
Development of Dibenzothiazepine Derivatives as Multifunctional Compounds for Neuropathic Pain

 ,
,
Abstract
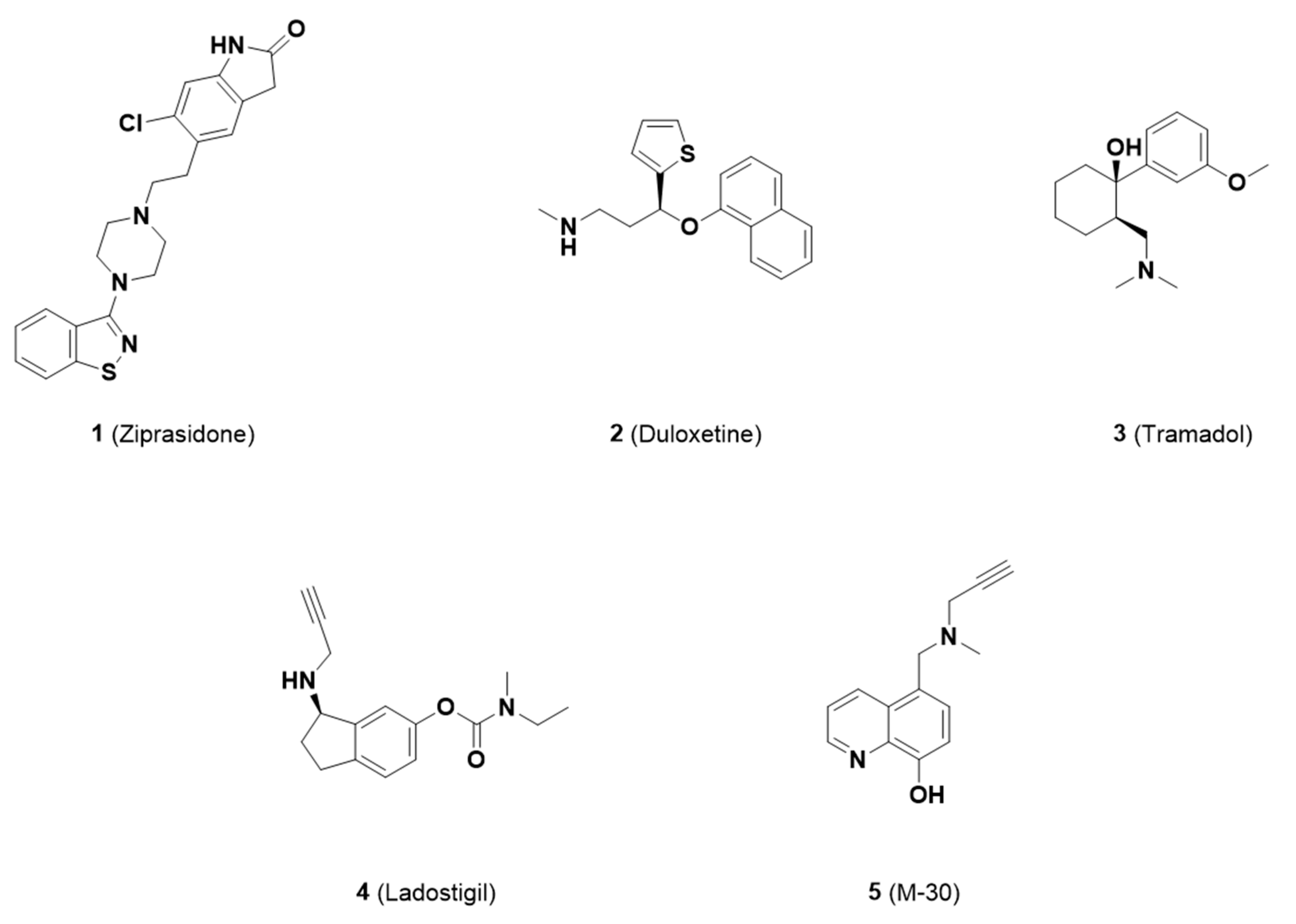
:1. Introduction
2. Results and Discussion
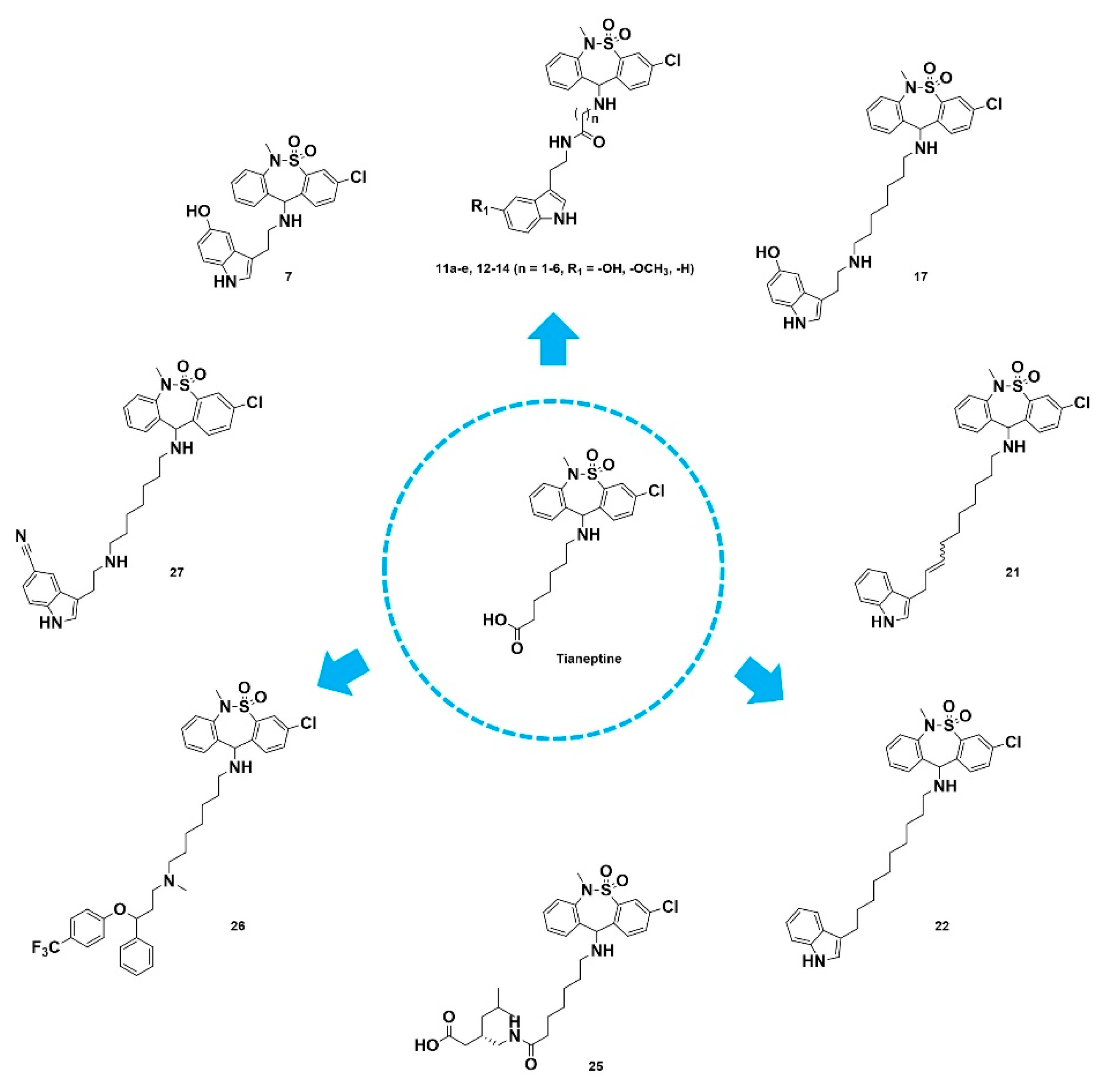
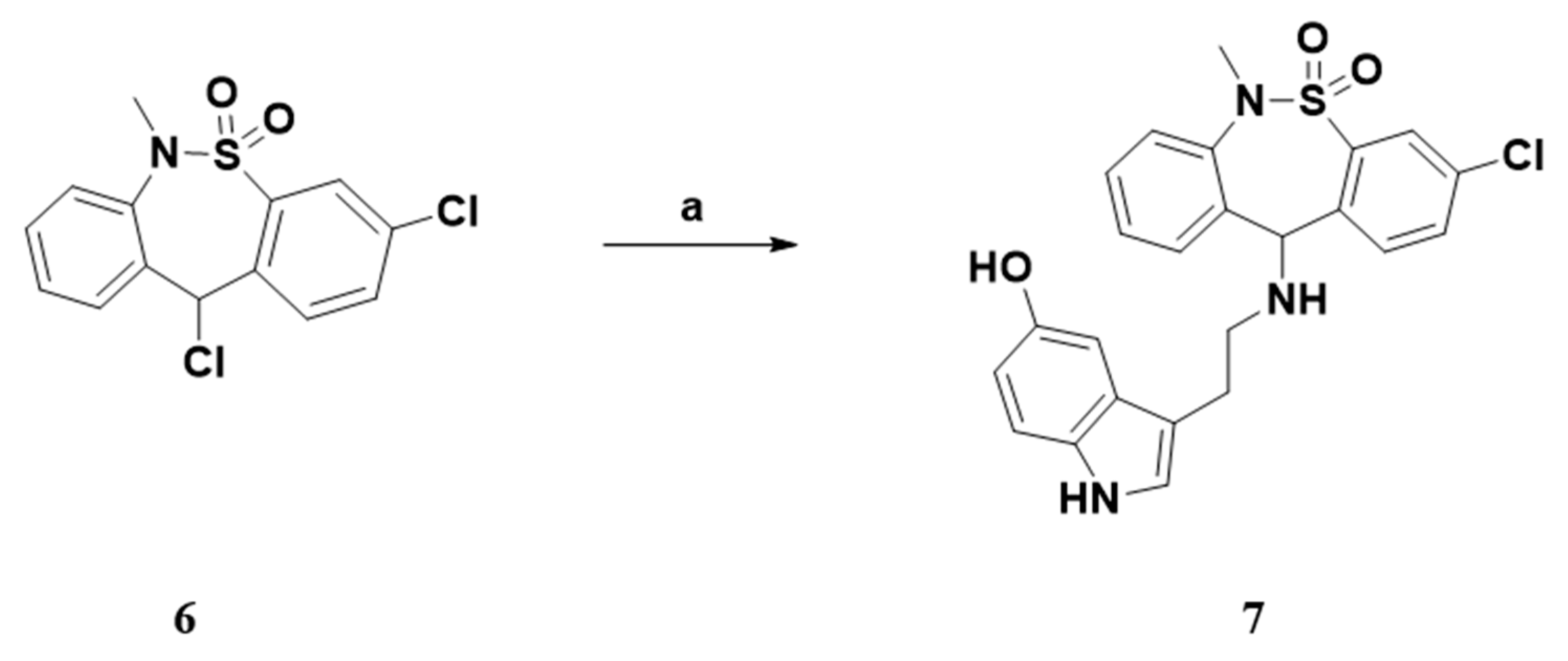
2.1. Chemistry
2.2. Biological Activity
2.2.1. In Vitro Assay for the Dibenzothiazepine Derivatives
2.2.2. In Vitro Assay for Opioid Receptors Agonisms
2.2.3. β-Arrestin Recruitment at the hOPRM1 Receptor
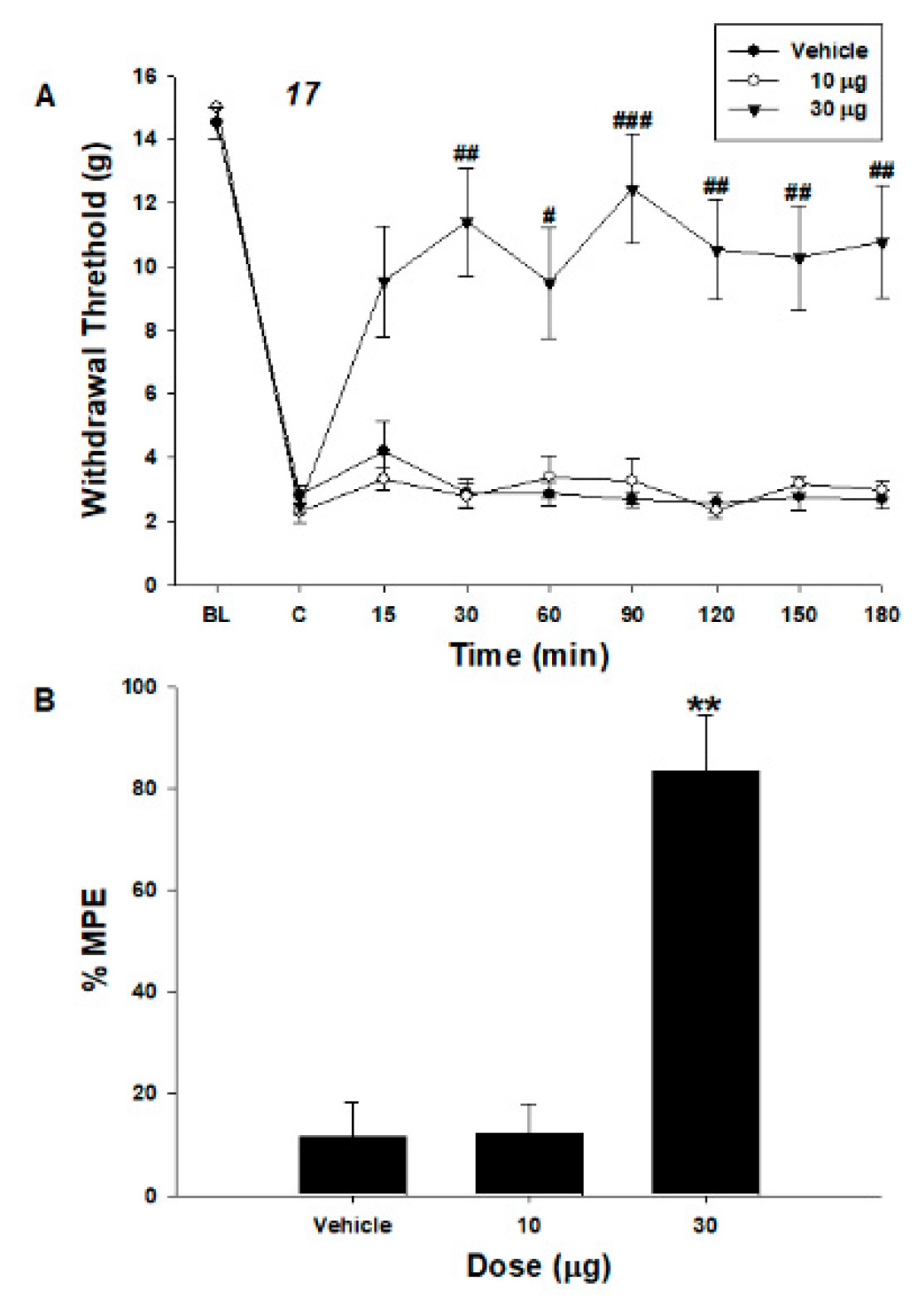
2.2.4. In Vivo Behavioral Responses
2.2.5. In Vivo Pharmacology of Tianeptine Derivatives in Neuropathic Pain Animal Models
2.2.6. Multiple Mechanisms of Action for Treatment of Neuropathic Pain
3. Materials and Methods
3.1. Chemical Synthesis
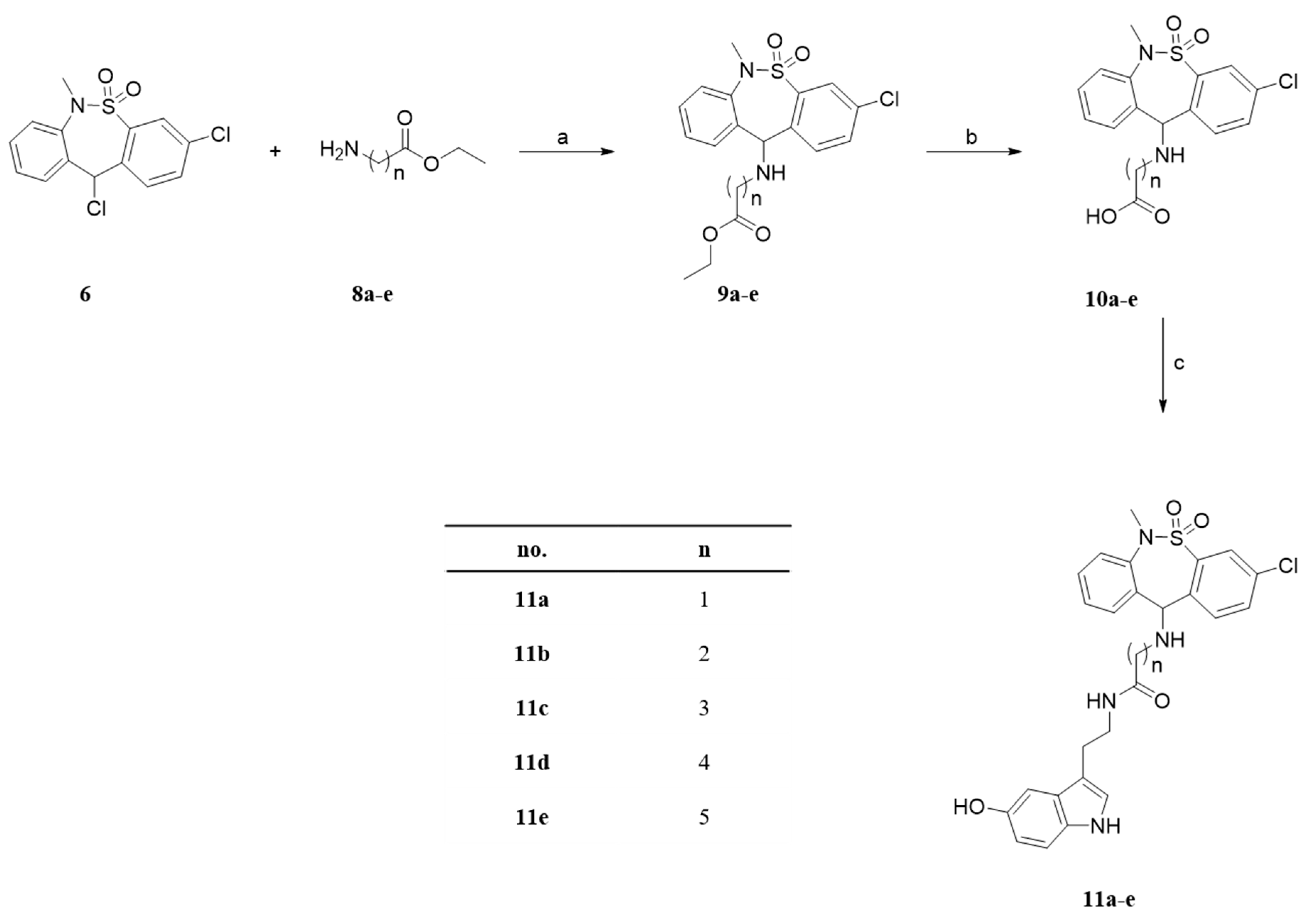
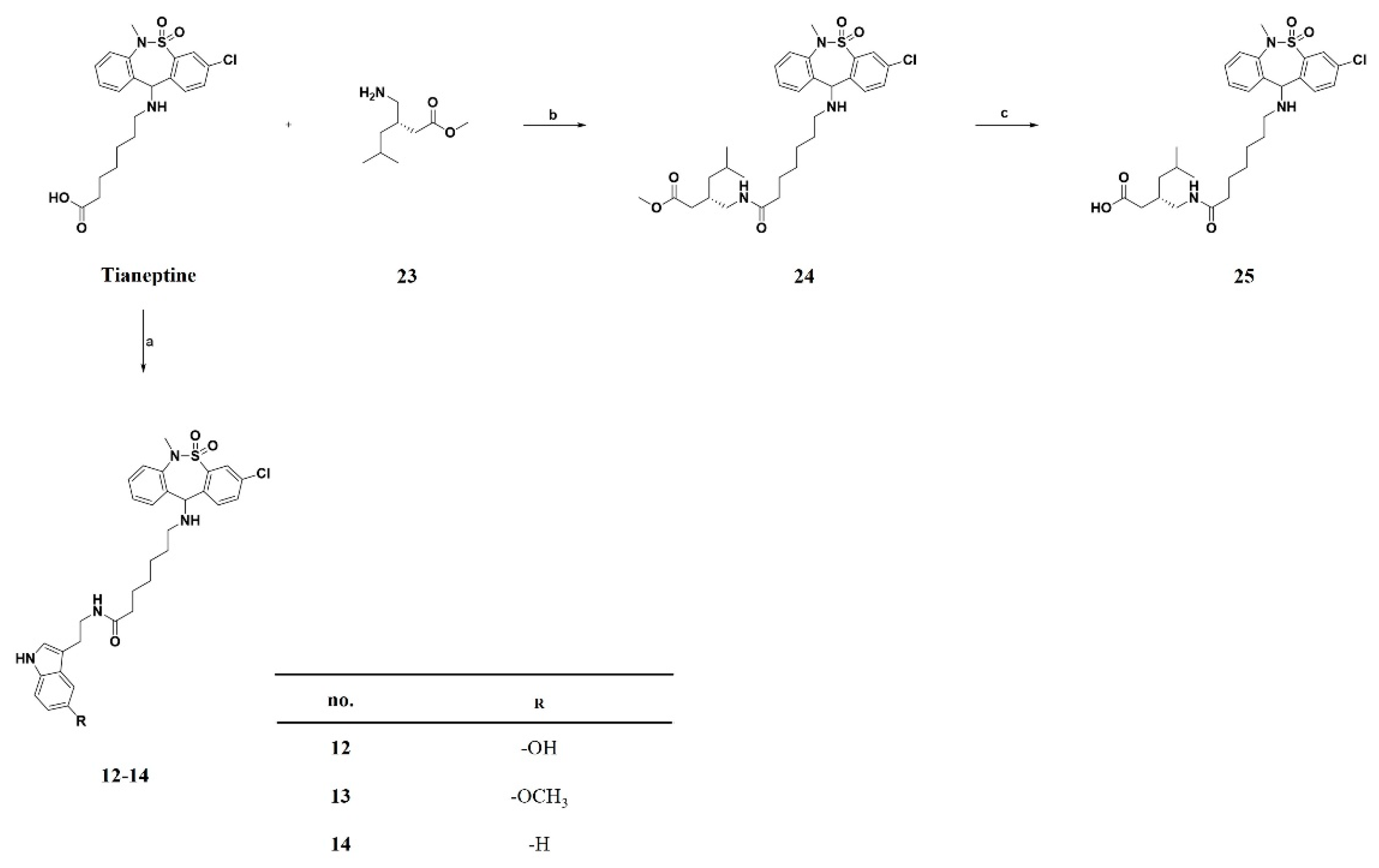
