and SERMs in endometrial carcinogenesis. Nat. Rev. Cancer 2006, 6, 360–368. [Google Scholar] [CrossRef]
- Gangwar, S.K.; Kumar, A.; Jose, S.; Alqahtani, M.S.; Abbas, M.; Sethi, G.; Kunnumakkara, A.B. Nuclear receptors in oral cancer-emerging players in tumorigenesis. Cancer Lett. 2022, 536, 215666. [Google Scholar] [CrossRef]
- Evans, R.M. The nuclear receptor superfamily: A rosetta stone for physiology. Mol. Endocrinol. 2005, 19, 1429–1438. [Google Scholar] [CrossRef]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Sonoda, J.; Pei, L.; Evans, R.M. Nuclear receptors: Decoding metabolic disease. FEBS Lett. 2008, 582, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.H.; Downes, M.; Evans, R.M. Nuclear receptors as modulators of the tumor microenvironment. Cancer Prev. Res. 2012, 5, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Nelson, P.S. The androgen/androgen receptor axis in prostate cancer. Curr. Opin. Oncol. 2012, 24, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Gustafsson, J.A. Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol. Metab. 2015, 26, 467–476. [Google Scholar] [CrossRef]
- Zhang, Y.; Hagedorn, C.H.; Wang, L. Role of nuclear receptor SHP in metabolism and cancer. Biochim. Biophys. Acta 2011, 1812, 893–908. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; DuBois, R.N. Inflammatory mediators and nuclear receptor signaling in colorectal cancer. Cell Cycle 2007, 6, 682–685. [Google Scholar] [CrossRef] [Green Version]
- Zacheis, D.; Dhar, A.; Lu, S.; Madler, M.M.; Klucik, J.; Brown, C.W.; Liu, S.; Clement, F.; Subramanian, S.; Weerasekare, G.M.; et al. Heteroarotinoids inhibit head and neck cancer cell lines in vitro and in vivo through both RAR and RXR retinoic acid receptors. J. Med. Chem. 1999, 42, 4434–4445. [Google Scholar] [CrossRef] [Green Version]
- Applegate, C.C.; Lane, M.A. Role of retinoids in the prevention and treatment of colorectal cancer. World J. Gastrointest. Oncol. 2015, 7, 184–203. [Google Scholar] [CrossRef]
- Chakravarti, N.; Lotan, R.; Diwan, A.H.; Warneke, C.L.; Johnson, M.M.; Prieto, V.G. Decreased expression of retinoid receptors in melanoma: Entailment in tumorigenesis and prognosis. Clin. Cancer Res. 2007, 13, 4817–4824. [Google Scholar] [CrossRef] [Green Version]
- Garattini, E.; Bolis, M.; Garattini, S.K.; Fratelli, M.; Centritto, F.; Paroni, G.; Gianni, M.; Zanetti, A.; Pagani, A.; Fisher, J.N.; et al. Retinoids and breast cancer: From basic studies to the clinic and back again. Cancer Treat. Rev. 2014, 40, 739–749. [Google Scholar] [CrossRef]
- Cesario, R.M.; Stone, J.; Yen, W.C.; Bissonnette, R.P.; Lamph, W.W. Differentiation and growth inhibition mediated via the RXR:PPARgamma heterodimer in colon cancer. Cancer Lett. 2006, 240, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Kollipara, R.K.; Singh, D.K.; Sudderth, J.; Hu, Z.; Nguyen, H.; Wang, S.; Humphries, C.G.; Carstens, R.; Huffman, K.E.; et al. Inhibition of cancer cell proliferation by PPARgamma is mediated by a metabolic switch that increases reactive oxygen species levels. Cell Metab. 2014, 20, 650–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papi, A.; Rocchi, P.; Ferreri, A.M.; Guerra, F.; Orlandi, M. Enhanced effects of PPARgamma ligands and RXR selective retinoids in combination to inhibit migration and invasiveness in cancer cells. Oncol. Rep. 2009, 21, 1083–1089. [Google Scholar] [CrossRef] [Green Version]
- Harris, G.; Ghazallah, R.A.; Nascene, D.; Wuertz, B.; Ondrey, F.G. PPAR activation and decreased proliferation in oral carcinoma cells with 4-HPR. Otolaryngol. Head Neck Surg. 2005, 133, 695–701. [Google Scholar] [CrossRef]
- Liu, Z.; Calderon, J.I.; Zhang, Z.; Sturgis, E.M.; Spitz, M.R.; Wei, Q. Polymorphisms of vitamin D receptor gene protect against the risk of head and neck cancer. Pharm. Genom. 2005, 15, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.L.; Tsou, Y.A.; Fan, S.R.; Tsai, M.H.; Chen, H.L.; Chang, N.W.; Cheng, J.C.; Chen, C.M. Methylation-associated gene silencing of RARB in areca carcinogens induced mouse oral squamous cell carcinoma. Biomed. Res. Int. 2014, 2014, 378358. [Google Scholar] [CrossRef] [Green Version]
- Elimrani, I.; Koenekoop, J.; Dionne, S.; Marcil, V.; Delvin, E.; Levy, E.; Seidman, E.G. Vitamin D Reduces Colitis- and Inflammation-Associated Colorectal Cancer in Mice Independent of NOD2. Nutr. Cancer 2017, 69, 276–288. [Google Scholar] [CrossRef]
- Mashhadi, R.; Pourmand, G.; Kosari, F.; Mehrsai, A.; Salem, S.; Pourmand, M.R.; Alatab, S.; Khonsari, M.; Heydari, F.; Beladi, L.; et al. Role of steroid hormone receptors in formation and progression of bladder carcinoma: A case-control study. Urol. J. 2014, 11, 1968–1973. [Google Scholar]
- Fu, Y.; Li, J.; Zhang, Y. Polymorphisms in the vitamin D receptor gene and the lung cancer risk. Tumour Biol. 2014, 35, 1323–1330. [Google Scholar] [CrossRef]
- Nilsson, S.; Koehler, K.F.; Gustafsson, J.A. Development of subtype-selective oestrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 2011, 10, 778–792. [Google Scholar] [CrossRef]
- Warner, M.; Gustafsson, J.A. On estrogen, cholesterol metabolism, and breast cancer. N. Engl. J. Med. 2014, 370, 572–573. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Gustafsson, J.A. Targeting liver X receptors in cancer therapeutics. Nat. Rev. Cancer 2015, 15, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z. Targeting the androgen receptor in prostate cancer. Expert. Opin. Pharmacother. 2014, 15, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Liang, C.; Wang, H.; Chen, Y.; Silberstein, J.L.; Piana, D.; Lai, Z.; Chen, Y.; et al. Germline DNA-repair Gene Mutations and Outcomes in Men with Metastatic Castration-resistant Prostate Cancer Receiving First-line Abiraterone and Enzalutamide. Eur. Urol. 2018, 74, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Beer, T.M.; Loriot, Y.; Higano, C.S.; Armstrong, A.J.; Sternberg, C.N.; de Bono, J.S.; Tombal, B.; Parli, T.; Bhattacharya, S.; et al. Radiographic Progression-Free Survival as a Clinically Meaningful End Point in Metastatic Castration-Resistant Prostate Cancer: The PREVAIL Randomized Clinical Trial. JAMA Oncol. 2018, 4, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Sottili, M.; Mangoni, M.; Gerini, C.; Salvatore, G.; Castiglione, F.; Desideri, I.; Bonomo, P.; Meattini, I.; Greto, D.; Loi, M.; et al. Peroxisome proliferator activated receptor-gamma stimulation for prevention of 5-fluorouracil-induced oral mucositis in mice. Head Neck 2018, 40, 577–583. [Google Scholar] [CrossRef]
- Smith, M.R.; Antonarakis, E.S.; Ryan, C.J.; Berry, W.R.; Shore, N.D.; Liu, G.; Alumkal, J.J.; Higano, C.S.; Chow Maneval, E.; Bandekar, R.; et al. Phase 2 Study of the Safety and Antitumor Activity of Apalutamide (ARN-509), a Potent Androgen Receptor Antagonist, in the High-risk Nonmetastatic Castration-resistant Prostate Cancer Cohort. Eur. Urol. 2016, 70, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Jordan, V.C. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr. Relat. Cancer 2014, 21, R235–R246. [Google Scholar] [CrossRef] [Green Version]
- Gingras, I.; Gebhart, G.; de Azambuja, E.; Piccart-Gebhart, M. HER2-positive breast cancer is lost in translation: Time for patient-centered research. Nat. Rev. Clin. Oncol. 2017, 14, 669–681. [Google Scholar] [CrossRef]
- Kono, M.; Fujii, T.; Lim, B.; Karuturi, M.S.; Tripathy, D.; Ueno, N.T. Androgen Receptor Function and Androgen Receptor-Targeted Therapies in Breast Cancer: A Review. JAMA Oncol. 2017, 3, 1266–1273. [Google Scholar] [CrossRef]
- Binkhorst, L.; Mathijssen, R.H.; Jager, A.; van Gelder, T. Individualization of tamoxifen therapy: Much more than just CYP2D6 genotyping. Cancer Treat. Rev. 2015, 41, 289–299. [Google Scholar] [CrossRef] [PubMed]
- McFall, T.; McKnight, B.; Rosati, R.; Kim, S.; Huang, Y.; Viola-Villegas, N.; Ratnam, M. Progesterone receptor A promotes invasiveness and metastasis of luminal breast cancer by suppressing regulation of critical microRNAs by estrogen. J. Biol. Chem. 2018, 293, 1163–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.H.; Indran, I.R.; Tan, H.M.; Li, Y.; Zhang, Z.; Li, J.; Yong, E.L. A Dietary Medium-Chain Fatty Acid, Decanoic Acid, Inhibits Recruitment of Nur77 to the HSD3B2 Promoter In Vitro and Reverses Endocrine and Metabolic Abnormalities in a Rat Model of Polycystic Ovary Syndrome. Endocrinology 2016, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Chopra, P.; Sethi, G.; Dastidar, S.G.; Ray, A. Polo-like kinase inhibitors: An emerging opportunity for cancer therapeutics. Expert. Opin. Investig. Drugs 2010, 19, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Yu, S.; Chen, D.; Jing, C.; Wang, Z.; Ma, R.; Liu, S.; Ni, J.; Feng, J.; Wu, J. Liver X receptor agonist T0901317 reverses resistance of A549 human lung cancer cells to EGFR-TKI treatment. FEBS Open Bio 2017, 7, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.; Xie, Y.; Xiao, G.; Behrens, C.; Girard, L.; Wistuba, I.I.; Minna, J.D.; Mangelsdorf, D.J. Nuclear receptor expression defines a set of prognostic biomarkers for lung cancer. PLoS Med. 2010, 7, e1000378. [Google Scholar] [CrossRef]
- Callewaert, L.; Van Tilborgh, N.; Claessens, F. Interplay between two hormone-independent activation domains in the androgen receptor. Cancer Res. 2006, 66, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Slagsvold, T.; Kraus, I.; Bentzen, T.; Palvimo, J.; Saatcioglu, F. Mutational analysis of the androgen receptor AF-2 (activation function 2) core domain reveals functional and mechanistic differences of conserved residues compared with other nuclear receptors. Mol. Endocrinol. 2000, 14, 1603–1617. [Google Scholar] [CrossRef]
- Wilson, E.M. Analysis of interdomain interactions of the androgen receptor. Methods Mol. Biol. 2011, 776, 113–129. [Google Scholar]
- Heinlein, C.A.; Chang, C. Androgen receptor (AR) coregulators: An overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef]
- Mikkonen, L.; Pihlajamaa, P.; Sahu, B.; Zhang, F.P.; Janne, O.A. Androgen receptor and androgen-dependent gene expression in lung. Mol. Cell. Endocrinol. 2010, 317, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattern, J.; Klinga, K.; Runnebaum, B.; Volm, M. Influence of hormone therapy on human lung tumors transplanted into nude mice. Oncology 1985, 42, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, U.; Hofmann, J.; Schilli, M.; Wegmann, B.; Klotz, U.; Wedel, S.; Virmani, A.K.; Wollmer, E.; Branscheid, D.; Gazdar, A.F.; et al. Steroid-hormone receptors in cell lines and tumor biopsies of human lung cancer. Int. J. Cancer 1996, 67, 357–364. [Google Scholar] [CrossRef]
- Dou, M.; Zhu, K.; Fan, Z.; Zhang, Y.; Chen, X.; Zhou, X.; Ding, X.; Li, L.; Gu, Z.; Guo, M.; et al. Shen, P.; Wang, S. Reproductive Hormones and Their Receptors May Affect Lung Cancer. Cell. Physiol. Biochem. 2017, 44, 1425–1434. [Google Scholar] [CrossRef]
- Maasberg, M.; Rotsch, M.; Jaques, G.; Enderle-Schmidt, U.; Weehle, R.; Havemann, K. Androgen receptors, androgen-dependent proliferation, and 5 alpha-reductase activity of small-cell lung cancer cell lines. Int. J. Cancer 1989, 43, 685–691. [Google Scholar] [CrossRef]
- Lin, P.; Chang, J.T.; Ko, J.L.; Liao, S.H.; Lo, W.S. Reduction of androgen receptor expression by benzo[alpha]pyrene and 7,8-dihydro-9,10-epoxy-7,8,9,10-tetrahydrobenzo[alpha]pyrene in human lung cells. Biochem. Pharmacol. 2004, 67, 1523–1530. [Google Scholar] [CrossRef]
