
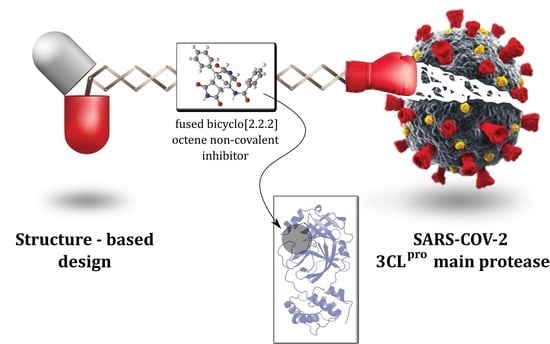
Design, Synthesis and Evaluation of Fused Bicyclo[2.2.2]octene as a Potential Core Scaffold for the Non-Covalent Inhibitors of SARS-CoV-2 3CLpro Main Protease

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
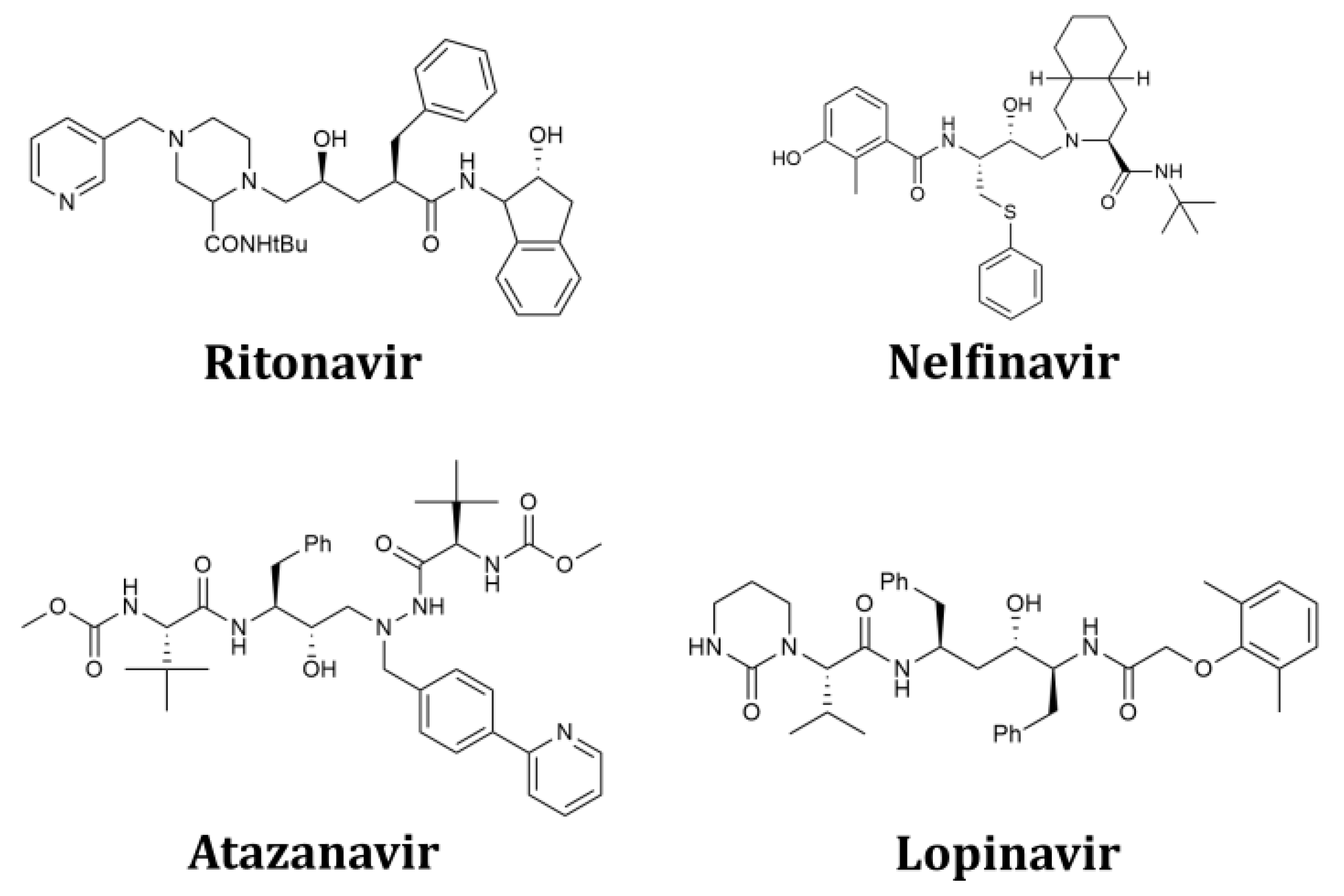
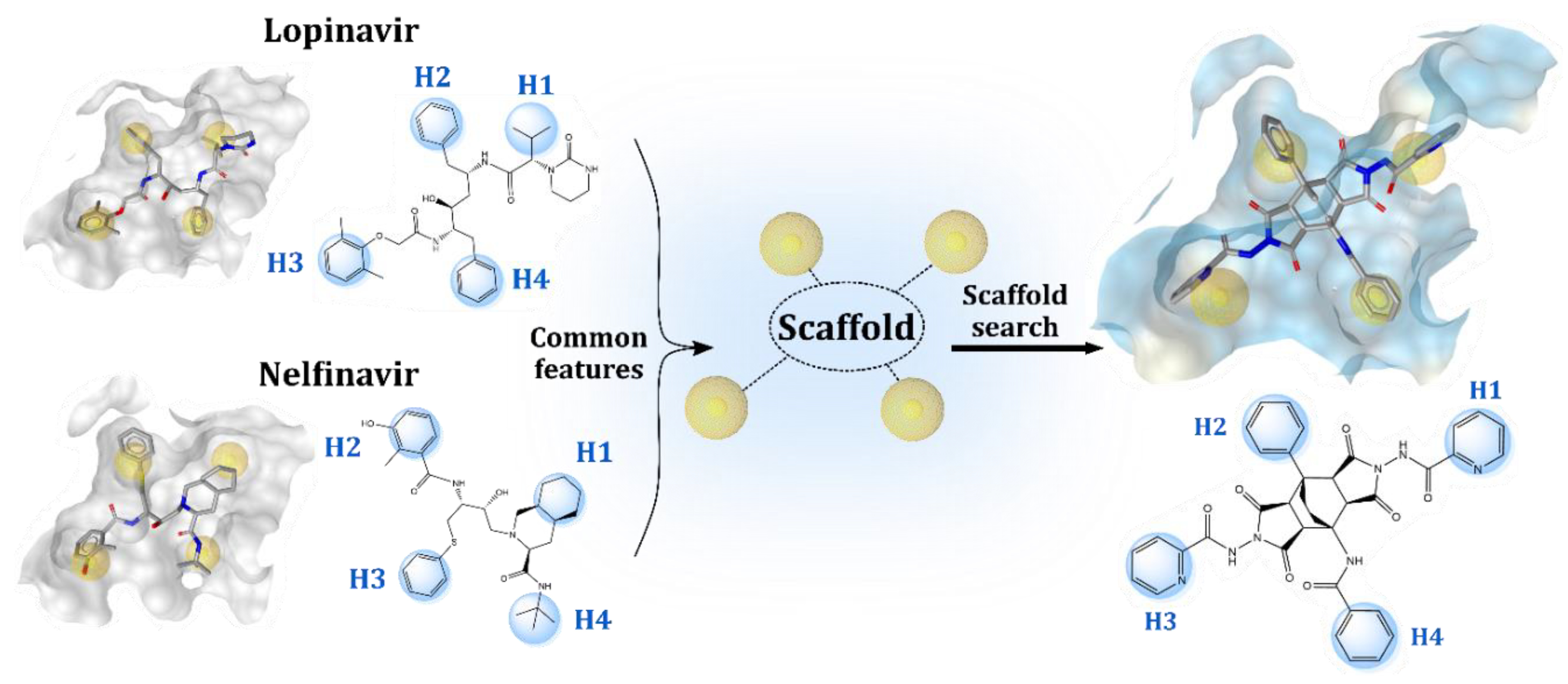