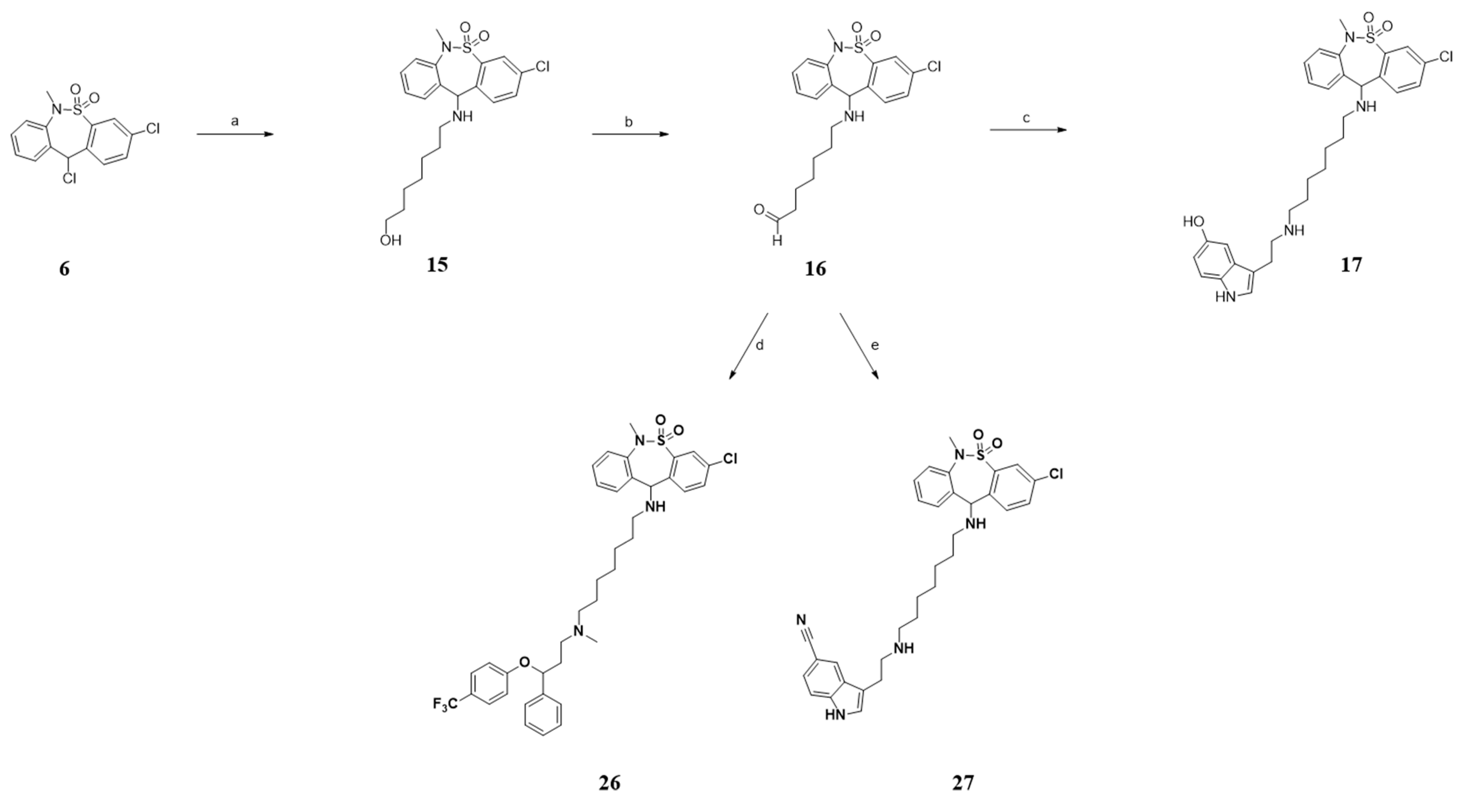
3.1.1. General Procedure A. Synthesis of Compounds 7, 9a–e, 15 and 21–22
3.1.2. General Procedure B. Synthesis of Compounds 10a–e and 25
3.1.3. General Procedure C. Synthesis of Compounds 11a–e, 12, 13, 14 and 24
3.2. Neurotransmitter Reuptake Assay and Data Analysis
3.3. Intracellular Calcium Mobilization Measurement by Using FLIPR Tetra System
3.4. β-Arrestin Recruitment Assay at hOPRM1 Receptor
3.5. In Vivo Efficacy
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jensen, T.S.; Baron, R.; Haanpaa, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.; Treede, R.D. A new definition of neuropathic pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Doth, A.H.; Hansson, P.T.; Jensen, M.P.; Taylor, R.S. The burden of neuropathic pain: A systematic review and meta-analysis of health utilities. Pain 2010, 149, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Langley, P.C.; Van Litsenburg, C.; Cappelleri, J.C.; Carroll, D. The burden associated with neuropathic pain in Western Europe. J. Med. Econ. 2013, 16, 85–95. [Google Scholar] [CrossRef]
- Szczudlik, A.; Dobrogowski, J.; Wordliczek, J.; Stepien, A.; Krajnik, M.; Leppert, W.; Woron, J.; Przeklasa-Muszynska, A.; Kocot-Kepska, M.; Zajaczkowska, R.; et al. Diagnosis and management of neuropathic pain: Review of literature and recommendations of the Polish Association for the Study of Pain and the Polish Neurological Society—Part Two. Neurol. Neurochir. Pol. 2014, 48, 423–435. [Google Scholar] [CrossRef]
- Vu, T.N. Current pharmacologic approaches to treating neuropathic pain. Curr. Pain. Headache Rep. 2004, 8, 15–18. [Google Scholar] [CrossRef]
- Wallace, J.M. Update on pharmacotherapy guidelines for treatment of neuropathic pain. Curr. Pain. Headache Rep. 2007, 11, 208–214. [Google Scholar] [CrossRef]
- Largent-Milnes, T.M.; Brookshire, S.W.; Skinner, D.P., Jr.; Hanlon, K.E.; Giuvelis, D.; Yamamoto, T.; Davis, P.; Campos, C.R.; Nair, P.; Deekonda, S.; et al. Building a better analgesic: Multifunctional compounds that address injury-induced pathology to enhance analgesic efficacy while eliminating unwanted side effects. J. Pharm. Exp. Ther. 2013, 347, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Attal, N.; Cruccu, G.; Baron, R.; Haanpaa, M.; Hansson, P.; Jensen, T.S.; Nurmikko, T. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur. J. Neurol. 2010, 17, 1113-e88. [Google Scholar] [CrossRef] [PubMed]
- Attal, N.; Finnerup, N.B. Pharmacological Management of Neuropathic Pain. IASP 2010, 18, 9. [Google Scholar]