- Recchia, A.G.; Musti, A.M.; Lanzino, M.; Panno, M.L.; Turano, E.; Zumpano, R.; Belfiore, A.; Ando, S.; Maggiolini, M. A cross-talk between the androgen receptor and the epidermal growth factor receptor leads to p38MAPK-dependent activation of mTOR and cyclinD1 expression in prostate and lung cancer cells. Int. J. Biochem. Cell Biol. 2009, 41, 603–614. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Bharathkumar, H.; Mohan, C.D.; Ananda, H.; Fuchs, J.E.; Li, F.; Rangappa, S.; Surender, M.; Bulusu, K.C.; Girish, K.S.; Sethi, G.; et al. Microwave-assisted synthesis, characterization and cytotoxic studies of novel estrogen receptor alpha ligands towards human breast cancer cells. Bioorg. Med. Chem. Lett. 2015, 25, 1804–1807. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kar, S.; Lai, X.; Cai, W.; Arfuso, F.; Sethi, G.; Lobie, P.E.; Goh, B.C.; Lim, L.H.K.; Hartman, M.; et al. Triple negative breast cancer in Asia: An insider’s view. Cancer Treat. Rev. 2018, 62, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ahn, K.S.; Shanmugam, M.K.; Wang, H.; Shen, H.; Arfuso, F.; Chinnathambi, A.; Alharbi, S.A.; Chang, Y.; Sethi, G.; et al. Oleuropein induces apoptosis via abrogating NF-kappaB activation cascade in estrogen receptor-negative breast cancer cells. J. Cell. Biochem. 2019, 120, 4504–4513. [Google Scholar] [CrossRef] [PubMed]
- Deroo, B.J.; Buensuceso, A.V. Minireview: Estrogen receptor-beta: Mechanistic insights from recent studies. Mol. Endocrinol. 2010, 24, 1703–1714. [Google Scholar] [CrossRef]
- Haldosen, L.A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor beta in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef]
- Green, S.; Kumar, V.; Krust, A.; Walter, P.; Chambon, P. Structural and functional domains of the estrogen receptor. Cold Spring Harb. Symp. Quant. Biol. 1986, 51 Pt 2, 751–758. [Google Scholar] [CrossRef]
- Kumar, V.; Green, S.; Stack, G.; Berry, M.; Jin, J.R.; Chambon, P. Functional domains of the human estrogen receptor. Cell 1987, 51, 941–951. [Google Scholar] [CrossRef]
- Kelly, M.J.; Levin, E.R. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2001, 12, 152–156. [Google Scholar] [CrossRef]
- Pietras, R.J.; Nemere, I.; Szego, C.M. Steroid hormone receptors in target cell membranes. Endocrine 2001, 14, 417–427. [Google Scholar] [CrossRef]
- Manolagas, S.C.; Kousteni, S.; Jilka, R.L. Sex steroids and bone. Recent Prog. Horm. Res. 2002, 57, 385–409. [Google Scholar] [CrossRef]
- Fishman, J. Biological action of catechol oestrogens. J. Endocrinol. 1981, 89, 59P–65P. [Google Scholar] [PubMed]
- Yager, J.D.; Liehr, J.G. Molecular mechanisms of estrogen carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 203–232. [Google Scholar] [CrossRef] [PubMed]
- Canver, C.C.; Memoli, V.A.; Vanderveer, P.L.; Dingivan, C.A.; Mentzer, R.M., Jr. Sex hormone receptors in non-small-cell lung cancer in human beings. J. Thorac. Cardiovasc. Surg. 1994, 108, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Ollayos, C.W.; Riordan, G.P.; Rushin, J.M. Estrogen receptor detection in paraffin sections of adenocarcinoma of the colon, pancreas, and lung. Arch. Pathol. Lab. Med. 1994, 118, 630–632. [Google Scholar] [PubMed]
- Su, J.M.; Hsu, H.K.; Chang, H.; Lin, S.L.; Chang, H.C.; Huang, M.S.; Tseng, H.H. Expression of estrogen and progesterone receptors in non-small-cell lung cancer: Immunohistochemical study. Anticancer Res. 1996, 16, 3803–3806. [Google Scholar]
- Brown, R.W.; Campagna, L.B.; Dunn, J.K.; Cagle, P.T. Immunohistochemical identification of tumor markers in metastatic adenocarcinoma. A diagnostic adjunct in the determination of primary site. Am. J. Clin. Pathol. 1997, 107, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Di Nunno, L.; Larsson, L.G.; Rinehart, J.J.; Beissner, R.S. Estrogen and progesterone receptors in non-small cell lung cancer in 248 consecutive patients who underwent surgical resection. Arch. Pathol. Lab. Med. 2000, 124, 1467–1470. [Google Scholar] [CrossRef]
- Omoto, Y.; Kobayashi, Y.; Nishida, K.; Tsuchiya, E.; Eguchi, H.; Nakagawa, K.; Ishikawa, Y.; Yamori, T.; Iwase, H.; Fujii, Y.; et al. Expression, function, and clinical implications of the estrogen receptor beta in human lung cancers. Biochem. Biophys. Res. Commun. 2001, 285, 340–347. [Google Scholar] [CrossRef]
- Gershtein, E.S.; Smirnova, K.D.; Polotskii, B.E.; Kuz’mina, Z.V.; Davydov, M.I. Steroid hormone receptors in lung cancer tissue. Vopr. Onkol. 1990, 36, 1439–1442. [Google Scholar]
- Yang, M.H. Estrogen receptor in female lung carcinoma. Zhonghua Jie He He Hu Xi Za Zhi 1992, 15, 138–140. [Google Scholar]
- Chen, X.Q.; Zheng, L.X.; Li, Z.Y.; Lin, T.Y. Clinicopathological significance of oestrogen receptor expression in non-small cell lung cancer. J. Int. Med. Res. 2017, 45, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Raso, M.G.; Behrens, C.; Herynk, M.H.; Liu, S.; Prudkin, L.; Ozburn, N.C.; Woods, D.M.; Tang, X.; Mehran, R.J.; Moran, C.; et al. Immunohistochemical expression of estrogen and progesterone receptors identifies a subset of NSCLCs and correlates with EGFR mutation. Clin. Cancer Res. 2009, 15, 5359–5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, H.; Ishii, A.; Washiya, K.; Konno, T.; Kon, H.; Yamaya, C.; Ono, I.; Minamiya, Y.; Ogawa, J. Estrogen receptor alpha and beta are prognostic factors in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 5084–5089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stabile, L.P.; Davis, A.L.; Gubish, C.T.; Hopkins, T.M.; Luketich, J.D.; Christie, N.; Finkelstein, S.; Siegfried, J.M. Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor alpha and beta and show biological responses to estrogen. Cancer Res. 2002, 62, 2141–2150. [Google Scholar]
- Ishibashi, H.; Suzuki, T.; Suzuki, S.; Niikawa, H.; Lu, L.; Miki, Y.; Moriya, T.; Hayashi, S.; Handa, M.; Kondo, T.; et al. Progesterone receptor in non-small cell lung cancer--a potent prognostic factor and possible target for endocrine therapy. Cancer Res. 2005, 65, 6450–6458. [Google Scholar] [CrossRef] [Green Version]
- Kawai, H.; Ishii, A.; Washiya, K.; Konno, T.; Kon, H.; Yamaya, C.; Ono, I.; Ogawa, J. Combined overexpression of EGFR and estrogen receptor alpha correlates with a poor outcome in lung cancer. Anticancer Res. 2005, 25, 4693–4698. [Google Scholar]
- Wu, C.T.; Chang, Y.L.; Shih, J.Y.; Lee, Y.C. The significance of estrogen receptor beta in 301 surgically treated non-small cell lung cancers. J. Thorac. Cardiovasc. Surg. 2005, 130, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Fasco, M.J.; Hurteau, G.J.; Spivack, S.D. Gender-dependent expression of alpha and beta estrogen receptors in human nontumor and tumor lung tissue. Mol. Cell. Endocrinol. 2002, 188, 125–140. [Google Scholar] [CrossRef]
- Abe, K.; Miki, Y.; Ono, K.; Mori, M.; Kakinuma, H.; Kou, Y.; Kudo, N.; Koguchi, M.; Niikawa, H.; Suzuki, S.; et al. Highly concordant coexpression of aromatase and estrogen receptor beta in non-small cell lung cancer. Hum. Pathol. 2010, 41, 190–198. [Google Scholar] [CrossRef]
- Wu, C.T.; Chang, Y.L.; Lee, Y.C. Expression of the estrogen receptor beta in 37 surgically treated pulmonary sclerosing hemangiomas in comparison with non-small cell lung carcinomas. Hum. Pathol. 2005, 36, 1108–1112. [Google Scholar] [CrossRef]
- Schwartz, A.G.; Prysak, G.M.; Murphy, V.; Lonardo, F.; Pass, H.; Schwartz, J.; Brooks, S. Nuclear estrogen receptor beta in lung cancer: Expression and survival differences by sex. Clin. Cancer Res. 2005, 11, 7280–7287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Zhang, Z.; Liao, Y.; Liu, C.; Fan, S.; Wei, X.; Ai, B.; Xiong, J. 17beta-estradiol upregulates IL6 expression through the ERbeta pathway to promote lung adenocarcinoma progression. J. Exp. Clin. Cancer Res. 2018, 37, 133. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Dacic, S.; Land, S.R.; Lenzner, D.E.; Dhir, R.; Acquafondata, M.; Landreneau, R.J.; Grandis, J.R.; Siegfried, J.M. Combined analysis of estrogen receptor beta-1 and progesterone receptor expression identifies lung cancer patients with poor outcome. Clin. Cancer Res. 2011, 17, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Liu, T.; Ji, H.; Yang, S.; Sui, J.; Yang, W.S.; Liang, G.Y.; Zhang, X.M. Expression pattern of estrogen receptor beta and its correlation with multidrug resistance in non-small cell lung cancer. Neoplasma 2019, 66, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xin, B.; Pang, H.; Han, L.; Shen, W.; Zhao, Z.; Duan, L.; Cao, P.; Liu, L.; Zhang, H. Downregulation of estrogen receptor beta inhibits lung adenocarcinoma cell growth. Oncol. Rep. 2019, 41, 2967–2974. [Google Scholar]
- Fu, S.; Liu, C.; Huang, Q.; Fan, S.; Tang, H.; Fu, X.; Ai, B.; Liao, Y.; Chu, Q. Estrogen receptor beta1 activation accelerates resistance to epidermal growth factor receptor-tyrosine kinase inhibitors in non-small cell lung cancer. Oncol. Rep. 2018, 39, 1313–1321. [Google Scholar]
- Zhang, G.; Yanamala, N.; Lathrop, K.L.; Zhang, L.; Klein-Seetharaman, J.; Srinivas, H. Ligand-independent antiapoptotic function of estrogen receptor-beta in lung cancer cells. Mol. Endocrinol. 2010, 24, 1737–1747. [Google Scholar] [CrossRef] [Green Version]
- Pietras, R.J.; Marquez, D.C.; Chen, H.W.; Tsai, E.; Weinberg, O.; Fishbein, M. Estrogen and growth factor receptor interactions in human breast and non-small cell lung cancer cells. Steroids 2005, 70, 372–381. [Google Scholar] [CrossRef]
- Lai, J.C.; Cheng, Y.W.; Chiou, H.L.; Wu, M.F.; Chen, C.Y.; Lee, H. Gender difference in estrogen receptor alpha promoter hypermethylation and its prognostic value in non-small cell lung cancer. Int. J. Cancer 2005, 117, 974–980. [Google Scholar] [CrossRef]
- Stabile, L.P.; Lyker, J.S.; Gubish, C.T.; Zhang, W.; Grandis, J.R.; Siegfried, J.M. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res. 2005, 65, 1459–1470. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Liao, Y.; Qiu, W.; Huang, Q.; Xiao, H.; Liu, C.; Li, D.; Cao, X.; Li, L.; Liang, H.; et al. Estrogen promotes the metastasis of nonsmall cell lung cancer via estrogen receptor beta by upregulation of Tolllike receptor 4 and activation of the myd88/NFkappaB/MMP2 pathway. Oncol. Rep. 2020, 43, 2105–2119. [Google Scholar]
- Marquez-Garban, D.C.; Chen, H.W.; Fishbein, M.C.; Goodglick, L.; Pietras, R.J. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids 2007, 72, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yuan, Y.; Sun, J.; Gao, W.; Shu, Y.Q. Combined tamoxifen and gefitinib in non-small cell lung cancer shows antiproliferative effects. Biomed. Pharmacother. 2010, 64, 88–92. [Google Scholar] [CrossRef]
- Sikka, S.; Chen, L.; Sethi, G.; Kumar, A.P. Targeting PPARgamma Signaling Cascade for the Prevention and Treatment of Prostate Cancer. PPAR Res. 2012, 2012, 968040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, V.M.; Wong, F.S.; Ramachandran, L.; Sethi, G.; Kumar, A.P.; Yap, C.W. Identification of novel peroxisome proliferator-activated receptor-gamma (PPARgamma) agonists using molecular modeling method. J. Comput. Aided Mol. Des. 2014, 28, 1143–1151. [Google Scholar] [CrossRef]
- Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 1995, 270, 1354–1357. [Google Scholar] [CrossRef]
- Gelman, L.; Zhou, G.; Fajas, L.; Raspe, E.; Fruchart, J.C.; Auwerx, J. p300 interacts with the N- and C-terminal part of PPARgamma2 in a ligand-independent and -dependent manner, respectively. J. Biol. Chem. 1999, 274, 7681–7688. [Google Scholar] [CrossRef] [Green Version]
