
Investigation of Direct and Retro Chromone-2-Carboxamides Based Analogs of Pseudomonas aeruginosa Quorum Sensing Signal as New Anti-Biofilm Agents


 , , ,
, , ,
Abstract
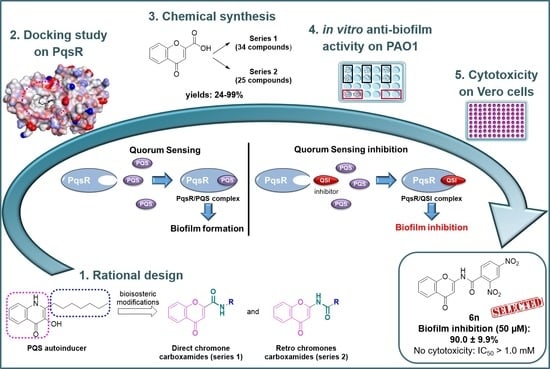
:
1. Introduction
2. Results and Discussion
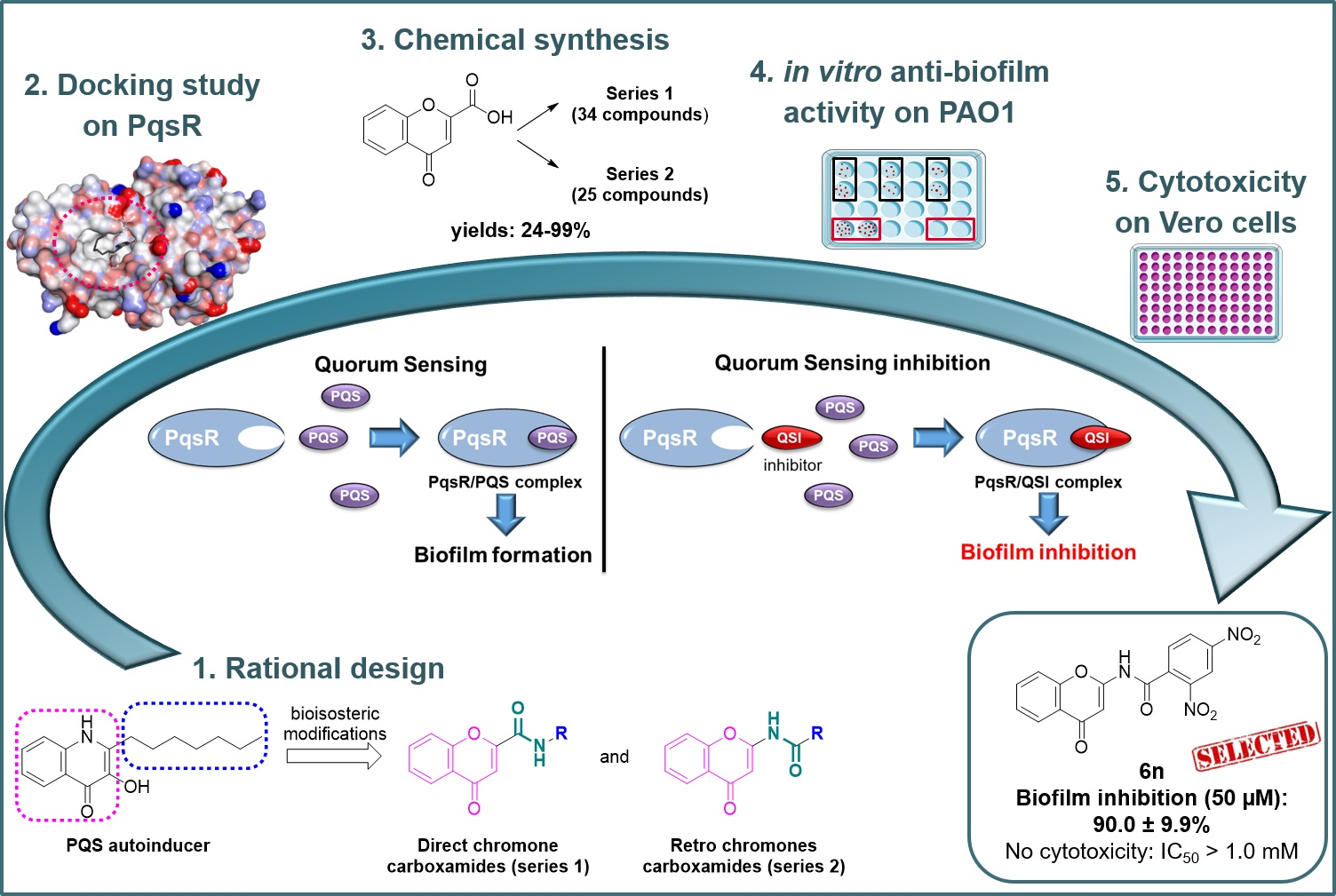
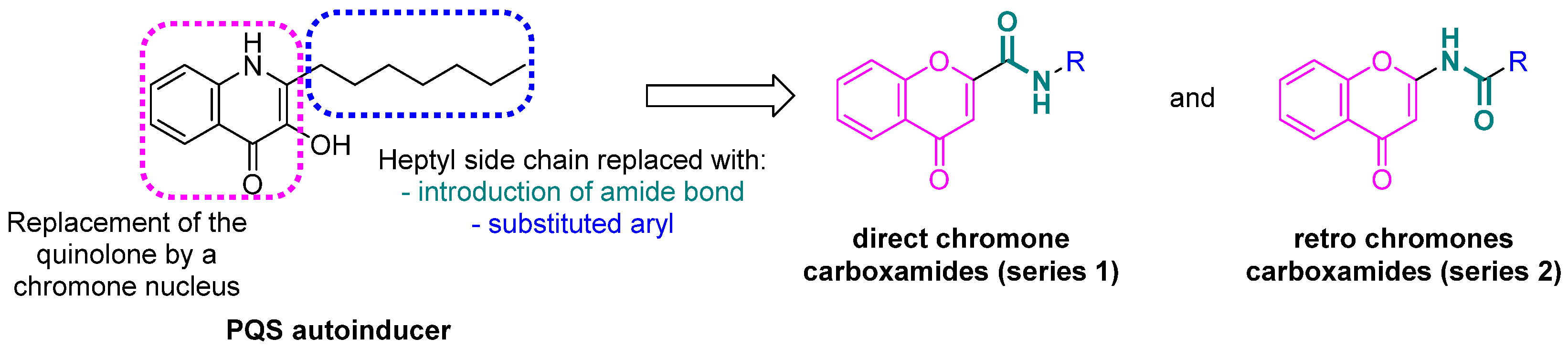
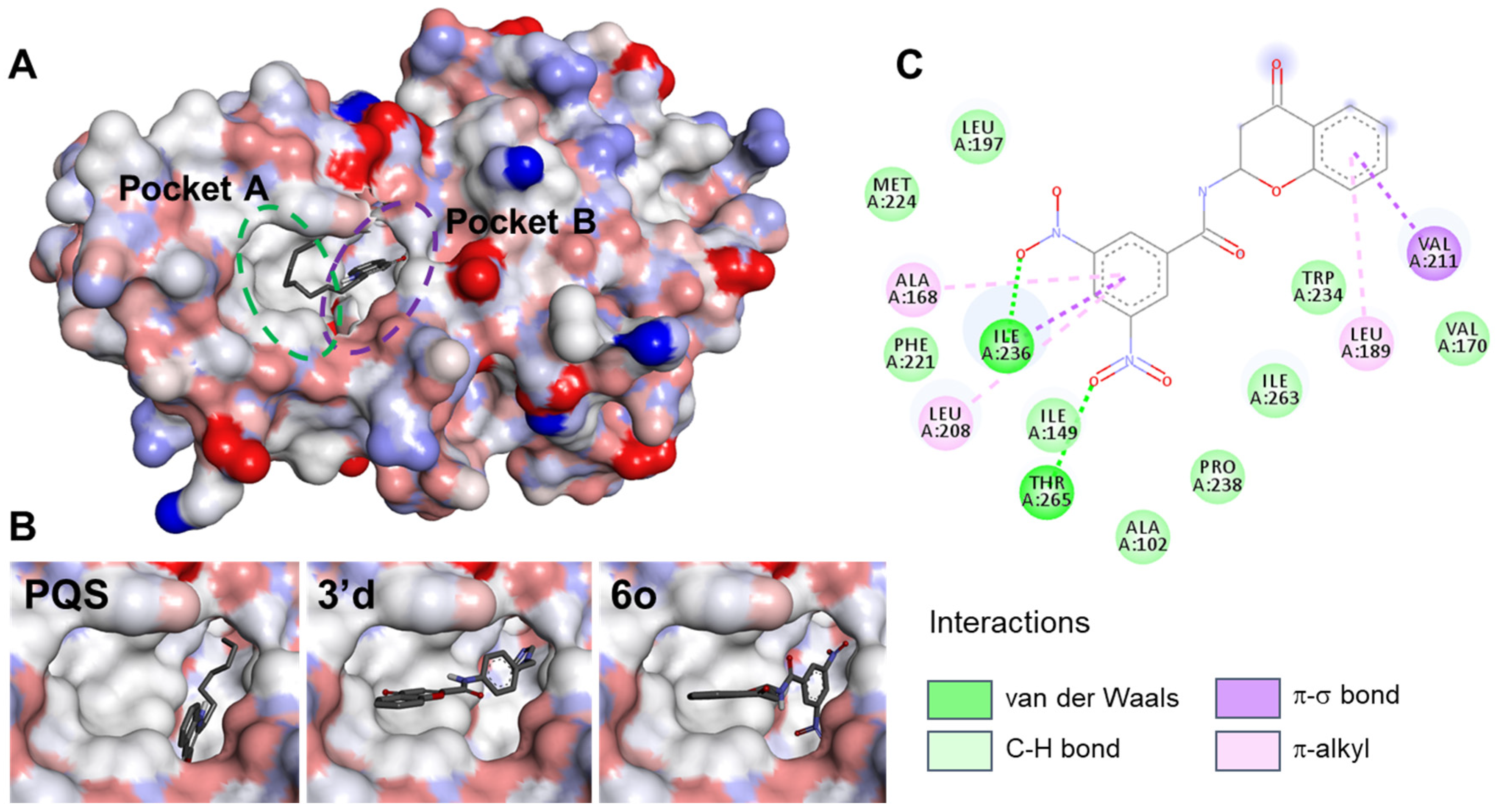
2.1. Docking of Chromone 2-Carboxamides with PqsR
2.2. Synthesis
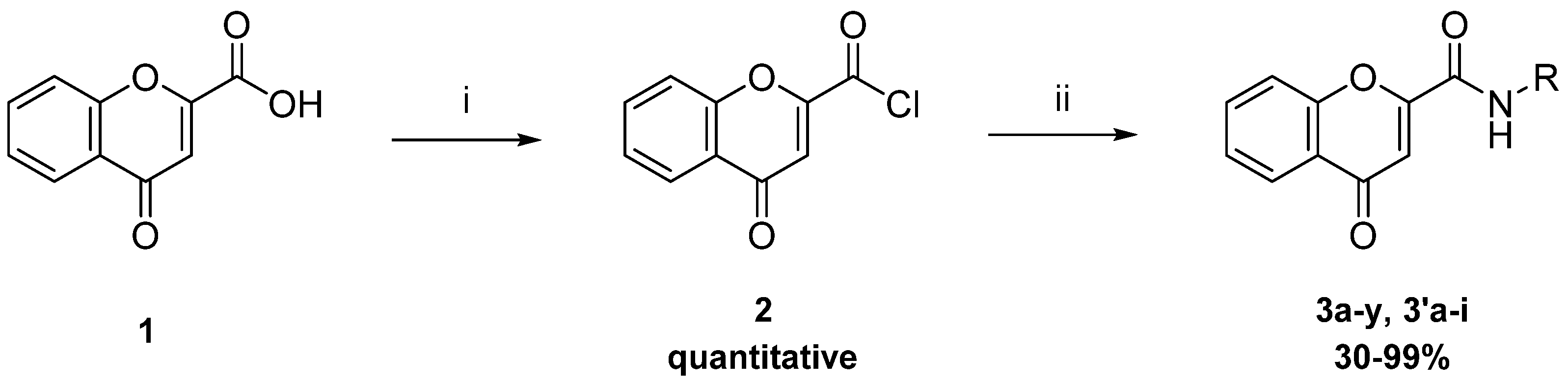
2.2.1. Synthesis of Direct Chromone Carboxamides Series 1
2.2.2. Synthesis of Retro Chromone Carboxamides Series 2
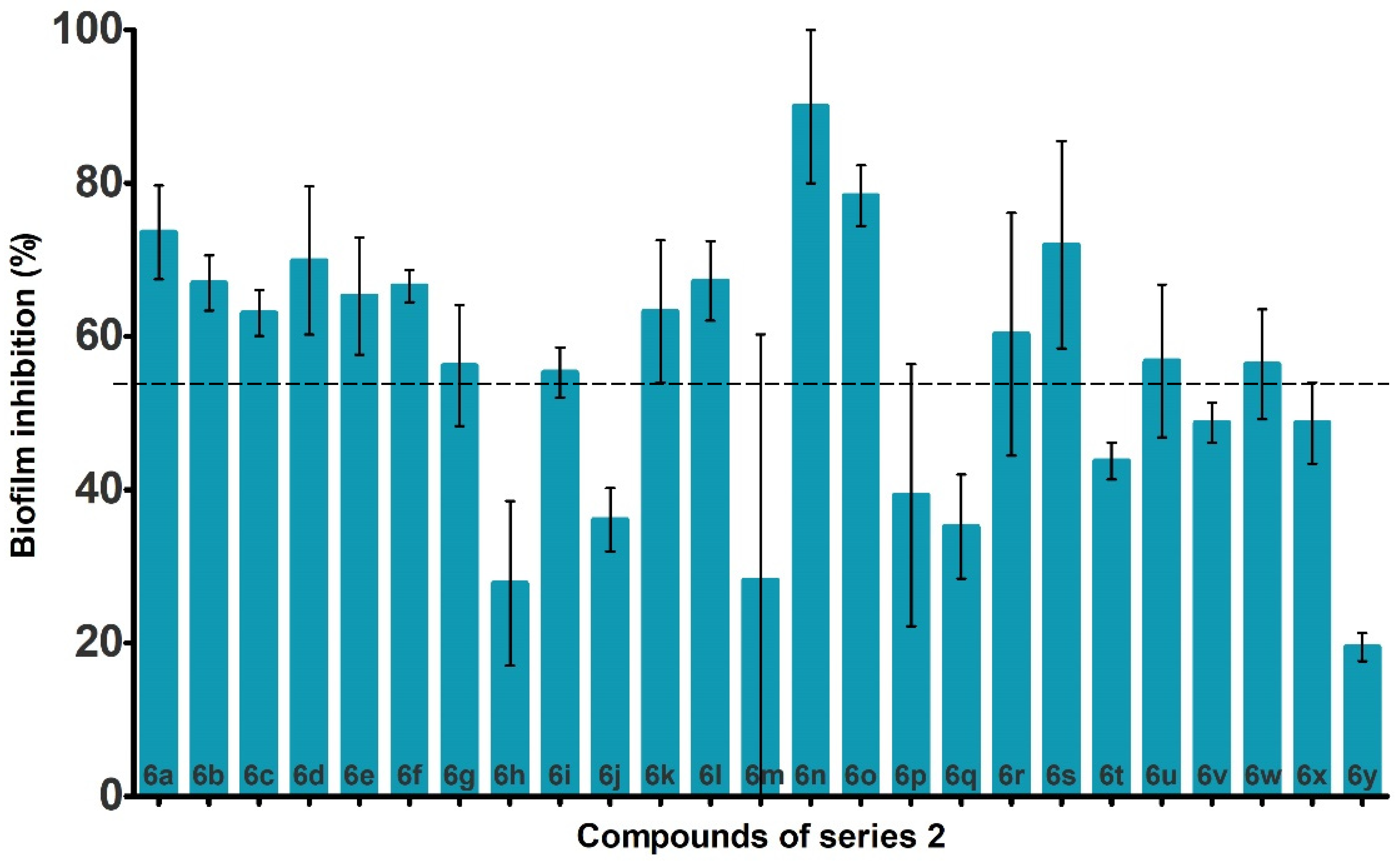
2.3. Anti-Biofilm Activity against P. aeruginosa
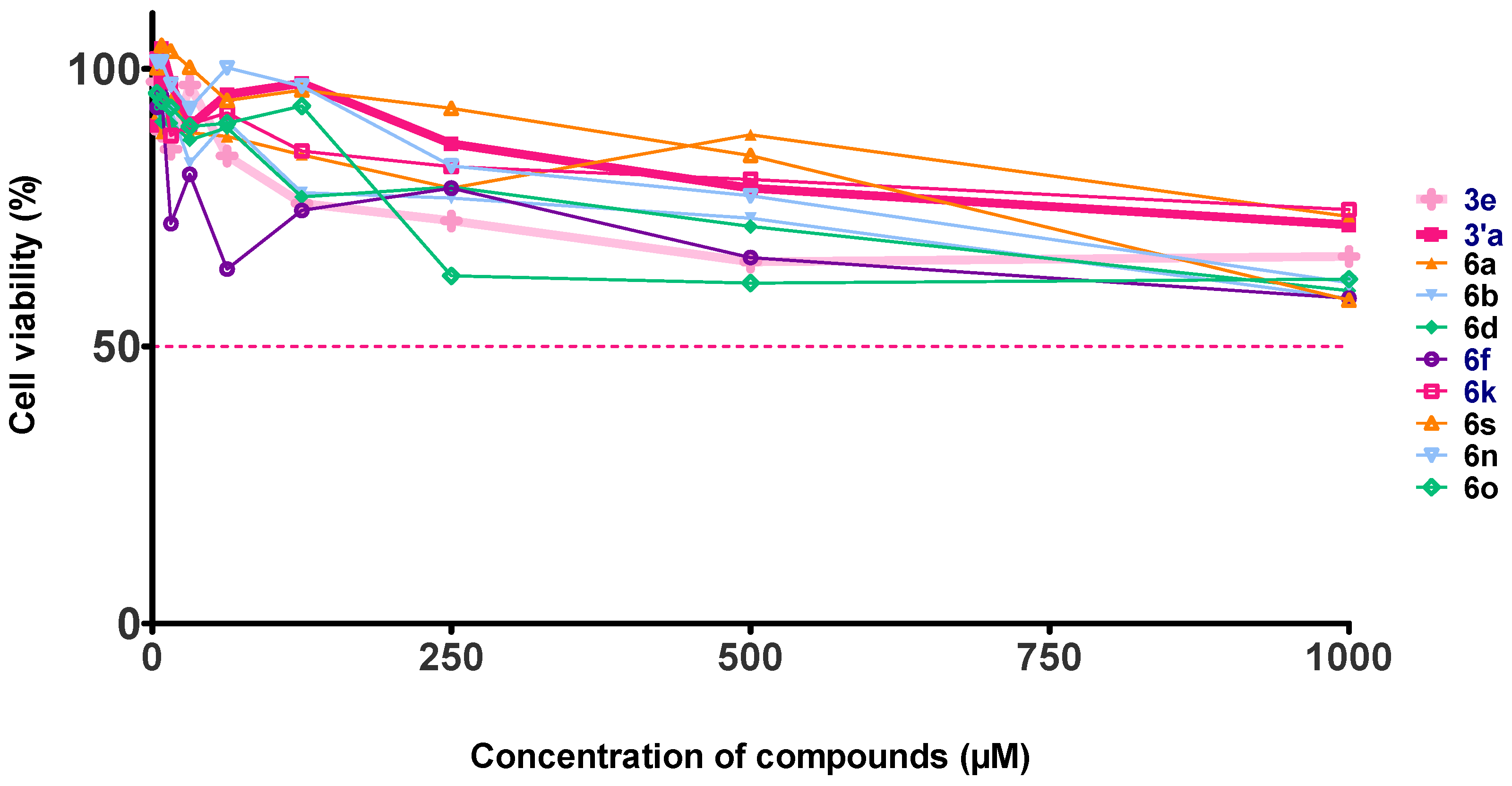
2.4. Evaluation of the Cytotoxic Effect on Vero Cells
3. Materials and Methods
3.1. Molecular Docking
3.1.1. Protein Structure File and Ligand Preparation
3.1.2. Structure-Based Virtual Screening
3.2. Chemistry
3.2.1. General
3.2.2. 4-Oxo-4H-chromene-2-carbonyl chloride (2)
3.2.3. General Procedure for the Synthesis of Direct Chromone Carboxamides (Series 1, 3a-y and 3′a-g)
3.2.4. General Procedure for the Synthesis of Retro Chromone Carboxamides
3.3. Bacterial Strains and Growth Conditions
3.4. Anti-Biofilm Assay
3.5. Cytotoxic Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Rickard, A.H.; Davies, D.G. Biofilms and biocomplexity. Microbe 2007, 2, 347. [Google Scholar] [CrossRef] [Green Version]
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-Related Infections: Bridging the Gap between Clinical Management and Fundamental Aspects of Recalcitrance toward Antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Health Organization Report 2017. Available online: http://www.who.int/en/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 25 October 2021).
- Lu, C.; Maurer, C.K.; Kirsch, B.; Steinbach, A.; Hartmann, R.W. Overcoming the Unexpected Functional Inversion of a PqsR Antagonist in Pseudomonas aeruginosa: An In Vivo Potent Antivirulence Agent Targeting pqs Quorum Sensing. Angew. Chem. Int. Ed. 2014, 53, 1109–1112. [Google Scholar] [CrossRef]