- Fattaccini, C.M.; Bolanos-Jimenez, F.; Gozlan, H.; Hamon, M. Tianeptine stimulates uptake of 5-hydroxytryptamine in vivo in the rat brain. Neuropharmacology 1990, 29, 1–8. [Google Scholar] [CrossRef]
- Mennini, T.; Mocaer, E.; Garattini, S. Tianeptine, a selective enhancer of serotonin uptake in rat brain. Naunyn Schmiedebergs Arch. Pharm. 1987, 336, 478–482. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Chattarji, S.; Diamond, D.M.; Jay, T.M.; Reagan, L.P.; Svenningsson, P.; Fuchs, E. The neurobiological properties of tianeptine (Stablon): From monoamine hypothesis to glutamatergic modulation. Mol. Psychiatry 2010, 15, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, S.; McEwen, B.S. Neurobiological and clinical effects of the antidepressant tianeptine. CNS Drugs 2008, 22, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Uzbay, I.T.; Cinar, M.G.; Aytemir, M.; Tuglular, I. Analgesic effect of tianeptine in mice. Life Sci. 1999, 64, 1313–1319. [Google Scholar] [CrossRef]
- Han, S.M.; Kim, Y.H.; Jo, H.U.; Kwak, J.A.; Park, H.J. Tianeptine Reduces Mechanical Allodynia in Spinal Nerve-ligated and Chemotherapy-induced Neuropathic Mice. Pain Physician 2017, 20, E593–E600. [Google Scholar]
- Lee, H.G.; Choi, J.I.; Yoon, M.H.; Obata, H.; Saito, S.; Kim, W.M. The antiallodynic effect of intrathecal tianeptine is exerted by increased serotonin and norepinephrine in the spinal dorsal horn. Neurosci. Lett. 2014, 583, 103–107. [Google Scholar] [CrossRef]
- Kim, W.M.; Lee, S.H.; Jeong, H.J.; Lee, H.G.; Choi, J.I.; Yoon, M.H. The analgesic activity of intrathecal tianeptine, an atypical antidepressant, in a rat model of inflammatory pain. Anesth. Analg. 2012, 114, 683–689. [Google Scholar] [CrossRef]