2.1. Structure-Based Design of Fused Bicyclo[2.2.2]octene as a Core Scaffold of SARS-CoV-2 3CLpro Inhibitors
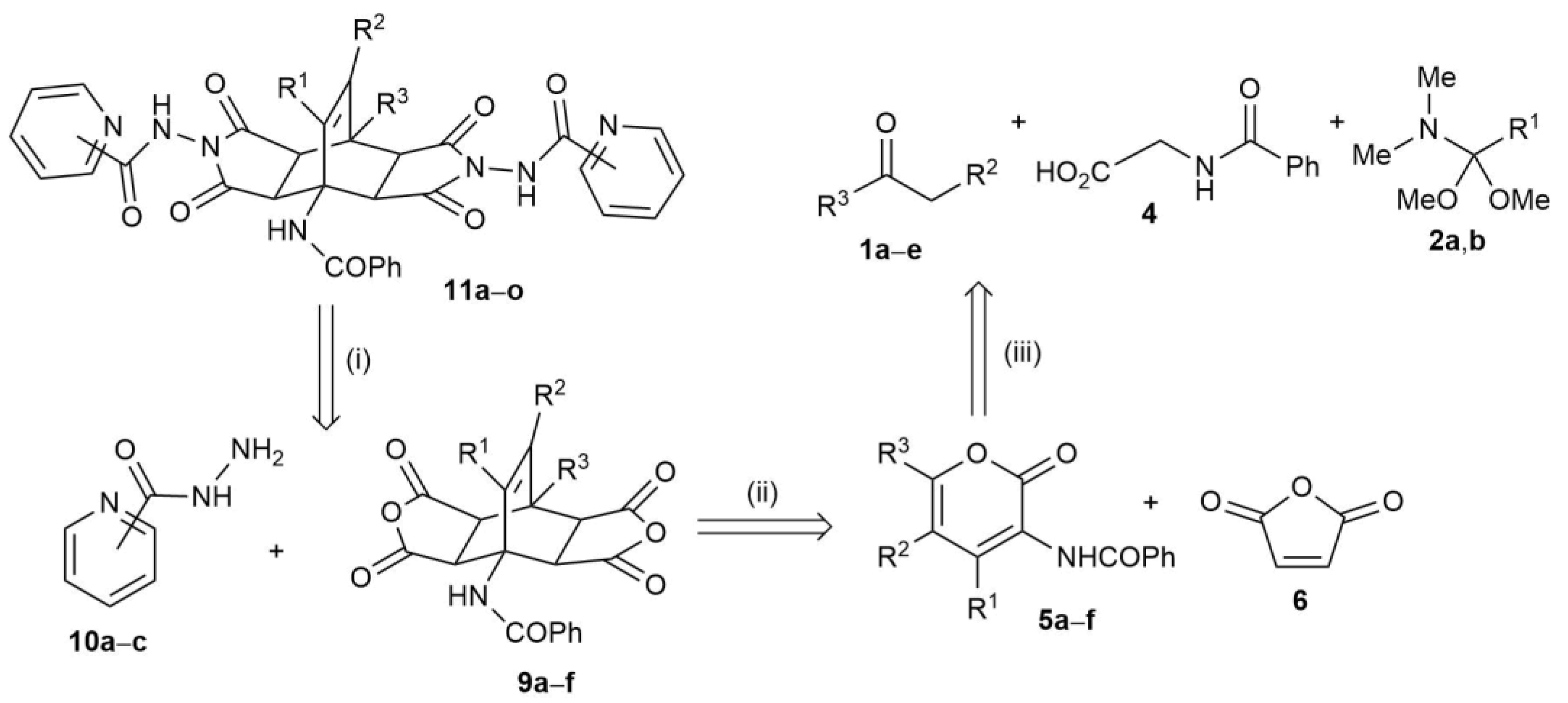
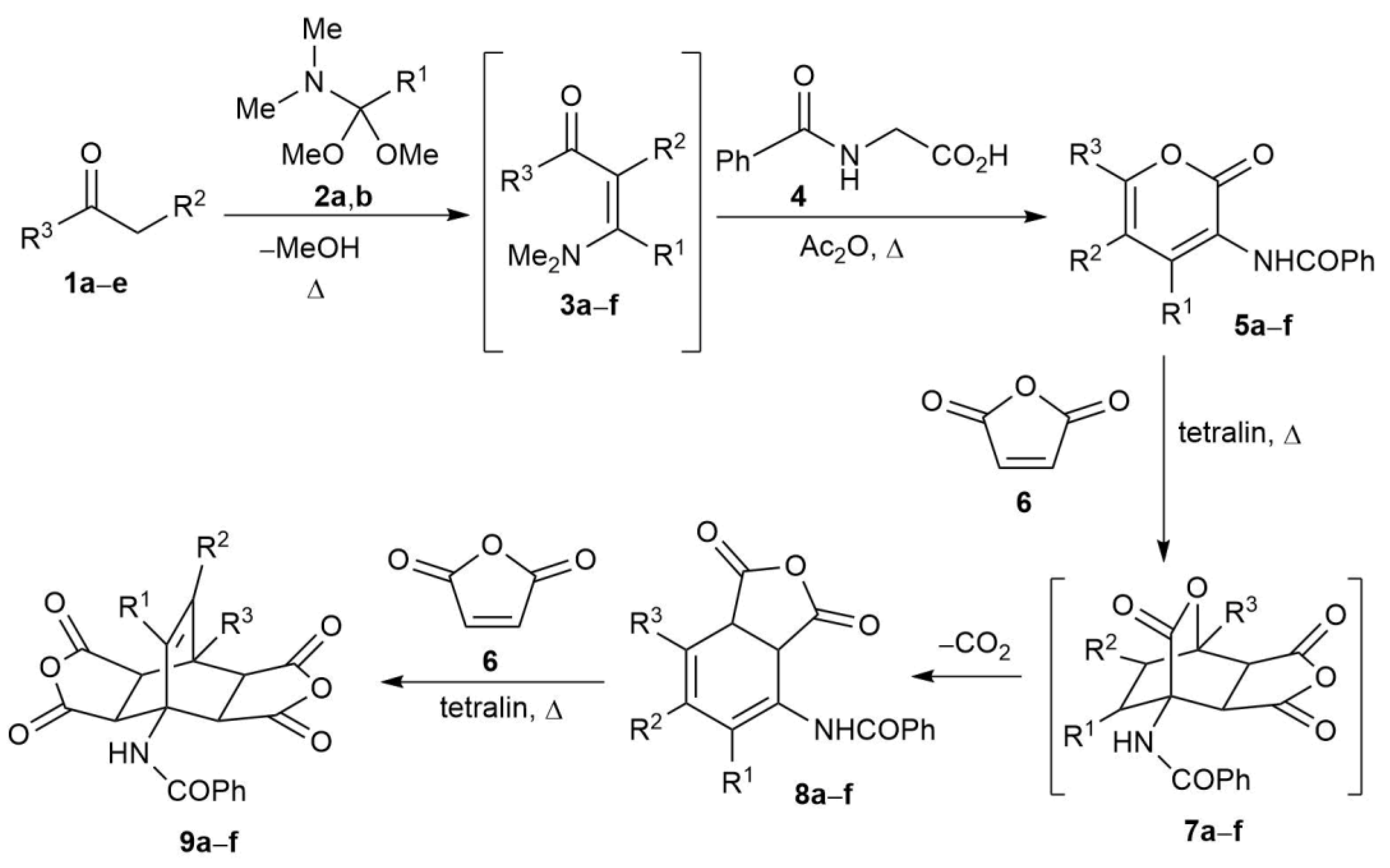
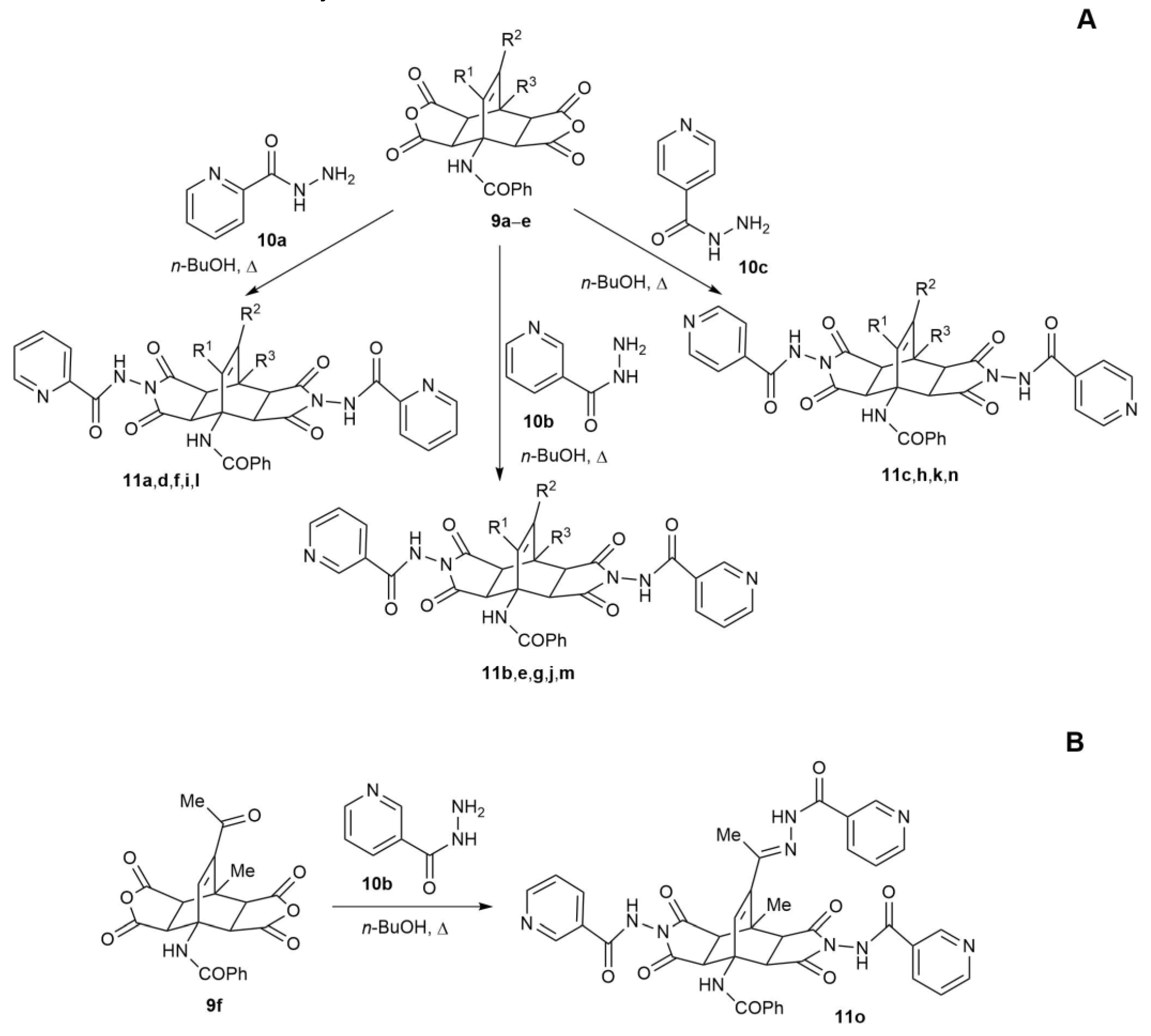
2.2. Synthesis of Fused Bicyclo[2.2.2]octenes
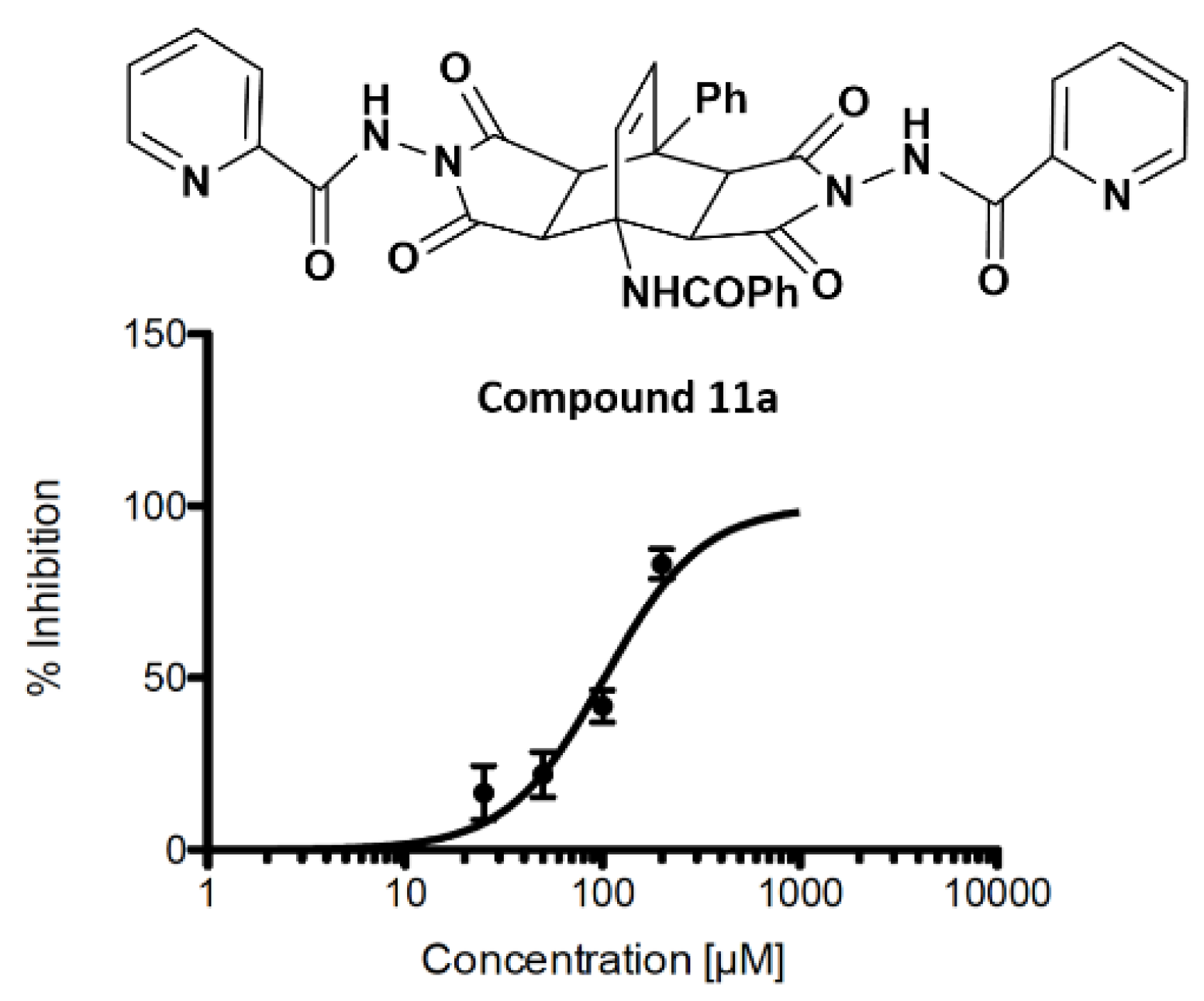
2.3. Inhibition of SARS-CoV-2 3CLpro Main Protease
2.4. Computational Evaluation of Inhibitor Binding to 3CLpro