- Brackman, G.; Cos, P.; Maes, L.; Nelis, H.J.; Coenye, T. Quorum Sensing Inhibitors Increase the Susceptibility of Bacterial Biofilms to Antibiotics In Vitro and In Vivo. Antimicrob. Agents Chemother. 2011, 55, 2655–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furiga, A.; Lajoie, B.; El Hage, S.; Baziard, G.; Roques, C. Impairment of Pseudomonas aeruginosa Biofilm Resistance to Antibiotics by Combining the Drugs with a New Quorum-Sensing Inhibitor. Antimicrob. Agents Chemother. 2016, 60, 1676–1686. [Google Scholar] [CrossRef] [Green Version]
- Kalia, V.C. Quorum sensing inhibitors: An overview. Biotechnol. Adv. 2013, 31, 224–245. [Google Scholar] [CrossRef]
- Frei, R.; Breitbach, A.S.; Blackwell, H.E. 2-Aminobenzimidazole Derivatives Strongly Inhibit and Disperse Pseudomonas aeruginosa Biofilms. Angew. Chem. Int. Ed. 2012, 51, 5226–5229. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhang, L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 2015, 6, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Déziel, E.; Lépine, F.; Milot, S.; He, J.; Mindrinos, M.N.; Tompkins, R.G.; Rahme, L.G. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc. Nat. Acad. Sci. USA 2004, 101, 1339–1344. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Krishnan, G.; Goumnerov, B.; Tsongalis, J.; Tompkins, R.; Rahme, L.G. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc. Nat. Acad. Sci. USA 2001, 98, 14613–14618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allesen-Holm, M.; Barken, K.B.; Yang, L.; Klausen, M.; Webb, J.S.; Kjelleberg, S.; Molin, S.; Givskov, M.; Tolker-Nielsen, T. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 2006, 59, 1114–1128. [Google Scholar] [CrossRef] [PubMed]
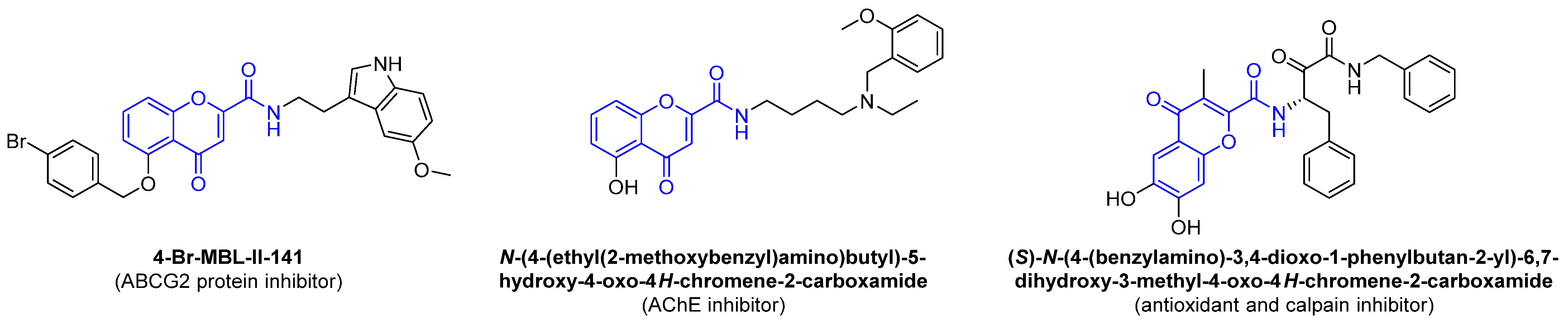
- Reis, J.; Gaspar, A.; Milhazes, N.; Borges, F. Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem. 2017, 60, 7941–7957. [Google Scholar] [CrossRef]
- Roussel, E.; Moréno, A.; Altounian, N.; Philouze, C.; Pérès, B.; Thomas, A.; Renaudet, O.; Falson, P.; Boumendjel, A. Chromones bearing amino acid residues: Easily accessible and potent inhibitors of the breast cancer resistance protein ABCG2. Eur. J. Med. Chem. 2020, 202, 112503. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qiang, X.; Li, Y.; Sang, Z.; Li, Y.; Tan, Z.; Deng, Y. Design, synthesis and evaluation of chromone-2-carboxamido-alkylbenzylamines as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, Y.H.; Jung, S.Y.; Kim, H.J.; Jin, C.; Lee, Y.S. Synthesis of chromone carboxamide derivatives with antioxidative and calpain inhibitory properties. Eur. J. Med. Chem. 2011, 46, 1721–1728. [Google Scholar] [CrossRef]