- Heinlein, C.A.; Ting, H.J.; Yeh, S.; Chang, C. Identification of ARA70 as a ligand-enhanced coactivator for the peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 1999, 274, 16147–16152. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, L.; Manu, K.A.; Shanmugam, M.K.; Li, F.; Siveen, K.S.; Vali, S.; Kapoor, S.; Abbasi, T.; Surana, R.; Smoot, D.T.; et al. Isorhamnetin inhibits proliferation and invasion and induces apoptosis through the modulation of peroxisome proliferator-activated receptor gamma activation pathway in gastric cancer. J. Biol. Chem. 2012, 287, 38028–38040. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yuan, Y.; Kar, S.; Kanchi, M.M.; Arora, S.; Kim, J.E.; Koh, P.F.; Yousef, E.; Samy, R.P.; Shanmugam, M.K.; et al. PPARgamma Ligand-induced Annexin A1 Expression Determines Chemotherapy Response via Deubiquitination of Death Domain Kinase RIP in Triple-negative Breast Cancers. Mol. Cancer Ther. 2017, 16, 2528–2542. [Google Scholar] [CrossRef] [Green Version]
- Cavailles, V.; Dauvois, S.; L’Horset, F.; Lopez, G.; Hoare, S.; Kushner, P.J.; Parker, M.G. Nuclear factor RIP140 modulates transcriptional activation by the estrogen receptor. EMBO J. 1995, 14, 3741–3751. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.D.; Umesono, K.; Evans, R.M. SMRT isoforms mediate repression and anti-repression of nuclear receptor heterodimers. Proc. Natl. Acad. Sci. USA 1996, 93, 7567–7571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzel, T.; Lavinsky, R.M.; Mullen, T.M.; Soderstrom, M.; Laherty, C.D.; Torchia, J.; Yang, W.M.; Brard, G.; Ngo, S.D.; Davie, J.R.; et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature 1997, 387, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Farioli-Vecchioli, S.; Ceru, M.P. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef]
- Heneka, M.T.; Landreth, G.E. PPARs in the brain. Biochim. Biophys. Acta 2007, 1771, 1031–1045. [Google Scholar] [CrossRef]
- Neschen, S.; Morino, K.; Dong, J.; Wang-Fischer, Y.; Cline, G.W.; Romanelli, A.J.; Rossbacher, J.C.; Moore, I.K.; Regittnig, W.; Munoz, D.S.; et al. n-3 Fatty acids preserve insulin sensitivity in vivo in a peroxisome proliferator-activated receptor-alpha-dependent manner. Diabetes 2007, 56, 1034–1041. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [Green Version]
- Ferre, P. The biology of peroxisome proliferator-activated receptors: Relationship with lipid metabolism and insulin sensitivity. Diabetes 2004, 53 (Suppl. S1), S43–S50. [Google Scholar] [CrossRef] [Green Version]
- Lehrke, M.; Lazar, M.A. The many faces of PPARgamma. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Medina-Gomez, G.; Gray, S.L.; Yetukuri, L.; Shimomura, K.; Virtue, S.; Campbell, M.; Curtis, R.K.; Jimenez-Linan, M.; Blount, M.; Yeo, G.S.; et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 2007, 3, e64. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Xiong Chen, Z.; Rane, G.; Satendra Singh, S.; Choo, Z.; Wang, C.; Yuan, Y.; Zea Tan, T.; Arfuso, F.; Yap, C.T.; et al. Wanted DEAD/H or Alive: Helicases Winding Up in Cancers. J. Natl. Cancer Inst. 2017, 109, djw278. [Google Scholar] [CrossRef] [PubMed]
- IJpenberg, A.; Jeannin, E.; Wahli, W.; Desvergne, B. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J. Biol. Chem. 1997, 272, 20108–20117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.G.; Shao, G.; Heyman, R.A. Transactivation by retinoid X receptor-peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimers: Intermolecular synergy requires only the PPARgamma hormone-dependent activation function. Mol. Cell. Biol. 1998, 18, 3483–3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zou, P.; Bai, M.; Jin, Y.; Tao, X. Peroxisome proliferator-activated receptor-gamma activated by ligands can inhibit human lung cancer cell growth through induction of apoptosis. J. Huazhong Univ. Sci. Technol. Med. Sci. 2003, 23, 138–140. [Google Scholar]
- Sasaki, H.; Tanahashi, M.; Yukiue, H.; Moiriyama, S.; Kobayashi, Y.; Nakashima, Y.; Kaji, M.; Kiriyama, M.; Fukai, I.; Yamakawa, Y.; et al. Decreased perioxisome proliferator-activated receptor gamma gene expression was correlated with poor prognosis in patients with lung cancer. Lung Cancer 2002, 36, 71–76. [Google Scholar] [CrossRef]
- Keshamouni, V.G.; Reddy, R.C.; Arenberg, D.A.; Joel, B.; Thannickal, V.J.; Kalemkerian, G.P.; Standiford, T.J. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor progression in non-small-cell lung cancer. Oncogene 2004, 23, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.W.; Chen, G.G.; Xu, H.; Yip, J.H.; Chak, E.C.; Mok, T.S.; Yim, A.P. Differential expression of inducible nitric oxide synthase and peroxisome proliferator-activated receptor gamma in non-small cell lung carcinoma. Eur. J. Cancer 2003, 39, 1296–1301. [Google Scholar] [CrossRef]
- Tsubouchi, Y.; Sano, H.; Kawahito, Y.; Mukai, S.; Yamada, R.; Kohno, M.; Inoue, K.; Hla, T.; Kondo, M. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-gamma agonists through induction of apoptosis. Biochem. Biophys. Res. Commun. 2000, 270, 400–405. [Google Scholar] [CrossRef]
- Theocharis, S.; Kanelli, H.; Politi, E.; Margeli, A.; Karkandaris, C.; Philippides, T.; Koutselinis, A. Expression of peroxisome proliferator activated receptor-gamma in non-small cell lung carcinoma: Correlation with histological type and grade. Lung Cancer 2002, 36, 249–255. [Google Scholar] [CrossRef]
- Li, M.Y.; Yuan, H.; Ma, L.T.; Kong, A.W.; Hsin, M.K.; Yip, J.H.; Underwood, M.J.; Chen, G.G. Roles of peroxisome proliferator-activated receptor-alpha and -gamma in the development of non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 2010, 43, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sato, M.; Choi, J.W.; Kim, H.W.; Yeh, B.I.; Larsen, J.E.; Minna, J.D.; Cha, J.H.; Jeong, Y. Nuclear Receptor Expression and Function in Human Lung Cancer Pathogenesis. PLoS ONE 2015, 10, e0134842. [Google Scholar] [CrossRef] [PubMed]
- Pedchenko, T.V.; Gonzalez, A.L.; Wang, D.; DuBois, R.N.; Massion, P.P. Peroxisome proliferator-activated receptor beta/delta expression and activation in lung cancer. Am. J. Respir. Cell Mol. Biol. 2008, 39, 689–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Borland, M.G.; Zhu, B.; Sharma, A.K.; Amin, S.; El-Bayoumy, K.; Gonzalez, F.J.; Peters, J.M. Effect of ligand activation of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) in human lung cancer cell lines. Toxicology 2008, 254, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Ritzenthaler, J.D.; Roman, J.; Han, S. PPARbeta/delta agonist increases the expression of PGE2 receptor subtype EP4 in human lung carcinoma cells. Methods Mol. Biol. 2009, 512, 309–323. [Google Scholar] [PubMed]
- Zhang, M.; Zou, P.; Bai, M.; Tao, X.N.; Jin, Y.; Guo, R. Apoptosis of human lung cancer cells induced by activated peroxisome proliferator-activated receptor-gamma and its mechanism. Zhonghua Yi Xue Za Zhi 2003, 83, 1169–1172. [Google Scholar] [PubMed]
- Hegele, A.; Heidenreich, A.; Kropf, J.; von Knobloch, R.; Varga, Z.; Hofmann, R.; Olbert, P. Plasma levels of cellular fibronectin in patients with localized and metastatic renal cell carcinoma. Tumour Biol. 2004, 25, 111–116. [Google Scholar] [CrossRef]
- Hu, M.; Carles-Kinch, K.L.; Zelinski, D.P.; Kinch, M.S. EphA2 induction of fibronectin creates a permissive microenvironment for malignant cells. Mol. Cancer Res. 2004, 2, 533–540. [Google Scholar] [CrossRef]
- Jakowlew, S.B.; Mariano, J.M.; You, L.; Mathias, A. Differential regulation of protease and extracellular matrix protein expression by transforming growth factor-beta 1 in non-small cell lung cancer cells and normal human bronchial epithelial cells. Biochim. Biophys. Acta 1997, 1353, 157–170. [Google Scholar] [CrossRef]
- Han, J.Y.; Kim, H.S.; Lee, S.H.; Park, W.S.; Lee, J.Y.; Yoo, N.J. Immunohistochemical expression of integrins and extracellular matrix proteins in non-small cell lung cancer: Correlation with lymph node metastasis. Lung Cancer 2003, 41, 65–70. [Google Scholar] [CrossRef]
- Rintoul, R.C.; Sethi, T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Clin. Sci. (Lond) 2002, 102, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Ritzenthaler, J.D.; Rivera, H.N.; Roman, J. Peroxisome proliferator-activated receptor-gamma ligands suppress fibronectin gene expression in human lung carcinoma cells: Involvement of both CRE and Sp1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L419–L428. [Google Scholar] [CrossRef] [PubMed]
- Wick, M.; Hurteau, G.; Dessev, C.; Chan, D.; Geraci, M.W.; Winn, R.A.; Heasley, L.E.; Nemenoff, R.A. Peroxisome proliferator-activated receptor-gamma is a target of nonsteroidal anti-inflammatory drugs mediating cyclooxygenase-independent inhibition of lung cancer cell growth. Mol. Pharmacol. 2002, 62, 1207–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bren-Mattison, Y.; Van Putten, V.; Chan, D.; Winn, R.; Geraci, M.W.; Nemenoff, R.A. Peroxisome proliferator-activated receptor-gamma (PPAR(gamma)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC). Oncogene 2005, 24, 1412–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Toyoda, M.; Hoshino, H.; Monden, T.; Yamada, M.; Shimizu, H.; Miyamoto, K.; Mori, M. Activation of peroxisome proliferator-activated receptor-gamma stimulates the growth arrest and DNA-damage inducible 153 gene in non-small cell lung carcinoma cells. Oncogene 2002, 21, 2171–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCicco, K.L.; Tanaka, T.; Andreola, F.; De Luca, L.M. The effect of thalidomide on non-small cell lung cancer (NSCLC) cell lines: Possible involvement in the PPARgamma pathway. Carcinogenesis 2004, 25, 1805–1812. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Lee, T.W.; Mok, T.S.; Warner, T.D.; Yim, A.P.; Chen, G.G. Activation of peroxisome proliferator-activated receptor-gamma by troglitazone (TGZ) inhibits human lung cell growth. J. Cell. Biochem. 2005, 96, 760–774. [Google Scholar] [CrossRef]
- Han, S.; Roman, J. Suppression of prostaglandin E2 receptor subtype EP2 by PPARgamma ligands inhibits human lung carcinoma cell growth. Biochem. Biophys. Res. Commun. 2004, 314, 1093–1099. [Google Scholar] [CrossRef]
- Han, S.; Sidell, N.; Fisher, P.B.; Roman, J. Up-regulation of p21 gene expression by peroxisome proliferator-activated receptor gamma in human lung carcinoma cells. Clin. Cancer Res. 2004, 10, 1911–1919. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Lee, T.W.; Yim, A.P.; Mok, T.S.; Chen, G.G. Apoptosis induced by troglitazone is both peroxisome proliferator-activated receptor-gamma- and ERK-dependent in human non-small lung cancer cells. J. Cell. Physiol. 2006, 209, 428–438. [Google Scholar] [CrossRef]
- Lee, S.Y.; Hur, G.Y.; Jung, K.H.; Jung, H.C.; Lee, S.Y.; Kim, J.H.; Shin, C.; Shim, J.J.; In, K.H.; Kang, K.H.; et al. PPAR-gamma agonist increase gefitinib’s antitumor activity through PTEN expression. Lung Cancer 2006, 51, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Winn, R.A.; Van Scoyk, M.; Hammond, M.; Rodriguez, K.; Crossno, J.T., Jr.; Heasley, L.E.; Nemenoff, R.A. Antitumorigenic effect of Wnt 7a and Fzd 9 in non-small cell lung cancer cells is mediated through ERK-5-dependent activation of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2006, 281, 26943–26950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulzele, S.V.; Chatterjee, A.; Shaik, M.S.; Jackson, T.; Ichite, N.; Singh, M. 15-Deoxy-Delta12,14-prostaglandin J2 enhances docetaxel anti-tumor activity against A549 and H460 non-small-cell lung cancer cell lines and xenograft tumors. Anticancer Drugs 2007, 18, 65–78. [Google Scholar] [PubMed]