3. Materials and Methods
3.1. Synthetic Procedures and Compounds Characterization Data
3.2. FRET-Based Assay of SARS-CoV-2 3CLpro Main Protease Inhibition Activity
3.3. Molecular Docking Calculations and Binding Site Analysis Using Molecular Probes
3.4. Molecular Dynamics Simulations
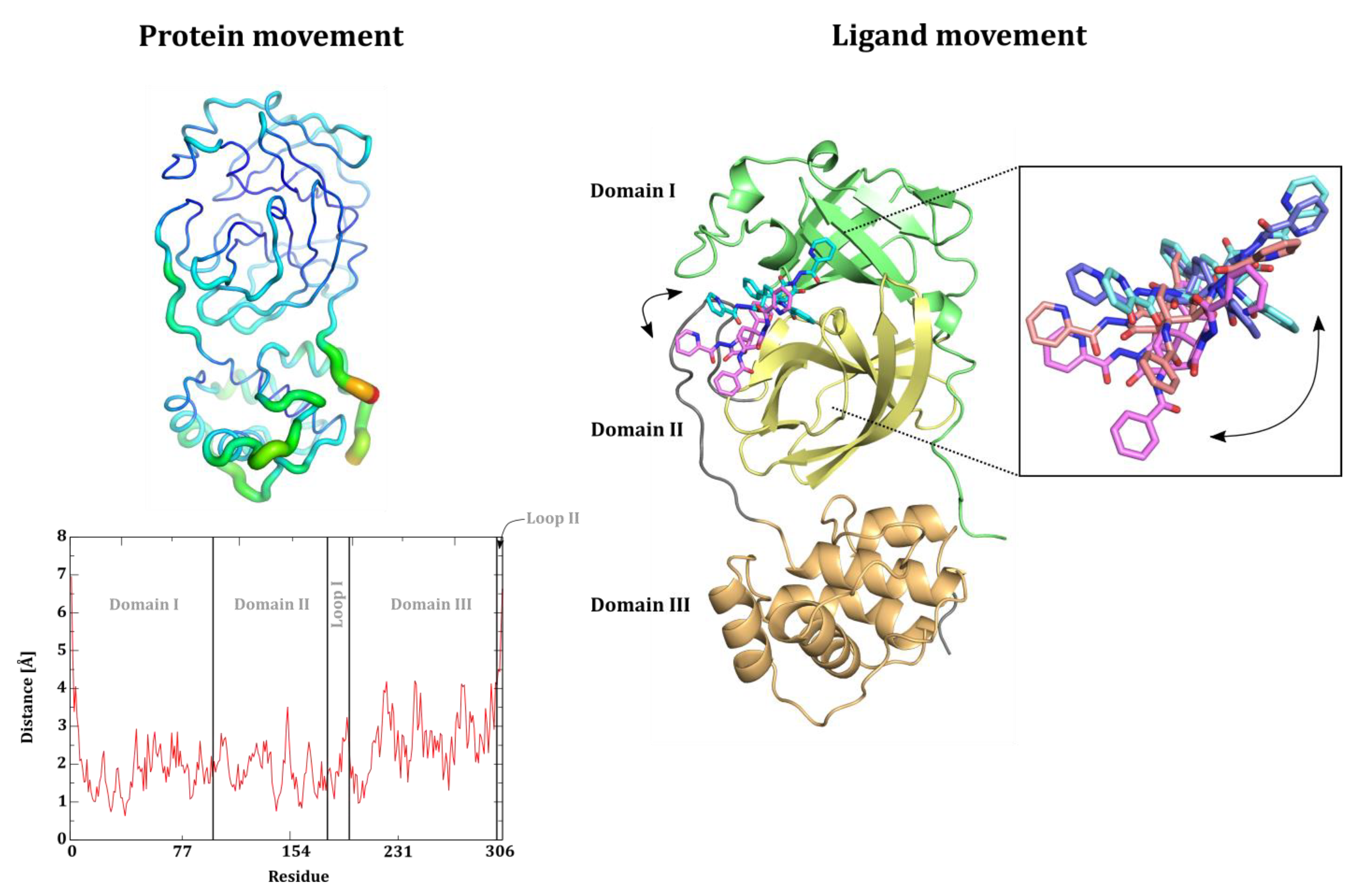