- Gaspar, A.; Matos, M.J.; Garrido, J.; Uriarte, E.; Borges, F. Chromone: A valid scaffold in medicinal chemistry. Chem. Rev. 2014, 114, 4960–4992. [Google Scholar] [CrossRef] [Green Version]
- Keri, R.S.; Budagumpi, S.; Pai, R.K.; Balakrishna, R.G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014, 78, 340–374. [Google Scholar] [CrossRef]
- Cai, Z.; Ding, Z.; Hao, Y.; Ni, T.; Xie, F.; Zhao, J.; Li, R.; Yu, S.; Wang, T.; Chai, X.; et al. Design, synthesis, and SAR study of 3-(benzo[d][1,3]dioxol-5-yl)-N-benzylpropanamide as novel potent synergists against fluconazole-resistant Candida albicans. Bioorg. Med. Chem. Lett. 2017, 27, 4571–4575. [Google Scholar] [CrossRef]
- Zitko, J.; Mindlová, A.; Valášek, O.; Jand’ourek, O.; Paterová, P.; Janoušek, J.; Konečná, K.; Doležal, M. Design, Synthesis and Evaluation of N-pyrazinylbenzamides as Potential Antimycobacterial Agents. Molecules 2018, 23, 2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalilzadeh, P.; Lajoie, B.; El Hage, S.; Furiga, A.; Baziard, G.; Berge, M.; Roques, C. Growth inhibition of adherent Pseudomonas aeruginosa by an N-butanoyl-l-homoserine lactone analog. Can. J. Microbiol. 2010, 56, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.M.; Forli, S. Using AutoDock 4 and AutoDock Vina with AutoDockTools: A Tutorial; The Scripps Research Institute: La Jolla, CA, USA, 2012. [Google Scholar]
- Ilangovan, A.; Fletcher, M.; Rampioni, G.; Pustelny, C.; Rumbaugh, K.; Heeb, S.; Cámara, M.; Truman, A.; Chhabra, S.R.; Emsley, J.; et al. Structural Basis for Native Agonist and Synthetic Inhibitor Recognition by the Pseudomonas aeruginosa Quorum Sensing Regulator PqsR (MvfR). PLoS Pathog. 2013, 9, e1003508. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Cheng, J.; Wang, Y.; Shen, X. The Pseudomonas Quinolone Signal (PQS): Not Just for Quorum Sensing Anymore. Front. Cell. Infect. Microbiol. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Kitao, T.; Lepine, F.; Babloudi, S.; Walte, F.; Steinbacher, S.; Maskos, K.; Blaesse, M.; Negri, M.; Pucci, M.; Zahler, B.; et al. Molecular Insights into Function and Competitive Inhibition of Pseudomonas aeruginosa Multiple Virulence Factor Regulator. mBio 2018, 9, e02158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellini, M.; Di Muzio, E.; D’Angelo, F.; Baldelli, V.; Ferrillo, S.; Visca, P.; Leoni, L.; Polticelli, F.; Rampioni, G. In silico Selection and Experimental Validation of FDA-Approved Drugs as Anti-quorum Sensing Agents. Front. Microbiol. 2019, 10, 2355. [Google Scholar] [CrossRef]