- Kim, K.Y.; Ahn, J.H.; Cheon, H.G. Apoptotic action of peroxisome proliferator-activated receptor-gamma activation in human non small-cell lung cancer is mediated via proline oxidase-induced reactive oxygen species formation. Mol. Pharmacol. 2007, 72, 674–685. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, T.; Motomura, W.; Tanno, S.; Kumei, S.; Yoshizaki, Y.; Tanno, S.; Okumura, T. Thiazolidinediones enhance vascular endothelial growth factor expression and induce cell growth inhibition in non-small-cell lung cancer cells. J. Exp. Clin. Cancer Res. 2010, 29, 22. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Y.; Wong, C. Oestrogen-related receptor alpha inverse agonist XCT-790 arrests A549 lung cancer cell population growth by inducing mitochondrial reactive oxygen species production. Cell Prolif. 2010, 43, 103–113. [Google Scholar] [CrossRef]
- Ciaramella, V.; Sasso, F.C.; Di Liello, R.; Corte, C.M.D.; Barra, G.; Viscardi, G.; Esposito, G.; Sparano, F.; Troiani, T.; Martinelli, E.; et al. Activity and molecular targets of pioglitazone via blockade of proliferation, invasiveness and bioenergetics in human NSCLC. J. Exp. Clin. Cancer Res. 2019, 38, 178. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, H.; Xing, L. Influence of ciglitazone on A549 cells growth in vitro and in vivo and mechanism. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 36–39. [Google Scholar]
- Kim, T.W.; Hong, D.W.; Hong, S.H. CB13, a novel PPARgamma ligand, overcomes radio-resistance via ROS generation and ER stress in human non-small cell lung cancer. Cell Death Dis. 2020, 11, 848. [Google Scholar] [CrossRef]
- Kim, T.W.; Hong, D.W.; Park, J.W.; Hong, S.H. CB11, a novel purine-based PPAR ligand, overcomes radio-resistance by regulating ATM signalling and EMT in human non-small-cell lung cancer cells. Br. J. Cancer 2020, 123, 1737–1748. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Skora, B.; Kryshchyshyn-Dylevych, A.; Kaminskyy, D.; Khyluk, D.; Lesyk, R. 4-thiazolidinone-based derivatives rosiglitazone and pioglitazone affect the expression of antioxidant enzymes in different human cell lines. Biomed. Pharmacother. 2021, 139, 111684. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Hong, D.W.; Kang, C.M.; Hong, S.H. A novel PPAR ligand, PPZ023, overcomes radioresistance via ER stress and cell death in human non-small-cell lung cancer cells. Exp. Mol. Med. 2020, 52, 1730–1743. [Google Scholar] [CrossRef] [PubMed]
- James, S.Y.; Lin, F.; Kolluri, S.K.; Dawson, M.I.; Zhang, X.K. Regulation of retinoic acid receptor beta expression by peroxisome proliferator-activated receptor gamma ligands in cancer cells. Cancer Res. 2003, 63, 3531–3538. [Google Scholar] [PubMed]
- Fukumoto, K.; Yano, Y.; Virgona, N.; Hagiwara, H.; Sato, H.; Senba, H.; Suzuki, K.; Asano, R.; Yamada, K.; Yano, T. Peroxisome proliferator-activated receptor delta as a molecular target to regulate lung cancer cell growth. FEBS Lett. 2005, 579, 3829–3836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaveri, N.T.; Sato, B.G.; Jiang, F.; Calaoagan, J.; Laderoute, K.R.; Murphy, B.J. A novel peroxisome proliferator-activated receptor delta antagonist, SR13904, has anti-proliferative activity in human cancer cells. Cancer Biol. Ther. 2009, 8, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eifert, C.; Sangster-Guity, N.; Yu, L.M.; Chittur, S.V.; Perez, A.V.; Tine, J.A.; McCormick, P.J. Global gene expression profiles associated with retinoic acid-induced differentiation of embryonal carcinoma cells. Mol. Reprod. Dev. 2006, 73, 796–824. [Google Scholar] [CrossRef]
- Delacroix, L.; Moutier, E.; Altobelli, G.; Legras, S.; Poch, O.; Choukrallah, M.A.; Bertin, I.; Jost, B.; Davidson, I. Cell-specific interaction of retinoic acid receptors with target genes in mouse embryonic fibroblasts and embryonic stem cells. Mol. Cell. Biol. 2010, 30, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoid nuclear receptors: Lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 451–480. [Google Scholar] [CrossRef]
- Niederreither, K.; Dolle, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef]
- Dolle, P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Gudas, L.J. Gene expression profiling elucidates a specific role for RARgamma in the retinoic acid-induced differentiation of F9 teratocarcinoma stem cells. Biochem. Pharmacol. 2008, 75, 1129–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette-Egly, C. Dynamic combinatorial networks in nuclear receptor-mediated transcription. J. Biol. Chem. 2005, 280, 32565–32568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette-Egly, C.; Germain, P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e005. [Google Scholar] [CrossRef]
- Germain, P.; Chambon, P.; Eichele, G.; Evans, R.M.; Lazar, M.A.; Leid, M.; De Lera, A.R.; Lotan, R.; Mangelsdorf, D.J.; Gronemeyer, H. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol. Rev. 2006, 58, 760–772. [Google Scholar] [CrossRef] [Green Version]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [CrossRef]
- Bushue, N.; Wan, Y.J. Retinoid pathway and cancer therapeutics. Adv. Drug Deliv. Rev. 2010, 62, 1285–1298. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.H.; Gudas, L.J. Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol. 2011, 6, 345–364. [Google Scholar] [CrossRef]
- Freemantle, S.J.; Spinella, M.J.; Dmitrovsky, E. Retinoids in cancer therapy and chemoprevention: Promise meets resistance. Oncogene 2003, 22, 7305–7315. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lee, M.O.; Wang, H.G.; Li, Y.; Hashimoto, Y.; Klaus, M.; Reed, J.C.; Zhang, X. Retinoic acid receptor beta mediates the growth-inhibitory effect of retinoic acid by promoting apoptosis in human breast cancer cells. Mol. Cell. Biol. 1996, 16, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Gebert, J.F.; Moghal, N.; Frangioni, J.V.; Sugarbaker, D.J.; Neel, B.G. High frequency of retinoic acid receptor beta abnormalities in human lung cancer. Oncogene 1991, 6, 1859–1868. [Google Scholar] [PubMed]
- Houle, B.; Rochette-Egly, C.; Bradley, W.E. Tumor-suppressive effect of the retinoic acid receptor beta in human epidermoid lung cancer cells. Proc. Natl. Acad. Sci. USA 1993, 90, 985–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.K.; Liu, Y.; Lee, M.O.; Pfahl, M. A specific defect in the retinoic acid response associated with human lung cancer cell lines. Cancer Res. 1994, 54, 5663–5669. [Google Scholar] [PubMed]
- Xu, X.C.; Sozzi, G.; Lee, J.S.; Lee, J.J.; Pastorino, U.; Pilotti, S.; Kurie, J.M.; Hong, W.K.; Lotan, R. Suppression of retinoic acid receptor beta in non-small-cell lung cancer in vivo: Implications for lung cancer development. J. Natl. Cancer Inst. 1997, 89, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Picard, E.; Seguin, C.; Monhoven, N.; Rochette-Egly, C.; Siat, J.; Borrelly, J.; Martinet, Y.; Martinet, N.; Vignaud, J.M. Expression of retinoid receptor genes and proteins in non-small-cell lung cancer. J. Natl. Cancer Inst. 1999, 91, 1059–1066. [Google Scholar] [CrossRef] [Green Version]
- Higashimoto, Y.; Ohata, M.; Nishio, K.; Iwamoto, Y.; Fujimoto, H.; Uetani, K.; Suruda, T.; Nakamura, Y.; Funasako, M.; Saijo, N. 1 alpha, 25-dihydroxyvitamin D3 and all-trans-retinoic acid inhibit the growth of a lung cancer cell line. Anticancer Res. 1996, 16, (5A), 2653–2659. [Google Scholar]
- Wan, H.; Dawson, M.I.; Hong, W.K.; Lotan, R. Enhancement of Calu-1 human lung carcinoma cell growth in serum-free medium by retinoids: Dependence on AP-1 activation, but not on retinoid response element activation. Oncogene 1997, 15, 2109–2118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.X.; Mills, K.J.; Dawson, M.I.; Collins, S.J.; Jetten, A.M. Evidence for the involvement of retinoic acid receptor RAR alpha-dependent signaling pathway in the induction of tissue transglutaminase and apoptosis by retinoids. J. Biol. Chem. 1995, 270, 6022–6029. [Google Scholar] [CrossRef] [Green Version]
- Inui, N.; Sasaki, S.; Suda, T.; Chida, K.; Nakamura, H. The loss of retinoic acid receptor alpha, beta and alcohol dehydrogenase3 expression in non-small cell lung cancer. Respirology 2003, 8, 302–309. [Google Scholar] [CrossRef]
- Brabender, J.; Metzger, R.; Salonga, D.; Danenberg, K.D.; Danenberg, P.V.; Holscher, A.H.; Schneider, P.M. Comprehensive expression analysis of retinoic acid receptors and retinoid X receptors in non-small cell lung cancer: Implications for tumor development and prognosis. Carcinogenesis 2005, 26, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Muniz-Hernandez, S.; Huerta-Yepez, S.; Hernandez-Pedro, N.; Ramirez-Tirado, L.A.; Aviles-Salas, A.; Maldonado, A.; Hernandez-Cueto, D.; Baay-Guzman, G.; Arrieta, O. Association between nuclear expression of retinoic acid receptor alpha and beta and clinicopathological features and prognosis of advanced non-small cell lung cancer. Int. J. Clin. Oncol. 2016, 21, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, E.S.; Zinovieva, O.L.; Oparina, N.Y.; Prokofjeva, M.M.; Spirin, P.V.; Favorskaya, I.A.; Zborovskaya, I.B.; Lisitsyn, N.A.; Prassolov, V.S.; Mashkova, T.D. Abnormal expression of genes that regulate retinoid metabolism and signaling in non-small-cell lung cancer. Mol. Biol. (Mosk) 2016, 50, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Geradts, J.; Chen, J.Y.; Russell, E.K.; Yankaskas, J.R.; Nieves, L.; Minna, J.D. Human lung cancer cell lines exhibit resistance to retinoic acid treatment. Cell Growth Differ. 1993, 4, 799–809. [Google Scholar] [PubMed]
- Petty, W.J.; Li, N.; Biddle, A.; Bounds, R.; Nitkin, C.; Ma, Y.; Dragnev, K.H.; Freemantle, S.J.; Dmitrovsky, E. A novel retinoic acid receptor beta isoform and retinoid resistance in lung carcinogenesis. J. Natl. Cancer Inst. 2005, 97, 1645–1651. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.S.; Chung, J.H.; Shin, D.H.; Chung, K.Y.; Kim, Y.S.; Chang, J.; Kim, S.K.; Kim, S.K. Retinoic acid receptor-beta expression in stage I non-small cell lung cancer and adjacent normal appearing bronchial epithelium. Yonsei Med. J. 2004, 45, 435–442. [Google Scholar] [CrossRef]
- Xu, X.C.; Lee, J.S.; Lee, J.J.; Morice, R.C.; Liu, X.; Lippman, S.M.; Hong, W.K.; Lotan, R. Nuclear retinoid acid receptor beta in bronchial epithelium of smokers before and during chemoprevention. J. Natl. Cancer Inst. 1999, 91, 1317–1321. [Google Scholar] [CrossRef] [Green Version]
- Avis, I.; Mathias, A.; Unsworth, E.J.; Miller, M.J.; Cuttitta, F.; Mulshine, J.L.; Jakowlew, S.B. Analysis of small cell lung cancer cell growth inhibition by 13-cis-retinoic acid: Importance of bioavailability. Cell Growth Differ. 1995, 6, 485–492. [Google Scholar]
- Ayoub, J.; Jean-Francois, R.; Cormier, Y.; Meyer, D.; Ying, Y.; Major, P.; Desjardins, C.; Bradley, W.E. Placebo-controlled trial of 13-cis-retinoic acid activity on retinoic acid receptor-beta expression in a population at high risk: Implications for chemoprevention of lung cancer. J. Clin. Oncol. 1999, 17, 3546–3552. [Google Scholar] [CrossRef]
- Sun, S.Y.; Yue, P.; Shroot, B.; Hong, W.K.; Lotan, R. Induction of apoptosis in human non-small cell lung carcinoma cells by the novel synthetic retinoid CD437. J. Cell. Physiol. 1997, 173, 279–284. [Google Scholar] [CrossRef]
- Nervi, C.; Vollberg, T.M.; George, M.D.; Zelent, A.; Chambon, P.; Jetten, A.M. Expression of nuclear retinoic acid receptors in normal tracheobronchial cells and in lung carcinoma cells. Exp. Cell Res. 1991, 195, 163–170. [Google Scholar] [CrossRef]