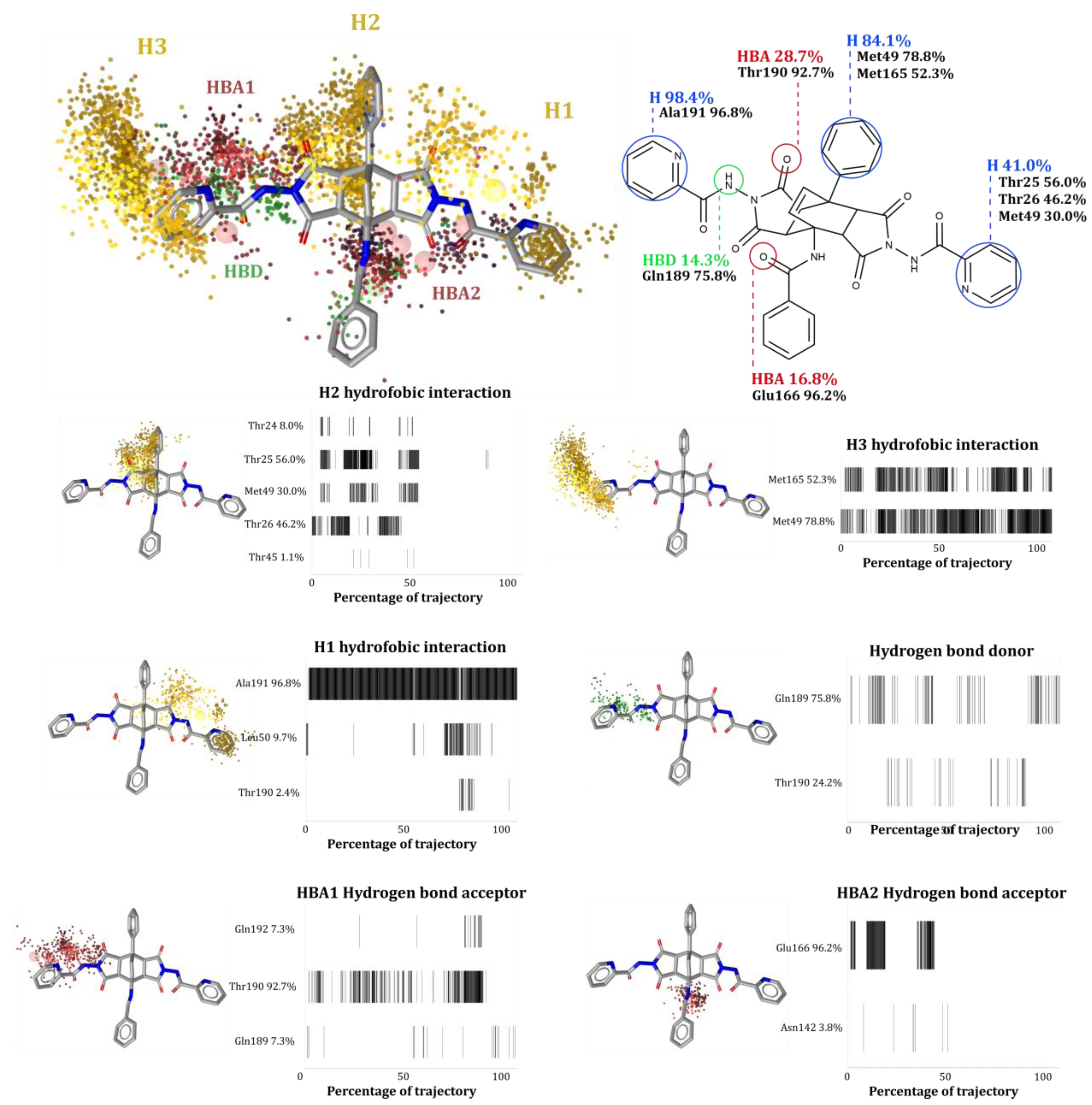
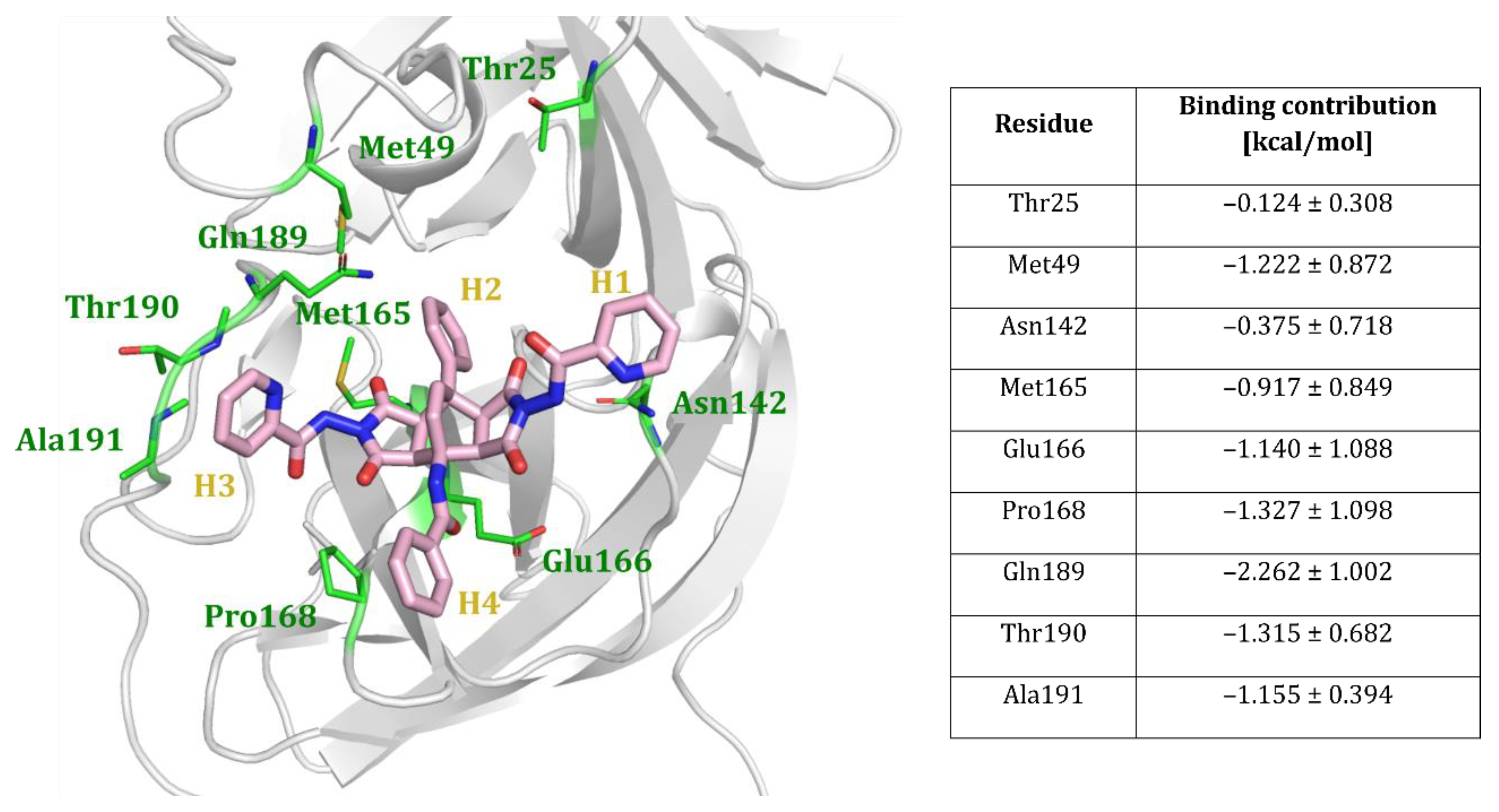
Analysis of the MD Trajectories
- (a)
- Cα RMSD and RMSF calculations
- (b)
- MM/GBSA binding free energy calculations
- (c)
- Dynamic pharmacophore model calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 589, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhar, Z.; Khan, S.; AlOmar, S.Y.; Alkhuriji, A.; Ahmad, A. ABBV-744 as a potential inhibitor of SARS-CoV-2 main protease enzyme against COVID-19. Sci. Rep. 2021, 11, 234. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramirez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- CDC—COVID-19. Available online: https://www.cdc.gov/coronavirus/2019-nCoV/index.html. (accessed on 29 September 2021).