- Soukarieh, F.; Vico Oton, E.; Dubern, J.-F.; Gomes, J.; Halliday, N.; De Pilar Crespo, M.; Ramírez-Prada, J.; Insuasty, B.; Abonia, R.; Quiroga, J.; et al. In Silico and in Vitro-Guided Identification of Inhibitors of Alkylquinolone-Dependent Quorum Sensing in Pseudomonas aeruginosa. Molecules 2018, 23, 257. [Google Scholar] [CrossRef] [Green Version]
- Soukarieh, F.; Mashabi, A.; Richardson, W.; Oton, E.V.; Romero, M.; Roberston, S.N.; Grossman, S.; Sou, T.; Liu, R.; Halliday, N.; et al. Design and Evaluation of New Quinazolin-4(3H)-one Derived PqsR Antagonists as Quorum Sensing Quenchers in Pseudomonas aeruginosa. ACS Infect. Dis. 2021, 7, 2666–2685. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Bousejra-ElGarah, F.; Lajoie, B.; Souchard, J.-P.; Baziard, G.; Bouajila, J.; El Hage, S. Synthesis and evaluation of chromone-2-carboxamide derivatives as cytotoxic agents and 5-lipoxygenase inhibitors. Med. Chem. Res. 2016, 25, 2547–2556. [Google Scholar] [CrossRef] [Green Version]
- Cagide, F.; Reis, J.; Gaspar, A.; Borges, F. Accelerating lead optimization of chromone carboxamide scaffold throughout microwave-assisted organic synthesis. Tetrahedron Lett. 2011, 52, 6446–6449. [Google Scholar] [CrossRef]
- Gaspar, A.; Reis, J.; Kachler, S.; Paoletta, S.; Uriarte, E.; Klotz, K.-N.; Moro, S.; Borges, F. Discovery of novel A3 adenosine receptor ligands based on chromone scaffold. Biochem. Pharmacol. 2012, 84, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Cagide, F.; Reis, J.; Gaspar, A.; Chavarria, D.; Kachler, S.; Klotz, K.N.; Gomes, L.R.; Low, J.N.; Vilar, S.; Hripcsak, G.; et al. Discovery of the first A1 adenosine receptor ligand based on the chromone scaffold. RSC Adv. 2016, 6, 46972–46976. [Google Scholar] [CrossRef]
- Reis, J.; Cagide, F.; Chavarria, D.; Silva, T.; Fernandes, C.; Gaspar, A.; Uriarte, E.; Remião, F.; Alcaro, S.; Ortuso, F.; et al. Discovery of New Chemical Entities for Old Targets: Insights on the Lead Optimization of Chromone-Based Monoamine Oxidase B (MAO-B) Inhibitors. J. Med. Chem. 2016, 59, 5879–5893. [Google Scholar] [CrossRef] [PubMed]