- Dahl, A.R.; Grossi, I.M.; Houchens, D.P.; Scovell, L.J.; Placke, M.E.; Imondi, A.R.; Stoner, G.D.; De Luca, L.M.; Wang, D.; Mulshine, J.L. Inhaled isotretinoin (13-cis retinoic acid) is an effective lung cancer chemopreventive agent in A/J mice at low doses: A pilot study. Clin. Cancer Res. 2000, 6, 3015–3024. [Google Scholar] [PubMed]
- Hsu, S.L.; Hsu, J.W.; Liu, M.C.; Chen, L.Y.; Chang, C.D. Retinoic acid-mediated G1 arrest is associated with induction of p27(Kip1) and inhibition of cyclin-dependent kinase 3 in human lung squamous carcinoma CH27 cells. Exp. Cell Res. 2000, 258, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.K.; Scott, F.M.; Cuttitta, F.; Weber, E.; Kalemkerian, G.P.; Nelkin, B.D.; Mabry, M. Induction of gastrin releasing peptide by all-trans retinoic acid in small cell lung cancer cells. Oncol. Rep. 1998, 5, 497–501. [Google Scholar] [CrossRef]
- Sun, S.Y.; Yue, P.; Shroot, B.; Hong, W.K.; Lotan, R. Implication of c-Myc in apoptosis induced by the retinoid CD437 in human lung carcinoma cells. Oncogene 1999, 18, 3894–3901. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.Y.; Yue, P.; Wu, G.S.; El-Deiry, W.S.; Shroot, B.; Hong, W.K.; Lotan, R. Mechanisms of apoptosis induced by the synthetic retinoid CD437 in human non-small cell lung carcinoma cells. Oncogene 1999, 18, 2357–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Wu, M.; Levi, G.; Ferrari, N. Inhibition of cancer cell growth by all-trans retinoic acid and its analog N-(4-hydroxyphenyl) retinamide: A possible mechanism of action via regulation of retinoid receptors expression. Int. J. Cancer 1998, 78, 248–254. [Google Scholar] [CrossRef]
- Fang, K.; Mukhopadhyay, T.; Shih, S.H. Retinoic Acid Modulates Epidermal Growth Factor Receptor Expression in Human Lung Epithelial Cancer Cells. J. Biomed. Sci. 1995, 2, 256–262. [Google Scholar] [CrossRef]
- Mukhopadhyay, T.; Fang, K.; Maxwell, S. Posttranscriptional regulation of epidermal growth-factor receptor expression by retinoic Acid in a human lung epithelial-cell line. Oncol. Rep. 1995, 2, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Hajnal, A.; Klemenz, R.; Schafer, R. Subtraction cloning of H-rev107, a gene specifically expressed in H-ras resistant fibroblasts. Oncogene 1994, 9, 479–490. [Google Scholar]
- Higuchi, E.; Chandraratna, R.A.; Hong, W.K.; Lotan, R. Induction of TIG3, a putative class II tumor suppressor gene, by retinoic acid in head and neck and lung carcinoma cells and its association with suppression of the transformed phenotype. Oncogene 2003, 22, 4627–4635. [Google Scholar] [CrossRef]
- Lian, F.; Hu, K.Q.; Russell, R.M.; Wang, X.D. Beta-cryptoxanthin suppresses the growth of immortalized human bronchial epithelial cells and non-small-cell lung cancer cells and up-regulates retinoic acid receptor beta expression. Int. J. Cancer 2006, 119, 2084–2089. [Google Scholar] [CrossRef] [PubMed]
- Kalitin, N.N.; Karamysheva, A.F. RARalpha mediates all-trans-retinoic acid-induced VEGF-C, VEGF-D, and VEGFR3 expression in lung cancer cells. Cell Biol. Int. 2016, 40, 456–464. [Google Scholar] [CrossRef]
- Zou, C.P.; Kurie, J.M.; Lotan, D.; Zou, C.C.; Hong, W.K.; Lotan, R. Higher potency of N-(4-hydroxyphenyl)retinamide than all-trans-retinoic acid in induction of apoptosis in non-small cell lung cancer cell lines. Clin. Cancer Res. 1998, 4, 1345–1355. [Google Scholar]
- Leblanc, B.P.; Stunnenberg, H.G. 9-cis retinoic acid signaling: Changing partners causes some excitement. Genes Dev. 1995, 9, 1811–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.I.; Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta 2012, 1821, 21–56. [Google Scholar] [CrossRef] [Green Version]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Gilardi, F.; Desvergne, B. RXRs: Collegial partners. Subcell. Biochem. 2014, 70, 75–102. [Google Scholar]
- Kojetin, D.J.; Matta-Camacho, E.; Hughes, T.S.; Srinivasan, S.; Nwachukwu, J.C.; Cavett, V.; Nowak, J.; Chalmers, M.J.; Marciano, D.P.; Kamenecka, T.M.; et al. Structural mechanism for signal transduction in RXR nuclear receptor heterodimers. Nat. Commun. 2015, 6, 8013. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, M.; Alvarez, S.; de Lera, A.R. Natural and Structure-based RXR Ligand Scaffolds and Their Functions. Curr. Top. Med. Chem. 2017, 17, 631–662. [Google Scholar] [CrossRef] [PubMed]
- Bourguet, W.; Germain, P.; Gronemeyer, H. Nuclear receptor ligand-binding domains: Three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci. 2000, 21, 381–388. [Google Scholar] [CrossRef]
- Brabender, J.; Danenberg, K.D.; Metzger, R.; Schneider, P.M.; Lord, R.V.; Groshen, S.; Tsao-Wei, D.D.; Park, J.; Salonga, D.; Holscher, A.H.; et al. The role of retinoid X receptor messenger RNA expression in curatively resected non-small cell lung cancer. Clin. Cancer Res. 2002, 8, 438–443. [Google Scholar]
- Mernitz, H.; Smith, D.E.; Zhu, A.X.; Wang, X.D. 9-cis-Retinoic acid inhibition of lung carcinogenesis in the A/J mouse model is accompanied by increased expression of RAR-beta but no change in cyclooxygenase-2. Cancer Lett. 2006, 244, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Ding, Y.; Huang, D.; Li, H.; Chen, X. The retinoid X receptor-selective ligand, LGD1069, inhibits tumor-induced angiogenesis via suppression of VEGF in human non-small cell lung cancer. Cancer Lett. 2007, 248, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.S.; Moerland, J.A.; Zhang, D.; Carapellucci, S.; Lockwood, B.; Krieger-Burke, T.; Aleiwi, B.; Ellsworth, E.; Liby, K.T. The RXR Agonist MSU42011 Is Effective for the Treatment of Preclinical HER2+ Breast Cancer and Kras-Driven Lung Cancer. Cancers 2021, 13, 5004. [Google Scholar] [CrossRef]
- Ai, X.; Mao, F.; Shen, S.; Shentu, Y.; Wang, J.; Lu, S. Bexarotene inhibits the viability of non-small cell lung cancer cells via slc10a2/PPARgamma/PTEN/mTOR signaling pathway. BMC Cancer 2018, 18, 407. [Google Scholar] [CrossRef]
- Bouillon, R.; Okamura, W.H.; Norman, A.W. Structure-function relationships in the vitamin D endocrine system. Endocr. Rev. 1995, 16, 200–257. [Google Scholar]
- DeLuca, H.F.; Zierold, C. Mechanisms and functions of vitamin D. Nutr. Rev. 1998, 56 Pt 2, S4–S10, discussion S54–S75. [Google Scholar] [CrossRef]
- Wurtz, J.M.; Bourguet, W.; Renaud, J.P.; Vivat, V.; Chambon, P.; Moras, D.; Gronemeyer, H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat. Struct. Biol. 1996, 3, 87–94. [Google Scholar] [CrossRef]
- Sone, T.; Kerner, S.; Pike, J.W. Vitamin D receptor interaction with specific DNA. Association as a 1,25-dihydroxyvitamin D3-modulated heterodimer. J. Biol. Chem. 1991, 266, 23296–23305. [Google Scholar] [CrossRef]
- Freedman, L.P. Increasing the complexity of coactivation in nuclear receptor signaling. Cell 1999, 97, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Bikle, D.D. Vitamin D: An ancient hormone. Exp. Dermatol. 2011, 20, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Thorne, J.; Campbell, M.J. The vitamin D receptor in cancer. Proc. Nutr. Soc. 2008, 67, 115–127. [Google Scholar] [CrossRef]
- Kaiser, U.; Schilli, M.; Wegmann, B.; Barth, P.; Wedel, S.; Hofmann, J.; Havemann, K. Expression of vitamin D receptor in lung cancer. J. Cancer Res. Clin. Oncol. 1996, 122, 356–359. [Google Scholar] [CrossRef]
- Sandgren, M.; Danforth, L.; Plasse, T.F.; DeLuca, H.F. 1,25-Dihydroxyvitamin D3 receptors in human carcinomas: A pilot study. Cancer Res. 1991, 51, 2021–2024. [Google Scholar]
- Zhou, W.; Suk, R.; Liu, G.; Park, S.; Neuberg, D.S.; Wain, J.C.; Lynch, T.J.; Giovannucci, E.; Christiani, D.C. Vitamin D is associated with improved survival in early-stage non-small cell lung cancer patients. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2303–2309. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Heist, R.S.; Liu, G.; Asomaning, K.; Neuberg, D.S.; Hollis, B.W.; Wain, J.C.; Lynch, T.J.; Giovannucci, E.; Su, L.; et al. Circulating 25-hydroxyvitamin D levels predict survival in early-stage non-small-cell lung cancer patients. J. Clin. Oncol. 2007, 25, 479–485. [Google Scholar] [CrossRef]
- Zhou, W.; Heist, R.S.; Liu, G.; Neuberg, D.S.; Asomaning, K.; Su, L.; Wain, J.C.; Lynch, T.J.; Giovannucci, E.; Christiani, D.C. Polymorphisms of vitamin D receptor and survival in early-stage non-small cell lung cancer patients. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2239–2245. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, M.; Parwani, A.V.; Hershberger, P.A.; Lenzner, D.E.; Weissfeld, J.L. Nuclear vitamin D receptor expression is associated with improved survival in non-small cell lung cancer. J. Steroid Biochem. Mol. Biol. 2011, 123, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Menezes, R.J.; Cheney, R.T.; Husain, A.; Tretiakova, M.; Loewen, G.; Johnson, C.S.; Jayaprakash, V.; Moysich, K.B.; Salgia, R.; Reid, M.E. Vitamin D receptor expression in normal, premalignant, and malignant human lung tissue. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Li, X.; Fu, Y.; Li, Y.; Lu, W.; Pan, Y.; Yang, J.; Kong, J. Inhibition of lung cancer by vitamin D depends on downregulation of histidine-rich calcium-binding protein. J. Adv. Res. 2021, 29, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Sasaki, Y.; Kato, S.; Kubodera, N.; Okano, T. 22-Oxa-1alpha,25-dihydroxyvitamin D3 inhibits metastasis and angiogenesis in lung cancer. Carcinogenesis 2005, 26, 1044–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investig. 2006, 116, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Jamroz-Wisniewska, A.; Wojcicka, G.; Horoszewicz, K.; Beltowski, J. Liver X receptors (LXRs). Part II: Non-lipid effects, role in pathology, and therapeutic implications. Postepy Hig Med Dosw (Online) 2007, 61, 760–785. [Google Scholar]
- Bovenga, F.; Sabba, C.; Moschetta, A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab. 2015, 21, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Chuu, C.P.; Lin, H.P. Antiproliferative effect of LXR agonists T0901317 and 22(R)-hydroxycholesterol on multiple human cancer cell lines. Anticancer Res. 2010, 30, 3643–3648. [Google Scholar]
- Fukuchi, J.; Kokontis, J.M.; Hiipakka, R.A.; Chuu, C.P.; Liao, S. Antiproliferative effect of liver X receptor agonists on LNCaP human prostate cancer cells. Cancer Res. 2004, 64, 7686–7689. [Google Scholar] [CrossRef] [Green Version]
- Chuu, C.P.; Hiipakka, R.A.; Kokontis, J.M.; Fukuchi, J.; Chen, R.Y.; Liao, S. Inhibition of tumor growth and progression of LNCaP prostate cancer cells in athymic mice by androgen and liver X receptor agonist. Cancer Res. 2006, 66, 6482–6486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Gong, H.; Khadem, S.; Lu, Y.; Gao, X.; Li, S.; Zhang, J.; Xie, W. Androgen deprivation by activating the liver X receptor. Endocrinology 2008, 149, 3778–3788. [Google Scholar] [PubMed] [Green Version]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Lou, R.; Cao, H.; Dong, S.; Shi, C.; Xu, X.; Ma, R.; Wu, J.; Feng, J. Liver X receptor agonist T0901317 inhibits the migration and invasion of non-small-cell lung cancer cells in vivo and in vitro. Anticancer Drugs 2019, 30, 495–500. [Google Scholar] [CrossRef]
- Chen, Q.J.; Shi, Y.; Shi, J.F.; Yuan, Z.H.; Ma, J.Y.; Fang, S.R.; Gu, W. Liver X receptors agonist T0901317 downregulates matrix metalloproteinase-9 expression in non-small-cell lung cancer by repressing nuclear factor-kappaB. Anticancer Drugs 2017, 28, 952–958. [Google Scholar] [CrossRef]
- Liang, H.; Shen, X. LXR activation radiosensitizes non-small cell lung cancer by restricting myeloid-derived suppressor cells. Biochem. Biophys. Res. Commun. 2020, 528, 330–335. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoon, K.A.; Yoon, H.J.; Hong, J.M.; Lee, M.J.; Lee, I.K.; Kim, S.Y. Liver X receptor activation inhibits osteoclastogenesis by suppressing NF-kappaB activity and c-Fos induction and prevents inflammatory bone loss in mice. J. Leukoc. Biol. 2013, 94, 99–107. [Google Scholar] [CrossRef]