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. Infez. Med. 2020, 28, 174–184. [Google Scholar]
- Herold, J.; Raabe, T.; Schelle-Prinz, B.; Siddell, S.G. Nucleotide sequence of the human coronavirus 229E RNA polymerase locus. Virology 1993, 195, 680–691. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Regulatory Approval of Lagevrio (Molnupiravir). Available online: https://www.gov.uk/government/publications/regulatory-approval-of-lagevrio-molnupiravir (accessed on 3 December 2021).
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. Febs. J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [Green Version]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemBioChem 2020, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why Are Lopinavir and Ritonavir Effective against the Newly Emerged Coronavirus 2019? Atomistic Insights into the Inhibitory Mechanisms. Biochemistry 2020, 59, 1769–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, H.; Watashi, K.; Saso, W.; Shionoya, K.; Iwanami, S.; Hirokawa, T.; Shirai, T.; Kanaya, S.; Ito, Y.; Kim, K.S.; et al. Potential anti-COVID-19 agents, cepharanthine and nelfinavir, and their usage for combination treatment. iScience 2021, 24, 102367. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, D.; Cattaneo, D.; Gervasoni, C.; Corbellino, M.; Galli, M.; Riva, A.; Gervasoni, C.; Clementi, E.; Clementi, E. Does lopinavir really inhibit SARS-CoV-2? Pharmacol. Res. 2020, 158, 104898. [Google Scholar] [CrossRef]
- Dorward, J.; Gbinigie, K. Lopinavir/Ritonavir: A Rapid Review of Effectiveness in COVID-19. 2020. Available online: https://www.cebm.net/covid-19/lopinavir-ritonavir-a-rapid-review-of-the-evidence-for-effectiveness-in-treating-covid/ (accessed on 12 November 2021).
- Fintelman-Rodrigues, N.; Sacramento, C.Q.; Ribeiro Lima, C.; Souza da Silva, F.; Ferreira, A.C.; Mattos, M.; de Freitas, C.S.; Cardoso Soares, V.; da Silva Gomes Dias, S.; Temerozo, J.R.; et al. Atazanavir, Alone or in Combination with Ritonavir, Inhibits SARS-CoV-2 Replication and Proinflammatory Cytokine Production. Antimicrob. Agents Chemother. 2020, 64, e00825-20. [Google Scholar] [CrossRef]
- Yamamoto, N.; Matsuyama, S.; Hoshino, T.; Yamamoto, N. Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; Tirado-Rives, J.; Deshmukh, M.; Ippolito, J.A.; Zhang, C.H.; Cabeza de Vaca, I.; Liosi, M.E.; Anderson, K.S.; Jorgensen, W.L. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2, ACS Med. Chem. Lett. 2020, 11, 2526–2533. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug. Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, J.J. Designing Peptidomimetics. Curr. Top. Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Song, Y.; Zhan, P.; Zhang, Q.; Liu, X. Conformational restriction: An effective tactic in ‘follow-on’-based drug discovery. Future. Med. Chem. 2014, 6, 885–901. [Google Scholar] [CrossRef]
- Ekar, J.; Kranjc, K. Synthesis of Hydrazinylpyridines via Nucleophilic Aromatic Substitution and Further Transformation to Bicyclo[2.2.2]octenes Fused with Two N-Aminosuccinimide Moieties. Synthesis 2020, 53, 1112–1120. [Google Scholar]
- Kranjc, K.; Juranovič, A.; Kočevar, M.; Perdih, F. Supramolecular Diversity of Oxabicyclo[2.2.2]octenes Formed between Substituted 2H-Pyran-2-ones and Vinyl-Moiety-Containing Dienophiles. Symmetry 2020, 12, 1714. [Google Scholar] [CrossRef]
- Kranjc, K.; Kočevar, M. Regio- and stereoselective syntheses and cycloadditions of substituted 2H-pyran-2-ones and their fused derivatives. Arkivoc 2013, (i), 333–363. [Google Scholar] [CrossRef]
- Wang, D.H.; Lee, K.M.; Lee, D.H.; Baczkowski, M.; Park, H.; McConney, M.E.; Tan, L.S. Role of Alicyclic Conformation-Isomerization in the Photomechanical Performance of Azobenzene-Functionalized Cross-Linked Polyimides Containing Tetra-Substituted Cyclohexane Moieties. Acs Macro. Lett. 2021, 10, 278–283. [Google Scholar] [CrossRef]
- Cantin, A.; Corma, A.; Diaz-Cabanas, M.J.; Jorda, J.L.; Moliner, M. Rational design and HT techniques allow the synthesis of new IWR zeolite polymorphs. J. Am. Chem. Soc. 2006, 128, 4216–4217. [Google Scholar] [CrossRef]
- Martín, N.; Paris, C.; Vennestrøm, P.N.R.; Thøgersen, J.R.; Moliner, M.; Corma, A. Cage-based small-pore catalysts for NH3-SCR prepared by combining bulky organic structure directing agents with modified zeolites as reagents. App. Catal. B 2017, 217, 125–136. [Google Scholar] [CrossRef]
- Moliner, M.; Serna, P.; Cantín, Á.; Sastre, G.; Díaz-Cabañas, M.J.; Corma, A. Synthesis of the Ti−Silicate Form of BEC Polymorph of β-Zeolite Assisted by Molecular Modeling. J. Phys. Chem. C 2008, 112, 19547–19554. [Google Scholar] [CrossRef]
- Sun, J.; Bonneau, C.; Cantin, A.; Corma, A.; Diaz-Cabanas, M.J.; Moliner, M.; Zhang, D.; Li, M.; Zou, X. The ITQ-37 mesoporous chiral zeolite. Nature 2009, 458, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. I. Mitteilung: Anlagerung von “Di-en”-kohlenwasserstoffen. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Diels, O.; Alder, K. Synthesen in der hydroaromatische Reihe; XIII. Mitteilung. “Dien-Synthesen” sauerstoffhaltiger Heteroringe. 3. Dien-Synthesen der Cumaline. Justus Liebigs Ann. Chem. 1931, 490, 257–266. [Google Scholar] [CrossRef]
- Karmarkar, K.S.; Samant, S.D. Comparative study of the Diels-Alder reactions of 4,6-bis(4-methoxyphenyl)-2H-pyran-2-one and 4-(4-methoxyphenyl)-6-methyl-2H-pyran-2-one and their thiones with some symmetrical dienophiles. Ind. J. Chem. Sect. B 1993, 32B, 1113–1118. [Google Scholar] [CrossRef]
- Abdulhamid, M.A.; Ma, X.; Ghanem, B.S.; Pinnau, I. Synthesis and Characterization of Organo-Soluble Polyimides Derived from Alicyclic Dianhydrides and a Dihydroxyl-Functionalized Spirobisindane Diamine. ACS App. Polym. Mater. 2018, 1, 63–69. [Google Scholar] [CrossRef]
- Nowak, I. Synthesis of polyfluorinated indacenes—A new type of sterically hindered alkenes. J. Fluor. Chem. 1999, 99, 59–66. [Google Scholar] [CrossRef]