- Vedachalam, S.; Zeng, J.; Gorityala, B.K.; Antonio, M.; Liu, X.-W. N-Heterocyclic Carbene-Catalyzed Intramolecular Aldehyde−Nitrile Cross Coupling: An Easy Access to 3- Aminochromones. Org. Lett. 2010, 12, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Murugesh, N.; Haribabu, J.; Arumugam, K.; Balachandran, C.; Swaathy, R.; Aoki, S.; Sreekanth, A.; Karvembu, R.; Vedachalam, S. NHC-catalyzed green synthesis of functionalized chromones: DFT mechanistic insights and in vitro activities in cancer cells. New J. Chem. 2019, 43, 13509–13525. [Google Scholar] [CrossRef]
- Payard, M.; Paris, J.; Couquelet, J.; Bastide, J.; Lapalus, P.; Alves, P.; Mongourd, N. Synthesis and pharmacological properties of new compounds related to 2-aminochromone. Eur. J. Med. Chem. 1976, 11, 13–18. [Google Scholar]
- Ghosh, T.; Saha, S.; Bandyopadhyay, C. Synthesis of 2,2′-Diaminobischromones Using a Modified Procedure for the Rearrangement of 5-(2-Hydroxyphenyl)isoxazole to 2-Aminochromone. Synthesis 2005, 2005, 1845–1849. [Google Scholar] [CrossRef]
- Petek, N.; Štefane, B.; Novinec, M.; Svete, J. Synthesis and biological evaluation of 7-(aminoalkyl)pyrazolo[1,5-a]pyrimidine derivatives as cathepsin K inhibitors. Bioorg. Chem. 2019, 84, 226–238. [Google Scholar] [CrossRef]
- Reddy, M.B.M.; Jayashankara, V.P.; Pasha, M.A. Aluminum-catalyzed efficient synthesis of anilides by the acylation of aryl amines under ultrasonic conditions. Green Chem. Lett. Rev. 2013, 6, 107–112. [Google Scholar] [CrossRef]
- Saito, S. Aluminum in Organic Synthesis. In Main Group Metals in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2004; pp. 189–306. [Google Scholar]
- Murugesh, N.; Karvembu, R.; Vedachalam, S. The base-induced regioselective radical arylation of 3-aminochromone with aryl hydrazine. Org. Biomol. Chem. 2020, 18, 7884–7891. [Google Scholar] [CrossRef] [PubMed]
- Ben Halima, T.; Vandavasi, J.K.; Shkoor, M.; Newman, S.G. A Cross-Coupling Approach to Amide Bond Formation from Esters. ACS Catal. 2017, 7, 2176–2180. [Google Scholar] [CrossRef]
- Shi, S.; Szostak, M. Pd–PEPPSI: A general Pd–NHC precatalyst for Buchwald–Hartwig cross-coupling of esters and amides (transamidation) under the same reaction conditions. Chem. Commun. 2017, 53, 10584–10587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Kumar, N.; Sharma, R.; Bhargava, G.; Mahajan, D. Direct Conversion of Carboxylic Acids to Various Nitrogen-Containing Compounds in the One-Pot Exploiting Curtius Rearrangement. J. Org. Chem. 2019, 84, 11323–11334. [Google Scholar] [CrossRef] [PubMed]