- Hu, Y.; Zang, J.; Cao, H.; Wu, Y.; Yan, D.; Qin, X.; Zhou, L.; Fan, F.; Ni, J.; Xu, X.; et al. Liver X receptors agonist GW3965 re-sensitizes gefitinib-resistant human non-small cell lung cancer cell to gefitinib treatment by inhibiting NF-kappaB in vitro. Oncotarget 2017, 8, 15802–15814. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Fu, J.; Korrer, M.; Gorbounov, M.; Murray, P.J.; Pardoll, D.; Masica, D.L.; Kim, Y.J. Caspase-1 from Human Myeloid-Derived Suppressor Cells Can Promote T Cell-Independent Tumor Proliferation. Cancer Immunol. Res. 2018, 6, 566–577. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Safari, E.; Ghorghanlu, S.; Ahmadi-Khiavi, H.; Mehranfar, S.; Rezaei, R.; Motallebnezhad, M. Myeloid-derived suppressor cells and tumor: Current knowledge and future perspectives. J. Cell. Physiol. 2019, 234, 9966–9981. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cao, H.; Chen, D.; Yu, S.; Sha, H.; Wu, J.; Ma, R.; Wang, Z.; Jing, C.; Zhang, J.; et al. LXR ligands induce apoptosis of EGFR-TKI-resistant human lung cancer cells in vitro by inhibiting Akt-NF-kappaB activation. Oncol. Lett. 2018, 15, 7168–7174. [Google Scholar]
- Ni, J.; Zhou, L.L.; Ding, L.; Zhang, X.Q.; Zhao, X.; Li, H.; Cao, H.; Liu, S.; Wang, Z.; Ma, R.; et al. Efatutazone and T0901317 exert synergistically therapeutic effects in acquired gefitinib-resistant lung adenocarcinoma cells. Cancer Med. 2018, 7, 1955–1966. [Google Scholar] [CrossRef]
- Deblois, G.; Giguere, V. Oestrogen-related receptors in breast cancer: Control of cellular metabolism and beyond. Nat. Rev. Cancer 2013, 13, 27–36. [Google Scholar] [CrossRef]
- Tora, L.; Gronemeyer, H.; Turcotte, B.; Gaub, M.P.; Chambon, P. The N-terminal region of the chicken progesterone receptor specifies target gene activation. Nature 1988, 333, 185–188. [Google Scholar] [CrossRef]
- Wärnmark, A.; Treuter, E.; Wright, A.P.; Gustafsson, J.-A. Activation functions 1 and 2 of nuclear receptors: Molecular strategies for transcriptional activation. Mol. Endocrinol. 2003, 17, 1901–1909. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Kraus, R.J.; Farrell, M.L.; Jordan, V.C.; Mertz, J.E. Estrogen-related receptor alpha1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol. Cancer Res. 2007, 5, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.S.; Mak, A.S.; Yam, J.W.; Cheung, A.N.; Ngan, H.Y.; Wong, A.S. Targeting estrogen-related receptor alpha inhibits epithelial-to-mesenchymal transition and stem cell properties of ovarian cancer cells. Mol. Ther. 2014, 22, 743–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariazi, E.A.; Clark, G.M.; Mertz, J.E. Estrogen-related receptor alpha and estrogen-related receptor gamma associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res. 2002, 62, 6510–6518. [Google Scholar]
- Fujimoto, J.; Alam, S.M.; Jahan, I.; Sato, E.; Sakaguchi, H.; Tamaya, T. Clinical implication of estrogen-related receptor (ERR) expression in ovarian cancers. J. Steroid Biochem. Mol. Biol. 2007, 104, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, J.; Sato, E. Clinical implication of estrogen-related receptor (ERR) expression in uterine endometrial cancers. J. Steroid Biochem. Mol. Biol. 2009, 116, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.W.; Guan, B.Z.; Yin, L.H.; Liu, F.N.; Hu, B.; Zheng, Q.Y.; Li, F.L.; Zhong, Y.X.; Chen, Y. Effects of estrogen-related receptor alpha (ERRalpha) on proliferation and metastasis of human lung cancer A549 cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, X.; Liang, N.; Li, S. Estrogen-related receptor alpha triggers the proliferation and migration of human non-small cell lung cancer via interleukin-6. Cell Biochem. Funct. 2018, 36, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Sethi, G.; Sung, B.; Goel, A.; Ralhan, R.; Aggarwal, B.B. Guggulsterone, a farnesoid X receptor antagonist, inhibits constitutive and inducible STAT3 activation through induction of a protein tyrosine phosphatase SHP-1. Cancer Res. 2008, 68, 4406–4415. [Google Scholar] [CrossRef] [Green Version]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [Green Version]
- Renga, B.; D’Amore, C.; Cipriani, S.; Mencarelli, A.; Carino, A.; Sepe, V.; Zampella, A.; Distrutti, E.; Fiorucci, S. FXR mediates a chromatin looping in the GR promoter thus promoting the resolution of colitis in rodents. Pharmacol. Res. 2013, 77, 1–10. [Google Scholar] [CrossRef]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, A.; Thomas, A.; Edwards, G.; Jaseja, R.; Guo, G.L.; Apte, U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J. Pharmacol. Exp. Ther. 2011, 338, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gottardi, A.; Touri, F.; Maurer, C.A.; Perez, A.; Maurhofer, O.; Ventre, G.; Bentzen, C.L.; Niesor, E.J.; Dufour, J.F. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig. Dis. Sci. 2004, 49, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Maran, R.R.; Thomas, A.; Roth, M.; Sheng, Z.; Esterly, N.; Pinson, D.; Gao, X.; Zhang, Y.; Ganapathy, V.; Gonzalez, F.J.; et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J. Pharmacol. Exp. Ther. 2009, 328, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, W.; Chen, B.; Liu, X.; Xue, S.; Qin, H.; Jiang, H. Farnesoid X receptor, a novel proto-oncogene in non-small cell lung cancer, promotes tumor growth via directly transactivating CCND1. Sci. Rep. 2017, 7, 591. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; He, L.; Bu, P.; Wan, Y.J. Pregnane X receptor is essential for normal progression of liver regeneration. Hepatology 2008, 47, 1277–1287. [Google Scholar] [CrossRef]
- Elcombe, C.R.; Elcombe, B.M.; Foster, J.R.; Chang, S.C.; Ehresman, D.J.; Butenhoff, J.L. Hepatocellular hypertrophy and cell proliferation in Sprague-Dawley rats from dietary exposure to potassium perfluorooctanesulfonate results from increased expression of xenosensor nuclear receptors PPARalpha and CAR/PXR. Toxicology 2012, 293, 16–29. [Google Scholar] [CrossRef]
- Gupta, D.; Venkatesh, M.; Wang, H.; Kim, S.; Sinz, M.; Goldberg, G.L.; Whitney, K.; Longley, C.; Mani, S. Expanding the roles for pregnane X receptor in cancer: Proliferation and drug resistance in ovarian cancer. Clin. Cancer Res. 2008, 14, 5332–5340. [Google Scholar] [CrossRef] [Green Version]
- Harmsen, S.; Meijerman, I.; Febus, C.L.; Maas-Bakker, R.F.; Beijnen, J.H.; Schellens, J.H. PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother. Pharmacol. 2010, 66, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Masuyama, H.; Hiramatsu, Y.; Kodama, J.; Kudo, T. Expression and potential roles of pregnane X receptor in endometrial cancer. J. Clin. Endocrinol. Metab. 2003, 88, 4446–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensah-Osman, E.J.; Thomas, D.G.; Tabb, M.M.; Larios, J.M.; Hughes, D.P.; Giordano, T.J.; Lizyness, M.L.; Rae, J.M.; Blumberg, B.; Hollenberg, P.F.; et al. Expression levels and activation of a PXR variant are directly related to drug resistance in osteosarcoma cell lines. Cancer 2007, 109, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tang, Y.; Wang, M.T.; Zeng, S.; Nie, D. Human pregnane X receptor and resistance to chemotherapy in prostate cancer. Cancer Res. 2007, 67, 10361–10367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Huang, W.; Chen, F.; Hu, G.; Li, F.; Li, J.; Xuan, A. Pregnane X receptors regulate CYP2C8 and P-glycoprotein to impact on the resistance of NSCLC cells to Taxol. Cancer Med. 2016, 5, 3564–3571. [Google Scholar] [CrossRef]
- Fayard, E.; Auwerx, J.; Schoonjans, K. LRH-1: An orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004, 14, 250–260. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. General molecular biology and architecture of nuclear receptors. Curr. Top. Med. Chem. 2012, 12, 486–504. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.B.; Molania, R.; Chand, A.; Knower, K.; Takano, E.A.; Byrne, D.J.; Mikeska, T.; Millar, E.K.A.; Lee, C.S.; O’Toole, S.A.; et al. LRH-1 expression patterns in breast cancer tissues are associated with tumour aggressiveness. Oncotarget 2017, 8, 83626–83636. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Aihara, A.; Chung, W.; Li, Y.; Chen, X.; Huang, Z.; Weng, S.; Carlson, R.I.; Nadolny, C.; Wands, J.R.; et al. LRH1 promotes pancreatic cancer metastasis. Cancer Lett. 2014, 350, 15–24. [Google Scholar] [CrossRef]
- Wu, C.; Feng, J.; Li, L.; Wu, Y.; Xie, H.; Yin, Y.; Ye, J.; Li, Z. Liver receptor homologue 1, a novel prognostic marker in colon cancer patients. Oncol. Lett. 2018, 16, 2833–2838. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Jin, J.; Woodfield, S.E.; Patel, R.H.; Jin, N.G.; Shi, Y.; Liu, B.; Sun, W.; Chen, X.; Yu, Y.; et al. Targeting LRH1 in hepatoblastoma cell lines causes decreased proliferation. Oncol. Rep. 2019, 41, 143–153. [Google Scholar]
- Annicotte, J.S.; Chavey, C.; Servant, N.; Teyssier, J.; Bardin, A.; Licznar, A.; Badia, E.; Pujol, P.; Vignon, F.; Maudelonde, T.; et al. The nuclear receptor liver receptor homolog-1 is an estrogen receptor target gene. Oncogene 2005, 24, 8167–8175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chand, A.L.; Herridge, K.A.; Thompson, E.W.; Clyne, C.D. The orphan nuclear receptor LRH-1 promotes breast cancer motility and invasion. Endocr. Relat. Cancer 2010, 17, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, T.; Li, J.; Sun, Z.; Liu, Y.; Kong, L.; Zhou, S.; Tang, J.; Wang, J.; Xing, H.R. Nr5a2 promotes cancer stem cell properties and tumorigenesis in nonsmall cell lung cancer by regulating Nanog. Cancer Med. 2019, 8, 1232–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xing, Y.; Wang, H.; Yan, S.; Wang, X.; Cai, L. LRH1 as a promising prognostic biomarker and predictor of metastasis in patients with non-small cell lung cancer. Thorac. Cancer 2018, 9, 1725–1732. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Tian, Y.; Jiang, S.; Liu, S.; Zhao, X.; Tian, D. MicroRNA-376c suppresses non-small-cell lung cancer cell growth and invasion by targeting LRH-1-mediated Wnt signaling pathway. Biochem. Biophys. Res. Commun. 2016, 473, 980–986. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Sadroddiny, E. MicroRNAs in lung diseases: Recent findings and their pathophysiological implications. Pulm Pharmacol. Ther. 2015, 34, 55–63. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Thompson, E.B. Gene regulation by the glucocorticoid receptor: Structure:function relationship. J. Steroid Biochem. Mol. Biol. 2005, 94, 383–394. [Google Scholar] [CrossRef]
- Bledsoe, R.K.; Montana, V.G.; Stanley, T.B.; Delves, C.J.; Apolito, C.J.; McKee, D.D.; Consler, T.G.; Parks, D.J.; Stewart, E.L.; Willson, T.M. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002, 110, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, Y.; Liao, S.; Tu, W.; Shen, Y.; Yan, Y.; Tao, D.; Lu, Y.; Ma, Y.; Yang, Y.; et al. Epigenetic modifications promote the expression of the orphan nuclear receptor NR0B1 in human lung adenocarcinoma cells. Oncotarget 2016, 7, 43162–43176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.; Xie, Y.; Lee, W.; Bookout, A.L.; Girard, L.; Raso, G.; Behrens, C.; Wistuba, I.I.; Gadzar, A.F.; Minna, J.D.; et al. ReseArch. resource: Diagnostic and therapeutic potential of nuclear receptor expression in lung cancer. Mol. Endocrinol. 2012, 26, 1443–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, L.A.; Nuzum, E.O.; Jones, B.C.; Malkinson, A.M.; Beer, D.G. Expression of the glucocorticoid receptor and K-ras genes in urethan-induced mouse lung tumors and transformed cell lines. Exp. Lung Res. 1991, 17, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.; Kaiser, U.; Maasberg, M.; Havemann, K. Glucocorticoid receptors and growth inhibitory effects of dexamethasone in human lung cancer cell lines. Eur. J. Cancer 1995, 31A, 2053–2058. [Google Scholar] [CrossRef]