- Westerhausen, M.; Stein, B.; Ossberger, M.W.; Görls, H.; Ruiz, J.C.G.; Nöth, P.M.H. Diels-Alder cycloaddition reactions of 1,1-dichloro-2,3,4,5-tetraethylgermole and 1-chloro-2,3,4,5-tetraethylphosphole with maleic anhydride and maleimide. Arkivoc 2007, 3, 46–59. [Google Scholar]
- Salakhov, M.S.; Umaeva, V.S.; Alikhanova, A.I. Synthesis of polychlorinated unsaturated cyclic dicarboxylic acid imides, Russ. J. Org. Chem. 2008, 44, 1438–1443. [Google Scholar]
- Mikhura, I.V.; Formanovsky, A.A.; Nozhevnikova, E.V.; Prokhorenko, I.A.; Korshun, V.A. Dianhydrides of 1(4)-substituted 7,8-diphenylbicyclo[2.2.2]oct-7-ene-2,3,5,6-tetracarboxylic acids. Mendeleev. Comm. 2017, 27, 446–447. [Google Scholar] [CrossRef]
- Huang, P.; Liu, L.; Chang, W.; Li, J. An unexpected double Diels-Alder reaction of (E)-2-bromo-4-aryl-1,3-pentadiene involving [1,5]-hydrogen migration and HBr elimination: Synthesis of bicyclo[2.2.2]octene derivatives. Chem. Asian. J. 2015, 10, 548–552. [Google Scholar] [CrossRef]
- Strübing, D.; von Wangelin, A.J.; Neumann, H.; Gördes, D.; Hübner, S.; Klaus, S.; Spannenberg, A.; Beller, M. Multicomponent reaction of aldehydes, anhydrides, and dienophiles: Synthesis of “butterfly”-like diazatetradecenes. Eur. J. Org. Chem. 2005, 2005, 107–113. [Google Scholar] [CrossRef]
- Požgan, F.; Kranjc, K.; Kepe, V.; Polanc, S.; Kočevar, M. Synthesis of 2H-pyran-2-ones and fused pyran-2-ones as useful building blocks. Arkivoc 2007, 8, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Kepe, V.; Kočevar, M.; Polanc, S. One-pot synthesis of some 2H-Pyran-2-one derivatives. J. Heterocycl. Chem. 1996, 33, 1707–1710. [Google Scholar] [CrossRef]
- Kepe, V.; Kočevar, M.; Polanc, S.; Verček, B.; Tišler, M. A simple and general one-pot synthesis of some 2H-pyran-2-ones and fused pyran-2-ones. Tetrahedron 1990, 46, 2081–2088. [Google Scholar] [CrossRef]
- Kepe, V.; Polanc, S.; Kočevar, M. A Simple Preparation of Some 4-Methyl-2H-pyran-2-ones. Heterocycles 1998, 48, 671–678. [Google Scholar]
- Kranjc, K.; Leban, I.; Polanc, S.; Kočevar, M. Diels-Alder Cycloaddition of Highly Substituted Pyran-2-ones with Maleic Anhydride. Heterocycles 2002, 58, 183–190. [Google Scholar]
- Hren, J.; Kranjc, K.; Polanc, S.; Kočevar, M. Aqueous versus Neat Reaction Conditions: The Microwave-Assisted, Selective Conversion of a Fused Anhydride Ring with Amines in the Presence of a Keto Group. Synthesis 2008, 3, 452-258. [Google Scholar]
- Kranjc, K.; Perdih, F.; Kočevar, M. Effect of Ring Size on the Exo/Endo Selectivity of a Thermal Double Cycloaddition of Fused Pyran-2-ones. J. Org. Chem. 2009, 74, 6303–6306. [Google Scholar] [CrossRef]
- Martelanc, M.; Kranjc, K.; Polanc, S.; Kočevar, M. An efficient microwave-assisted green transformation of fused succinic ahydrides into N-aminosuccinimide derivatives of bicyclo[2.2.2]octene in water. Green Chem. 2005, 7, 737–741. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Creighton, A.M.; Kozlowska, H.; Thampatty, P.; Allan, W.P.; Yalowich, J.C. Mitindomide is a catalytic inhibitor of DNA topoisomerase II that acts at the bisdioxopiperazine binding site. Mol. Pharmacol. 1997, 52, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.J.; Powis, G.; Melder, D.C.; Deutsch, H.M.; Zalkow, L.H. Cross-linking of DNA by diimide antitumor agents and the relationship to cytotoxicity in A204 rhabdomyosarcoma cells. J. Cell. Pharmacol. 1990, 1, 103–108. [Google Scholar]
- Deutsch, H.W.; Gelbaum, L.T.; McLaughlin, M.; Fleischmann, T.J.; Earnhart, L.L.; Haugwitz, R.D.; Zalkow, L.H. Synthesis of congeners and prodrugs of the benzene maleimide photoadduct mitindomide as potential antitumor agents. 2. J. Med. Chem. 1990, 29, 2164–2170. [Google Scholar] [CrossRef]
- Turło, J.; Suski, S.; Zawadowski, T. Synthesis of N,N’-bis-substituted diimides related to tricyclo[6.2.2.01,6]dodecane with an expected activity on the central nervous system. Il Farmaco 1998, 53, 451–454. [Google Scholar] [CrossRef]
- Buonfiglio, R.; Recanatini, M.; Masetti, M. Protein Flexibility in Drug Discovery: From Theory to Computation. ChemMedChem 2015, 10, 1141–1148. [Google Scholar] [CrossRef]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [Green Version]
- Mortier, J.; Rakers, C.; Bermudez, M.; Murgueitio, M.S.; Riniker, S.; Wolber, G. The impact of molecular dynamics on drug design: Applications for the characterization of ligand–macromolecule complexes. Drug Discov. Today 2015, 20, 686–702. [Google Scholar] [CrossRef]
- Bock, A.; Bermudez, M.; Krebs, F.; Matera, C.; Chirinda, B.; Sydow, D.; Dallanoce, C.; Holzgrabe, U.; De Amici, M.; Lohse, M.J.; et al. Ligand Binding Ensembles Determine Graded Agonist Efficacies at a G Protein-coupled Receptor. J. Biol. Chem. 2016, 291, 16375–16389. [Google Scholar] [CrossRef] [Green Version]
- Klingele, M.H.; Brooker, S. From N-Substituted Thioamides to Symmetrical and Unsymmetrical 3,4,5-Trisubstituted 4H-1,2,4-Triazoles: Synthesis and Characterisation of New Chelating Ligands, Eur. J. Org. Chem. 2004, 16, 3422–3434. [Google Scholar]
- Revanasiddappa, B.C.; Subrahmanyam, E.V.S. Synthesis and biological studies of some novel thiazolidinones. J. Pharm. Res. 2009, 8, 122–124. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, D.; Khan, S.A.; Chawla, G. Design & synthesis of 2-(substituted aryloxy)-5-(substituted benzylidene)-3-phenyl-2,5-dihydro-1H-[1,2,4] triazin-6-one as potential anticonvulsant agents. Eur. J. Med. Chem. 2010, 45, 3960–3969. [Google Scholar]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. WIREs Comput Mol Sci. 2020, 10, e1468. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Case, D.H.M.; Aktulga, K.; Belfon, I.Y.; Ben-Shalom, S.R.; Brozell, D.S.; Cerutti, T.E.; Cheatham, G.A., III; Cisneros, V.W.D.; Cruzeiro, T.A.; Darden, R.E.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2017, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.0; Schrodinger, LCC: New York, NY, USA, 2015.