- Starkey, M.; Lepine, F.; Maura, D.; Bandyopadhaya, A.; Lesic, B.; He, J.; Kitao, T.; Righi, V.; Milot, S.; Tzika, A.; et al. Identification of anti-virulence compounds that disrupt quorum-sensing regulated acute and persistent pathogenicity. PLoS Pathog. 2014, 10, e1004321. [Google Scholar] [CrossRef]
- Lu, C.; Kirsch, B.; Maurer, C.K.; de Jong, J.C.; Braunshausen, A.; Steinbach, A.; Hartmann, R.W. Optimization of anti-virulence PqsR antagonists regarding aqueous solubility and biological properties resulting in new insights in structure–activity relationships. Eur. J. Med. Chem. 2014, 79, 173–183. [Google Scholar] [CrossRef]
- Rampioni, G.; Falcone, M.; Heeb, S.; Frangipani, E.; Fletcher, M.P.; Dubern, J.-F.; Visca, P.; Leoni, L.; Cámara, M.; Williams, P. Unravelling the Genome-Wide Contributions of Specific 2-Alkyl-4-Quinolones and PqsE to Quorum Sensing in Pseudomonas aeruginosa. PLoS Pathog. 2016, 12, e1006029. [Google Scholar] [CrossRef]
- Maura, D.; Hazan, R.; Kitao, T.; Ballok, A.E.; Rahme, L.G. Evidence for Direct Control of Virulence and Defense Gene Circuits by the Pseudomonas aeruginosa Quorum Sensing Regulator, MvfR. Sci. Rep. 2016, 6, 34083. [Google Scholar] [CrossRef]
- Wilson, C.; Lukowicz, R.; Merchant, S.; Valquier-Flynn, H.; Caballero, J.; Sandoval, J.; Okuom, M.; Huber, C.; Brooks, T.D.; Wilson, E.; et al. Quantitative and Qualitative Assessment Methods for Biofilm Growth: A Mini-review. Res. Rev. J. Eng. Technol. 2017, 6, 1–25. [Google Scholar]
- Allkja, J.; van Charante, F.; Aizawa, J.; Reigada, I.; Guarch-Pérez, C.; Vazquez-Rodriguez, J.A.; Cos, P.; Coenye, T.; Fallarero, A.; Zaat, S.A.J.; et al. Interlaboratory study for the evaluation of three microtiter plate-based biofilm quantification methods. Sci. Rep. 2021, 11, 13779. [Google Scholar] [CrossRef]
- Moura-Alves, P.; Puyskens, A.; Stinn, A.; Klemm, M.; Guhlich-Bornhof, U.; Dorhoi, A.; Furkert, J.; Kreuchwig, A.; Protze, J.; Lozza, L.; et al. Host monitoring of quorum sensing during Pseudomonas aeruginosa infection. Science 2019, 366, eaaw1629. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M. Using Autodock with AutodockTools: A tutorial; The Scripps Research Institute: La Jolla, CA, USA, 2005. [Google Scholar]