- Greenberg, A.K.; Hu, J.; Basu, S.; Hay, J.; Reibman, J.; Yie, T.A.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am. J. Respir. Cell Mol. Biol. 2002, 27, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Tai, H.H. 15-Hydroxyprostaglandin dehydrogenase can be induced by dexamethasone and other glucocorticoids at the therapeutic level in A549 human lung adenocarcinoma cells. Arch. Biochem. Biophys. 2005, 435, 50–55. [Google Scholar] [CrossRef]
- Anggard, E. The biological activities of three metabolites of prostaglandin E 1. Acta Physiol. Scand. 1966, 66, 509–510. [Google Scholar] [CrossRef]
- Ferreira, S.H.; Vane, J.R. Prostaglandins: Their disappearance from and release into the circulation. Nature 1967, 216, 868–873. [Google Scholar] [CrossRef]
- Moore, P.K.; Hoult, J.R. Anti-inflammatory steroids reduce tissue PG synthetase activity and enhance PG breakdown. Nature 1980, 288, 269–270. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Ohsaki, Y.; Nishigaki, Y.; Toyoshima, E.; Kikuchi, K. Inhibition of human small cell lung cancer cell growth by methylprednisolone. Int. J. Oncol. 1999, 15, 1185–1190. [Google Scholar] [CrossRef]
- Sommer, P.; Cowen, R.L.; Berry, A.; Cookson, A.; Telfer, B.A.; Williams, K.J.; Stratford, I.J.; Kay, P.; White, A.; Ray, D.W. Glucocorticoid receptor over-expression promotes human small cell lung cancer apoptosis in vivo and thereby slows tumor growth. Endocr. Relat. Cancer 2010, 17, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Y.; Carlstedt-Duke, J.; Weigel, N.L.; Dahlman, K.; Gustafsson, J.A.; Tsai, M.J.; O’Malley, B.W. Molecular interactions of steroid hormone receptor with its enhancer element: Evidence for receptor dimer formation. Cell 1988, 55, 361–369. [Google Scholar] [CrossRef]
- Huse, B.; Verca, S.B.; Matthey, P.; Rusconi, S. Definition of a negative modulation domain in the human progesterone receptor. Mol. Endocrinol. 1998, 12, 1334–1342. [Google Scholar] [CrossRef]
- Sartorius, C.A.; Melville, M.Y.; Hovland, A.R.; Tung, L.; Takimoto, G.S.; Horwitz, K.B. A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol. Endocrinol. 1994, 8, 1347–1360. [Google Scholar] [PubMed]
- Vegeto, E.; Shahbaz, M.M.; Wen, D.X.; Goldman, M.E.; O’Malley, B.W.; McDonnell, D.P. Human progesterone receptor A form is a cell- and promoter-specific repressor of human progesterone receptor B function. Mol. Endocrinol. 1993, 7, 1244–1255. [Google Scholar]
- Mote, P.A.; Graham, J.D.; Clarke, C.L. Progesterone receptor isoforms in normal and malignant breast. In Ernst Schering Foundation Symposium Proceedings; Springer: Berlin/Heidelberg, Germany, 2007; pp. 77–107. [Google Scholar]
- Ali, I.U.; Lidereau, R.; Callahan, R. Presence of two members of c-erbA receptor gene family (c-erbA beta and c-erbA2) in smallest region of somatic homozygosity on chromosome 3p21-p25 in human breast carcinoma. J. Natl. Cancer Inst. 1989, 81, 1815–1820. [Google Scholar] [CrossRef]
- Leduc, F.; Brauch, H.; Hajj, C.; Dobrovic, A.; Kaye, F.; Gazdar, A.; Harbour, J.W.; Pettengill, O.S.; Sorenson, G.D.; van den Berg, A.; et al. Loss of heterozygosity in a gene coding for a thyroid hormone receptor in lung cancers. Am. J. Hum. Genet. 1989, 44, 282–287. [Google Scholar]
- Sisley, K.; Curtis, D.; Rennie, I.G.; Rees, R.C. Loss of heterozygosity of the thyroid hormone receptor B in posterior uveal melanoma. Melanoma Res. 1993, 3, 457–461. [Google Scholar] [CrossRef]
- Chen, L.C.; Matsumura, K.; Deng, G.; Kurisu, W.; Ljung, B.M.; Lerman, M.I.; Waldman, F.M.; Smith, H.S. Deletion of two separate regions on chromosome 3p in breast cancers. Cancer Res. 1994, 54, 3021–3024. [Google Scholar]
- Huber-Gieseke, T.; Pernin, A.; Huber, O.; Burger, A.G.; Meier, C.A. Lack of loss of heterozygosity at the c-erbA beta locus in gastrointestinal tumors. Oncology 1997, 54, 214–219. [Google Scholar] [CrossRef]
- Gonzalez-Sancho, J.M.; Garcia, V.; Bonilla, F.; Munoz, A. Thyroid hormone receptors/THR genes in human cancer. Cancer Lett. 2003, 192, 121–132. [Google Scholar] [CrossRef]
- Mohamed, F.; Abdelaziz, A.O.; Kasem, A.H.; Ellethy, T.; Gayyed, M.F. Thyroid hormone receptor alpha1 acts as a new squamous cell lung cancer diagnostic marker and poor prognosis predictor. Sci. Rep. 2021, 11, 7944. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Sunaga, N.; Tomizawa, Y.; Imai, H.; Iijima, H.; Yanagitani, N.; Horiguchi, K.; Yamada, M.; Mori, M. Epigenetic inactivation of the thyroid hormone receptor beta1 gene at 3p24.2 in lung cancer. Ann Surg. Oncol. 2010, 17, 2222–2228. [Google Scholar] [CrossRef]
- Joly-Pharaboz, M.O.; Ruffion, A.; Roch, A.; Michel-Calemard, L.; Andre, J.; Chantepie, J.; Nicolas, B.; Panaye, G. Inhibition of growth and induction of apoptosis by androgens of a variant of LNCaP cell line. J. Steroid Biochem. Mol. Biol. 2000, 73, 237–249. [Google Scholar] [CrossRef]
- Clarke, R.; Liu, M.C.; Bouker, K.B.; Gu, Z.; Lee, R.Y.; Zhu, Y.; Skaar, T.C.; Gomez, B.; O’Brien, K.; Wang, Y.; et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003, 22, 7316–7339. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lee, E.S.; Gajdos, C.; Pearce, S.T.; Chen, B.; Osipo, C.; Loweth, J.; McKian, K.; De Los Reyes, A.; Wing, L.; et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J. Natl. Cancer Inst. 2003, 95, 1586–1597. [Google Scholar] [CrossRef]
- Osipo, C.; Gajdos, C.; Liu, H.; Chen, B.; Jordan, V.C. Paradoxical action of fulvestrant in estradiol-induced regression of tamoxifen-stimulated breast cancer. J. Natl. Cancer Inst. 2003, 95, 1597–1608. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, H.; Messing, E.M.; Chang, C. Androgen deprivation therapy for prostate cancer: Current status and future prospects. Prostate 2004, 61, 332–353. [Google Scholar] [CrossRef]
- Holbeck, S.; Chang, J.; Best, A.M.; Bookout, A.L.; Mangelsdorf, D.J.; Martinez, E.D. Expression profiling of nuclear receptors in the NCI60 cancer cell panel reveals receptor-drug and receptor-gene interactions. Mol. Endocrinol. 2010, 24, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Sethi, G.; Ahn, K.S.; Chaturvedi, M.M.; Aggarwal, B.B. Epidermal growth factor (EGF) activates nuclear factor-kappaB through IkappaBalpha kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IkappaBalpha. Oncogene 2007, 26, 7324–7332. [Google Scholar] [CrossRef] [Green Version]
- Dumitrescu, R.G. Epigenetic markers of early tumor development. Methods Mol. Biol. 2012, 863, 3–14. [Google Scholar]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Ranjbar, A.; Seyed Saleh, S.H.; Bagherian, M.; Sharifzadeh, S.O.; Hushmandi, K.; et al. Regulation of Nuclear Factor-KappaB (NF-kappaB) signaling pathway by non-coding RNAs in cancer: Inhibiting or promoting carcinogenesis? Cancer Lett. 2021, 509, 63–80. [Google Scholar] [CrossRef]
- Brzezianska, E.; Dutkowska, A.; Antczak, A. The significance of epigenetic alterations in lung carcinogenesis. Mol. Biol. Rep. 2013, 40, 309–325. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G.; et al. Role of novel histone modifications in cancer. Oncotarget 2018, 9, 11414–11426. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, M.K.; Dharmarajan, A.; Warrier, S.; Bishayee, A.; Kumar, A.P.; Sethi, G.; Ahn, K.S. Role of histone acetyltransferase inhibitors in cancer therapy. Adv. Protein Chem. Struct Biol. 2021, 125, 149–191. [Google Scholar]
- Zochbauer-Muller, S.; Minna, J.D.; Gazdar, A.F. Aberrant DNA methylation in lung cancer: Biological and clinical implications. Oncologist 2002, 7, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Belinsky, S.A.; Klinge, D.M.; Dekker, J.D.; Smith, M.W.; Bocklage, T.J.; Gilliland, F.D.; Crowell, R.E.; Karp, D.D.; Stidley, C.A.; Picchi, M.A. Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin. Cancer Res. 2005, 11, 6505–6511. [Google Scholar] [CrossRef] [Green Version]
- Licchesi, J.D.; Westra, W.H.; Hooker, C.M.; Herman, J.G. Promoter hypermethylation of hallmark cancer genes in atypical adenomatous hyperplasia of the lung. Clin. Cancer Res. 2008, 14, 2570–2578. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Lee, H.J.; Kim, B.H.; Cho, N.Y.; Kang, G.H. DNA methylation profile during multistage progression of pulmonary adenocarcinomas. Virchows Arch. 2011, 459, 201–211. [Google Scholar] [CrossRef]
- Herceg, Z.; Vaissiere, T. Epigenetic mechanisms and cancer: An interface between the environment and the genome. Epigenetics 2011, 6, 804–819. [Google Scholar] [CrossRef]
- Behera, A.K.; Kumar, M.; Shanmugam, M.K.; Bhattacharya, A.; Rao, V.J.; Bhat, A.; Vasudevan, M.; Gopinath, K.S.; Mohiyuddin, A.; Chatterjee, A.; et al. Functional interplay between YY1 and CARM1 promotes oral carcinogenesis. Oncotarget 2019, 10, 3709–3724. [Google Scholar] [CrossRef] [Green Version]
- Zochbauer-Muller, S.; Fong, K.M.; Virmani, A.K.; Geradts, J.; Gazdar, A.F.; Minna, J.D. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001, 61, 249–255. [Google Scholar]
- Oshita, F.; Sekiyama, A.; Suzuki, R.; Ikehara, M.; Yamada, K.; Saito, H.; Noda, K.; Miyagi, Y. Detection of occult tumor cells in peripheral blood from patients with small cell lung cancer by promoter methylation and silencing of the retinoic acid receptor-beta. Oncol. Rep. 2003, 10, 105–108. [Google Scholar] [CrossRef]
- Grote, H.J.; Schmiemann, V.; Geddert, H.; Rohr, U.P.; Kappes, R.; Gabbert, H.E.; Bocking, A. Aberrant promoter methylation of p16(INK4a), RARB2 and SEMA3B in bronchial aspirates from patients with suspected lung cancer. Int. J. Cancer 2005, 116, 720–725. [Google Scholar] [CrossRef]
- Kim, Y.T.; Lee, S.H.; Sung, S.W.; Kim, J.H. Can aberrant promoter hypermethylation of CpG islands predict the clinical outcome of non-small cell lung cancer after curative resection? Ann. Thorac. Surg. 2005, 79, 1180–1188, discussion 1180–1188. [Google Scholar] [CrossRef]
- Feng, Q.; Hawes, S.E.; Stern, J.E.; Wiens, L.; Lu, H.; Dong, Z.M.; Jordan, C.D.; Kiviat, N.B.; Vesselle, H. DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiol. Biomark. Prev. 2008, 17, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Dutkowska, A.; Antczak, A.; Pastuszak-Lewandoska, D.; Migdalska-Sek, M.; Czarnecka, K.H.; Gorski, P.; Kordiak, J.; Nawrot, E.; Brzezianska-Lasota, E. RARbeta Promoter Methylation as an Epigenetic Mechanism of Gene Silencing in Non-small Cell Lung Cancer. Adv. Exp. Med. Biol. 2016, 878, 29–38. [Google Scholar]
- Feng, H.; Zhang, Z.; Qing, X.; Wang, X.; Liang, C.; Liu, D. Promoter methylation of APC and RAR-beta genes as prognostic markers in non-small cell lung cancer (NSCLC). Exp. Mol. Pathol. 2016, 100, 109–113. [Google Scholar] [CrossRef]
- Walter, R.F.H.; Rozynek, P.; Casjens, S.; Werner, R.; Mairinger, F.D.; Speel, E.J.M.; Zur Hausen, A.; Meier, S.; Wohlschlaeger, J.; Theegarten, D.; et al. Methylation of L1RE1, RARB, and RASSF1 function as possible biomarkers for the differential diagnosis of lung cancer. PLoS ONE 2018, 13, e0195716. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, H.; Kim, H.; Shim, Y.M.; Han, J.; Park, J.; Kim, D.H. Promoter methylation of retinoic acid receptor beta 2 and the development of second primary lung cancers in non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 3443–3450. [Google Scholar] [CrossRef]