- Sydow, D. Dynophores: Novel Dynamic Pharmacophores, Masterarbeit. Master’s Thesis, Universität zu Berlin, Lebenswissenschaftliche Fakultät, Berlin, Germany, 2015. [Google Scholar]
- Pogorelčnik, B.; Janežič, M.; Sosič, I.; Gobec, S.; Solmajer, T.; Perdih, A. 4,6-Substituted-1,3,5-triazin-2(1H)-ones as monocyclic catalytic inhibitors of human DNA topoisomerase IIα targeting the ATP binding site. Bioorg. Med. Chem. 2015, 23, 4218–4229. [Google Scholar] [CrossRef]
- Pfizer Unveils Its Oral SARS-CoV-2 Inhibitor. Available online: https://cen.acs.org/acs-news/acs-meeting-news/Pfizer-unveils-oral-SARS-CoV/99/i13 (accessed on 3 December 2021).
- Aljoundi, A.; Bjij, I.; El Rashedy, A.; Soliman, M.E.S. Covalent Versus Non-covalent Enzyme Inhibition: Which Route Should We Take? A Justification of the Good and Bad from Molecular Modelling Perspective. Protein J. 2020, 39, 97–105. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Bicyclo[2.2.2]octenes 9 | Hydrazide 10 | n(10) in mmol | Product 11 | Yield (%) b | |||
|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||||
| 1 | H | H | Ph | 9a | 10a | 2.0 | 11a | 69 |
| 2 | H | H | Ph | 9a | 10b | 2.0 | 11b | 70 |
| 3 | H | H | Ph | 9a | 10c | 2.0 | 11c | 72 |
| 4 | Me | H | Ph | 9b | 10a | 2.0 | 11d c | 62 |
| 5 | Me | H | Ph | 9b | 10b | 2.0 | 11e c | 84 |
| 6 | H | H | 2-thienyl | 9c | 10a | 1.5 | 11f | 72 |
| 7 | H | H | 2-thienyl | 9c | 10b | 1.5 | 11g | 79 |
| 8 | H | H | 2-thienyl | 9c | 10c | 1.5 | 11h | 77 |
| 9 | H | H | 2-furyl | 9d | 10a | 1.5 | 11i | 68 |
| 10 | H | H | 2-furyl | 9d | 10b | 1.5 | 11j | 70 |
| 11 | H | H | 2-furyl | 9d | 10c | 1.5 | 11k | 73 |
| 12 | H | 4-MeO-C6H4- | Me | 9e | 10a | 1.1 | 11l | 64 |
| 13 | H | 4-MeO-C6H4- | Me | 9e | 10b | 1.1 | 11m | 67 |
| 14 | H | 4-MeO-C6H4- | Me | 9e | 10c | 1.1 | 11n | 64 |
| 15 | H | COMe | Me | 9f | 10b | 1.65 | 11o d | 48 |
 | ||||
| R1 | R2 | R3 | IC50 [μM] | |
| 11a | H | H | Ph | 102.2 |
| 11d | Me | H | Ph | <1000 |
| 11f | H | H | 2-thienyl | <1000 |
| 11i | H | H | 2-furyl | <1000 |
| 11l | H | 4-MeO-C6H4- | Me | <1000 |
 | ||||
| R1 | R2 | R3 | ||
| 11b | H | H | Ph | <1000 |
| 11e | Me | H | Ph | ~200.0 |
| 11g | H | H | 2-thienyl | <1000 |
| 11j | H | H | 2-furyl | <1000 |
| 11m | H | 4-MeO-C6H4- | Me | <1000 |
| 11o | H | -C(Me)=NNH-CO-(3-pyridyl) | Me | <1000 |
 | ||||
| R1 | R2 | R3 | ||
| 11c | H | H | Ph | <1000 |
| 11h | H | H | 2-thienyl | <1000 |
| 11k | H | H | 2-furyl | <1000 |
| 11n | H | 4-MeO-C6H4- | Me | <1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herlah, B.; Hoivik, A.; Jamšek, L.; Valjavec, K.; Yamamoto, N.; Hoshino, T.; Kranjc, K.; Perdih, A. Design, Synthesis and Evaluation of Fused Bicyclo[2.2.2]octene as a Potential Core Scaffold for the Non-Covalent Inhibitors of SARS-CoV-2 3CLpro Main Protease. Pharmaceuticals 2022, 15, 539. https://doi.org/10.3390/ph15050539
Herlah B, Hoivik A, Jamšek L, Valjavec K, Yamamoto N, Hoshino T, Kranjc K, Perdih A. Design, Synthesis and Evaluation of Fused Bicyclo[2.2.2]octene as a Potential Core Scaffold for the Non-Covalent Inhibitors of SARS-CoV-2 3CLpro Main Protease. Pharmaceuticals. 2022; 15(5):539. https://doi.org/10.3390/ph15050539
Chicago/Turabian StyleHerlah, Barbara, Andrej Hoivik, Luka Jamšek, Katja Valjavec, Norio Yamamoto, Tyuji Hoshino, Krištof Kranjc, and Andrej Perdih. 2022. "Design, Synthesis and Evaluation of Fused Bicyclo[2.2.2]octene as a Potential Core Scaffold for the Non-Covalent Inhibitors of SARS-CoV-2 3CLpro Main Protease" Pharmaceuticals 15, no. 5: 539. https://doi.org/10.3390/ph15050539