- Gaspar, A.; Reis, J.; Matos, M.J.; Uriarte, E.; Borges, F. In search for new chemical entities as adenosine receptor ligands: Development of agents based on benzo-γ-pyrone skeleton. Eur. J. Med. Chem. 2012, 54, 914–918. [Google Scholar] [CrossRef]
- Payard, M. Nouveau mode de préparation du chlorure d’acide chromone carboxylique-2. Bull. Soc. Chim. Fr. 1973, 7–8, 2392–2394. [Google Scholar]
- Campanac, C.; Pineau, L.; Payard, A.; Baziard-Mouysset, G.; Roques, C. Interactions between biocide cationic agents and bacterial biofilms. Antimicrob. Agents Chemother. 2002, 46, 1469–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
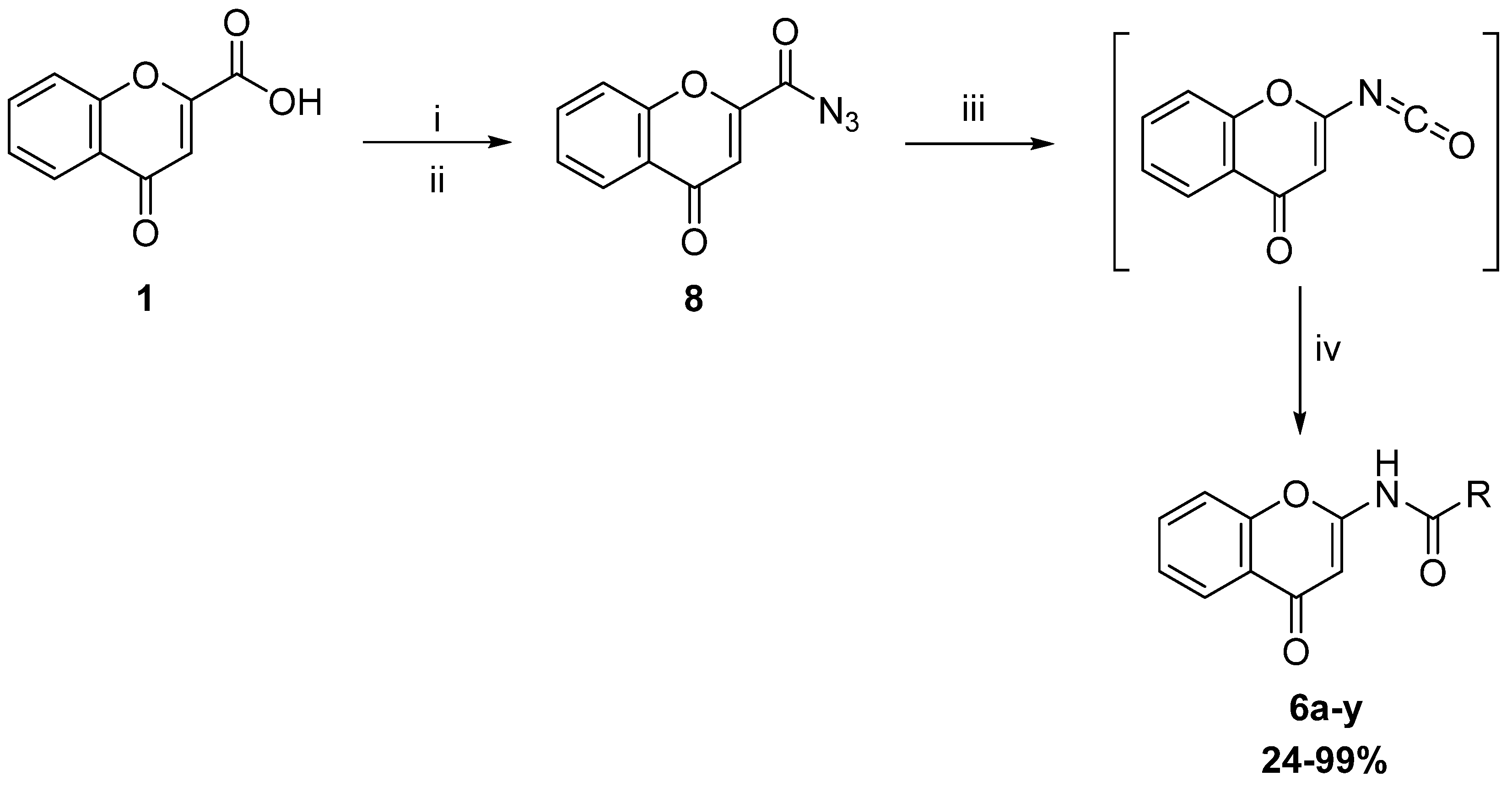{kind=link}
 General route for the synthesis of 6 according to the coupling conditions given below | |||
| Entry | R | Coupling Conditions | Result Compound (Global Yield) |
| 1 | NH2 | POCl3, DIPEA, dry DMF, 18 h, r.t | 6 (traces) |
| 2 | NH2 | (a) SOCl2, 0 to 100 °C (b) TEA, acetonitrile, r.t, 12 h | 6 (32%) |
| 3 | NH2 | (a) CDI, dry THF, r.t to 60 °C, 2 h (b) Et3N, 60 °C, 12 h | 6 (25%) |
| 4 | NH2 | (a) PyBOP, dry DMF, DIPEA, 0 °C, 45 min (b) r.t, overnight | 6 (23%) |
| 5 | NH2 | (a) PCl5, dry cyclohexane, reflux, 2 h (b) Al powder, acetonitrile, ultrasound, 1 h | E.P.N.I |
| 6 | NH2 | (a) SOCl2, dry toluene, reflux, 3 h (b) DMAC, 0 °C to r.t, 18 h | 6 (31%) |
| 7 | NH2 | (a) SOCl2, dry toluene, reflux, 3 h (b) TEA, DMAC, 0 °C to r.t, 18 h | 6 (22%) + 7 (35%)  |
| 8 | NH2 | (a) CDI, dry DMF, r.t to 65 °C, 1 h (b) Phenol, DMAP, 12 h (c) Pd(PEPSI), K2CO3, DME, 110 °C, 18 h | 6 (70%) |
| 9 | COOH | (a) PCl5, dry cyclohexane, reflux, 2 h (b) NaN3, dry cyclohexane, 0 °C to r.t, 1.5 h (c) cyclohexane, reflux, 18h | 6 (90%) |
| 10 | COOH | (a) Ph2POCl, DBU, dry toluene, 0 °C, 5 min (b) NaN3, DMAP, dry DMF, 100 °C, 3 h (c) 100 °C, 12 h | 6 (traces) |
 | ||||||
| R | Cpd | Ebind (kcal·mol−1) | Biofilm Inhibition (%) | Cpd | Ebind (kcal·mol−1) | Biofilm Inhibition (%) |
| PQS | - | −8.1 | - | |||
 | 3a | −8.0 | 21.4 ± 17.0 | 6a | −9.1 | 73.6 ± 6.1 |
 | 3b | −8.4 | 39.1 ± 5.4 | 6b | −8.8 | 67.0 ± 3.6 |
 | 3c | −8.6 | 43.3 ± 7.2 | 6c | −8.8 | 63.1 ± 3.0 |
 | 3d | −8.7 | 55.6 ± 18.3 | 6d | −9.2 | 69.9 ± 9.7 |
 | 3e | −8.2 | 73.3 ± 11.0 | 6e | −8.5 | 65.3 ± 7.6 |
 | 3f | −8.2 | 25.9 ± 4.3 | 6f | −8.6 | 66.6 ± 2.1 |