- Tomizawa, Y.; Iijima, H.; Nomoto, T.; Iwasaki, Y.; Otani, Y.; Tsuchiya, S.; Saito, R.; Dobashi, K.; Nakajima, T.; Mori, M. Clinicopathological significance of aberrant methylation of RARbeta2 at 3p24, RASSF1A at 3p21.3, and FHIT at 3p14.2 in patients with non-small cell lung cancer. Lung Cancer 2004, 46, 305–312. [Google Scholar] [CrossRef]
- Suh, Y.A.; Lee, H.Y.; Virmani, A.; Wong, J.; Mann, K.K.; Miller, W.H., Jr.; Gazdar, A.; Kurie, J.M. Loss of retinoic acid receptor beta gene expression is linked to aberrant histone H3 acetylation in lung cancer cell lines. Cancer Res. 2002, 62, 3945–3949. [Google Scholar]
- Vuillemenot, B.R.; Pulling, L.C.; Palmisano, W.A.; Hutt, J.A.; Belinsky, S.A. Carcinogen exposure differentially modulates RAR-beta promoter hypermethylation, an early and frequent event in mouse lung carcinogenesis. Carcinogenesis 2004, 25, 623–629. [Google Scholar] [CrossRef]
- Teraishi, F.; Kadowaki, Y.; Tango, Y.; Kawashima, T.; Umeoka, T.; Kagawa, S.; Tanaka, N.; Fujiwara, T. Ectopic p21sdi1 gene transfer induces retinoic acid receptor beta expression and sensitizes human cancer cells to retinoid treatment. Int. J. Cancer 2003, 103, 833–839. [Google Scholar] [CrossRef]
- Suga, Y.; Miyajima, K.; Oikawa, T.; Maeda, J.; Usuda, J.; Kajiwara, N.; Ohira, T.; Uchida, O.; Tsuboi, M.; Hirano, T.; et al. Quantitative p16 and ESR1 methylation in the peripheral blood of patients with non-small cell lung cancer. Oncol. Rep. 2008, 20, 1137–1142. [Google Scholar]
- Lin, Q.; Geng, J.; Ma, K.; Yu, J.; Sun, J.; Shen, Z.; Bao, G.; Chen, Y.; Zhang, H.; He, Y.; et al. RASSF1A, APC, ESR1, ABCB1 and HOXC9, but not p16INK4A, DAPK1, PTEN and MT1G genes were frequently methylated in the stage I non-small cell lung cancer in China. J. Cancer Res. Clin. Oncol. 2009, 135, 1675–1684. [Google Scholar] [CrossRef]
- Tekpli, X.; Skaug, V.; Baera, R.; Phillips, D.H.; Haugen, A.; Mollerup, S. Estrogen receptor expression and gene promoter methylation in non-small cell lung cancer—A short report. Cell. Oncol. 2016, 39, 583–589. [Google Scholar] [CrossRef]
- Issa, J.P.; Baylin, S.B.; Belinsky, S.A. Methylation of the estrogen receptor CpG island in lung tumors is related to the specific type of carcinogen exposure. Cancer Res. 1996, 56, 3655–3658. [Google Scholar]
- Belinsky, S.A.; Snow, S.S.; Nikula, K.J.; Finch, G.L.; Tellez, C.S.; Palmisano, W.A. Aberrant CpG island methylation of the p16(INK4a) and estrogen receptor genes in rat lung tumors induced by particulate carcinogens. Carcinogenesis 2002, 23, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Lee, J.Y.; Choi, J.E.; Lee, S.Y.; Park, J.Y.; Kim, D.S. Epigenetic inactivation of retinoid X receptor genes in non-small cell lung cancer and the relationship with clinicopathologic features. Cancer Genet. Cytogenet. 2010, 197, 39–45. [Google Scholar] [CrossRef]
- Mazieres, J.; Barlesi, F.; Rouquette, I.; Molinier, O.; Besse, B.; Monnet, I.; Audigier-Valette, C.; Toffart, A.C.; Renault, P.A.; Fraboulet, S.; et al. Randomized Phase II Trial Evaluating Treatment with EGFR-TKI Associated with Antiestrogen in Women with Nonsquamous Advanced-Stage NSCLC: IFCT-1003 LADIE Trial. Clin. Cancer Res. 2020, 26, 3172–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Li, Y.; Liu, R.; Agadir, A.; Lee, M.O.; Liu, Y.; Zhang, X. Modulation of retinoic acid sensitivity in lung cancer cells through dynamic balance of orphan receptors nur77 and COUP-TF and their heterodimerization. EMBO J. 1997, 16, 1656–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochhaus, G.; Rohdewald, P.; Mollmann, H.; Greschuchna, D. Identification of glucocorticoid receptors in normal and neoplastic adult human lung. Res. Exp. Med. 1983, 182, 71–78. [Google Scholar]
- Kaiser, A.; Stier, U.; Riecken, E.O.; Rosewicz, S. Glucocorticoid receptor concentration modulates glucocorticoid-regulated gene expression in rat pancreatic AR42J cells. Digestion 1996, 57, 149–160. [Google Scholar] [CrossRef]
- Lu, Y.S.; Lien, H.C.; Yeh, P.Y.; Kuo, S.H.; Chang, W.C.; Kuo, M.L.; Cheng, A.L. Glucocorticoid receptor expression in advanced non-small cell lung cancer: Clinicopathological correlation and in vitro effect of glucocorticoid on cell growth and chemosensitivity. Lung Cancer 2006, 53, 303–310. [Google Scholar] [CrossRef]
- Suzuki, S.; Suzuki, T.; Tsubochi, H.; Koike, K.; Tateno, H.; Krozowski, Z.S.; Sasano, H. Expression of 11 beta-hydroxysteroid dehydrogenase type 2 and mineralocorticoid receptor in primary lung carcinomas. Anticancer Res. 2000, 20, 323–328. [Google Scholar]
- Chang, T.H.; Szabo, E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor gamma in non-small cell lung cancer. Cancer Res. 2000, 60, 1129–1138. [Google Scholar]
- Marquez-Garban, D.C.; Mah, V.; Alavi, M.; Maresh, E.L.; Chen, H.W.; Bagryanova, L.; Horvath, S.; Chia, D.; Garon, E.; Goodglick, L.; et al. Progesterone and estrogen receptor expression and activity in human non-small cell lung cancer. Steroids 2011, 76, 910–920. [Google Scholar]
- Lu, X.; Guan, A.; Chen, X.; Xiao, J.; Xie, M.; Yang, B.; He, S.; You, S.; Li, W.; Chen, Q. mPRalpha mediates P4/Org OD02-0 to improve the sensitivity of lung adenocarcinoma to EGFR-TKIs via the EGFR-SRC-ERK1/2 pathway. Mol. Carcinog. 2020, 59, 179–192. [Google Scholar] [CrossRef]
- Huang, Q.; Fan, J.; Qian, X.; Lv, Z.; Zhang, X.; Han, J.; Wu, F.; Chen, C.; Du, J.; Guo, M.; et al. Retinoic acid-related orphan receptor C isoform 2 expression and its prognostic significance for non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 263–272. [Google Scholar] [CrossRef]
- Kolluri, S.K.; Bruey-Sedano, N.; Cao, X.; Lin, B.; Lin, F.; Han, Y.H.; Dawson, M.I.; Zhang, X.K. Mitogenic effect of orphan receptor TR3 and its regulation by MEKK1 in lung cancer cells. Mol. Cell. Biol. 2003, 23, 8651–8667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.O.; Andey, T.; Jin, U.H.; Kim, K.; Singh, M.; Safe, S. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene 2012, 31, 3265–3276. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.Z.; Chen, Z.; Tahir, S.K.; Rosenberg, S.H.; Ng, S.C. Synergistic effect of paclitaxel and 4-hydroxytamoxifen on estrogen receptor-negative colon cancer and lung cancer cell lines. Anticancer Drugs 1999, 10, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Tian, T.; Inoue, M.; Ikeda, J.; Qiu, Y.; Okumura, M.; Aozasa, K.; Morii, E. Tumorigenic role of orphan nuclear receptor NR0B1 in lung adenocarcinoma. Am. J. Pathol. 2009, 175, 1235–1245. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Wang, W.; Dai, M.; Li, H.; Chen, C.; Wang, D. PPARalpha ligand, AVE8134, and cyclooxygenase inhibitor therapy synergistically suppress lung cancer growth and metastasis. BMC Cancer 2019, 19, 1166. [Google Scholar] [CrossRef]
- Avis, I.; Martinez, A.; Tauler, J.; Zudaire, E.; Mayburd, A.; Abu-Ghazaleh, R.; Ondrey, F.; Mulshine, J.L. Inhibitors of the arachidonic acid pathway and peroxisome proliferator-activated receptor ligands have superadditive effects on lung cancer growth inhibition. Cancer Res. 2005, 65, 4181–4190. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.L.; Zhang, Z.X.; Xu, Y.J. Ciglitazone inhibits growth of lung cancer cells A549 in vitro and in vivo: An experimental study. Zhonghua Zhong Liu Za Zhi 2004, 26, 531–534. [Google Scholar]
- Zou, W.; Liu, X.; Yue, P.; Khuri, F.R.; Sun, S.Y. PPARgamma ligands enhance TRAIL-induced apoptosis through DR5 upregulation and c-FLIP downregulation in human lung cancer cells. Cancer Biol. Ther. 2007, 6, 99–106. [Google Scholar] [CrossRef]
- Yan, K.H.; Yao, C.J.; Chang, H.Y.; Lai, G.M.; Cheng, A.L.; Chuang, S.E. The synergistic anticancer effect of troglitazone combined with aspirin causes cell cycle arrest and apoptosis in human lung cancer cells. Mol. Carcinog. 2010, 49, 235–246. [Google Scholar] [CrossRef]
- Reka, A.K.; Kurapati, H.; Narala, V.R.; Bommer, G.; Chen, J.; Standiford, T.J.; Keshamouni, V.G. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol. Cancer Ther. 2010, 9, 3221–3232. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; James, M.; Wen, W.; Lu, Y.; Szabo, E.; Lubet, R.A.; You, M. Chemopreventive effects of pioglitazone on chemically induced lung carcinogenesis in mice. Mol. Cancer Ther. 2010, 9, 3074–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ritzenthaler, J.D.; Zheng, Y.; Roman, J.; Han, S. Rosiglitazone inhibits alpha4 nicotinic acetylcholine receptor expression in human lung carcinoma cells through peroxisome proliferator-activated receptor gamma-independent signals. Mol. Cancer Ther. 2009, 8, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, L.N.; Yan, L.; Li, C.; Cheng, K. Bavachinin exhibits antitumor activity against nonsmall cell lung cancer by targeting PPARgamma. Mol. Med. Rep. 2019, 20, 2805–2811. [Google Scholar] [PubMed]
- Kaur, S.; Nag, A.; Gangenahalli, G.; Sharma, K. Peroxisome Proliferator Activated Receptor Gamma Sensitizes Non-small Cell Lung Carcinoma to Gamma Irradiation Induced Apoptosis. Front. Genet. 2019, 10, 554. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.N.M.; Namkung, J.; Phan, A.N.H.; Vo, V.T.A.; Kim, M.K.; Jeong, Y.; Choi, J.W. PPARgamma-mediated ALDH1A3 suppression exerts anti-proliferative effects in lung cancer by inducing lipid peroxidation. J. Recept Signal Transduct Res. 2018, 38, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Ravi, R.K.; Knudsen, E.S.; Williams, J.R.; Dillehay, L.E.; Nelkin, B.D.; Kalemkerian, G.P.; Feramisco, J.R.; Mabry, M. Retinoic acid-mediated growth inhibition of small cell lung cancer cells is associated with reduced myc and increased p27Kip1 expression. Int. J. Cancer 1999, 80, 935–943. [Google Scholar] [CrossRef]
- Ahn, M.J.; Langenfeld, J.; Moasser, M.M.; Rusch, V.; Dmitrovsky, E. Growth suppression of transformed human bronchial epithelial cells by all-trans-retinoic acid occurs through specific retinoid receptors. Oncogene 1995, 11, 2357–2364. [Google Scholar]
- Fazely, F.; Ledinko, N. Retinoic acid and epidermal growth factor binding in retinoid-mediated invasion suppressed human lung carcinoma cells. Anticancer Res. 1990, 10, 667–670. [Google Scholar]
- Zhang, X.K.; Liu, Y.; Lee, M.O. Retinoid receptors in human lung cancer and breast cancer. Mutat. Res. 1996, 350, 267–277. [Google Scholar] [CrossRef]
- Li, Y.; Dawson, M.I.; Agadir, A.; Lee, M.O.; Jong, L.; Hobbs, P.D.; Zhang, X.K. Regulation of RAR beta expression by RAR- and RXR-selective retinoids in human lung cancer cell lines: Effect on growth inhibition and apoptosis induction. Int. J. Cancer 1998, 75, 88–95. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gangwar, S.K.; Kumar, A.; Yap, K.C.-H.; Jose, S.; Parama, D.; Sethi, G.; Kumar, A.P.; Kunnumakkara, A.B. Targeting Nuclear Receptors in Lung Cancer—Novel Therapeutic Prospects. Pharmaceuticals 2022, 15, 624. https://doi.org/10.3390/ph15050624
Gangwar SK, Kumar A, Yap KC-H, Jose S, Parama D, Sethi G, Kumar AP, Kunnumakkara AB. Targeting Nuclear Receptors in Lung Cancer—Novel Therapeutic Prospects. Pharmaceuticals. 2022; 15(5):624. https://doi.org/10.3390/ph15050624
Chicago/Turabian StyleGangwar, Shailendra Kumar, Aviral Kumar, Kenneth Chun-Hong Yap, Sandra Jose, Dey Parama, Gautam Sethi, Alan Prem Kumar, and Ajaikumar B. Kunnumakkara. 2022. "Targeting Nuclear Receptors in Lung Cancer—Novel Therapeutic Prospects" Pharmaceuticals 15, no. 5: 624. https://doi.org/10.3390/ph15050624