 | 3g | −8.3 | 47.2 ± 2.8 | 6g | −8.4 | 56.2 ± 7.9 |
 | 3h | −8.5 | 42.4 ± 10.4 | 6h | −8.5 | 27.8 ± 10.7 |
 | 3i | −8.5 | 35.2 ± 14.8 | 6i | −9.2 | 59.3 ± 3.3 |
 | 3j | −8.2 | 46.1 ± 10.2 | 6j | −9.0 | 36.1 ± 4.1 |
 | 3k | −8.7 | 44.7 ± 9.4 | 6k | −8.8 | 63.2 ± 9.3 |
 | 3l | −8.4 | NA | 6l | −8.9 | 67.2 ± 5.2 |
 | 3m | −8.3 | 58.7 ± 2.5 | 6m | −8.4 | NA |
 | 3n | −8.8 | NA | 6n | −8.4 | 90.0 ± 9.9 |
 | 3o | −8.8 | 43.8 ± 24.2 | 6o | −9.1 | 78.3 ± 4.0 |
 | 3p | −7.9 | 47.5 ± 5.3 | 6p | −8.4 | 39.3 ± 17.1 |
 | 3q | −8.3 | 41.3 ± 5.7 | 6q | −8.3 | 35.2 ± 6.8 |
 | 3r | −8.1 | 47.8 ± 5.7 | 6r | −8.2 | 60.3 ± 15.8 |
 | 3s | −7.9 | NA | 6s | −8.4 | 71.9 ± 13.5 |
 | 3t | −9.0 | NA | 6t | −8.5 | 43.8 ± 2.4 |
 | 3u | −9.1 | 51.2 ± 11.7 | 6u | −9.1/−8.9 | 56.8 ± 10.0 |
 | 3v | −8.3 | 46.0 ± 7.3 | 6v | −8.1 | 48.8 ± 2.6 |
 | 3w | −7.7 | NA | 6w | −7.8 | 56.4 ± 7.2 |
 | 3x | −8.9 | 32.0 ± 10.3 | 6x | −8.3 | 48.7 ± 5.3 |
 | 3y | −9.5 | 24.4 ± 8.8 | 6y | −9.4 | 19.5 ± 1.8 |
| R | Cpd | Ebind (kcal·mol−1) | Biofilm Inhibition (%) |
|---|---|---|---|
 | 3′a | −9.2 | 70.5 ± 5.6 |
 | 3′b | −8.4 | 35.5 ± 5.8 |
 | 3′c | −8.8/−9.0 | 39.7 ± 4.5 |
 | 3′d | −9.5 | NA |
 | 3′e | −9.3 | 36.0 ± 28.6 |
 | 3′f | −8.2 | NA |
 | 3′g | −7.5 | 14.7 ± 2.1 |
 | 3′h | −8.0 | 55.3 ± 5.5 |
 | 3′i | −9.3 | 10.8 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trognon, J.; Vera, G.; Rima, M.; Stigliani, J.-L.; Amielet, L.; El Hage, S.; Lajoie, B.; Roques, C.; El Garah, F. Investigation of Direct and Retro Chromone-2-Carboxamides Based Analogs of Pseudomonas aeruginosa Quorum Sensing Signal as New Anti-Biofilm Agents. Pharmaceuticals 2022, 15, 417. https://doi.org/10.3390/ph15040417
Trognon J, Vera G, Rima M, Stigliani J-L, Amielet L, El Hage S, Lajoie B, Roques C, El Garah F. Investigation of Direct and Retro Chromone-2-Carboxamides Based Analogs of Pseudomonas aeruginosa Quorum Sensing Signal as New Anti-Biofilm Agents. Pharmaceuticals. 2022; 15(4):417. https://doi.org/10.3390/ph15040417
Chicago/Turabian StyleTrognon, Jeanne, Gonzalo Vera, Maya Rima, Jean-Luc Stigliani, Laurent Amielet, Salomé El Hage, Barbora Lajoie, Christine Roques, and Fatima El Garah. 2022. "Investigation of Direct and Retro Chromone-2-Carboxamides Based Analogs of Pseudomonas aeruginosa Quorum Sensing Signal as New Anti-Biofilm Agents" Pharmaceuticals 15, no. 4: 417. https://doi.org/10.3390/ph15040417