- Gassaway, M.M.; Rives, M.L.; Kruegel, A.C.; Javitch, J.A.; Sames, D. The atypical antidepressant and neurorestorative agent tianeptine is a mu-opioid receptor agonist. Transl. Psychiatry 2014, 4, e411. [Google Scholar] [CrossRef] [PubMed]
- Sansone, R.A.; Sansone, L.A. Pain, Pain, Go Away: Antidepressants and Pain Management. Psychiatry 2008, 5, 16–19. [Google Scholar] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hubner, H.; et al. Structure-Based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.H.; Choi, J.I.; Jeong, S.W. Antinociception of intrathecal cholinesterase inhibitors and cholinergic receptors in rats. Acta Anaesthesiol. Scand. 2003, 47, 1079–1084. [Google Scholar] [CrossRef]
- Yaksh, T.L.; Rudy, T.A. Chronic catheterization of the spinal subarachnoid space. Physiol. Behav. 1976, 17, 1031–1036. [Google Scholar] [CrossRef]
- Kim, S.H.; Chung, J.M. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992, 50, 355–363. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compounds | R 1 | n | IC50 a or % Inhibition at 10 μM b | ||
|---|---|---|---|---|---|
| hDAT | hSERT | hNET | |||
| GBR12909 c | 85.9 ± 0.27% b | 70.6 ± 1.3% b | 79.1 ± 0.71% b | ||
| Fluoxetined | 29.7 ± 2.1 μM a | 0.034 ± 0.008 μM a | 5.58 ± 0.16 μM a | ||
| Nisoxetinee | 79.7 ± 1.4% b | 79.8 ± 0.32% b | 84.1 ± 1.1% b | ||
| Tianeptine | 20.3 ± 6.2% b | 16.6 ± 10% b | 27.2 ± 6.2% b | ||
| 7 | 36.2 ± 2.7% b | 37.5 ± 2.5% b | 30.8 ± 2.8% b | ||
| 11a | −OH | 1 | 33.2 ± 6.5% b | 50.9 ± 4.1% b | 13.2 ± 2.3% b |
| 11b | −OH | 2 | 44.9 ± 4.2% b | 4.08 ± 0.032 μM a | 54.8 ± 3.7% b |
| 11c | −OH | 3 | 41.3 ± 5.7% b | 35.5 ± 3.7% b | 24.3 ± 7.4% b |
| 11d | −OH | 4 | 41.3 ± 7.6% b | 10.1 ± 0.50 μM a | 21.1 ± 4.7% b |
| 11e | −OH | 5 | 40.2 ± 0.12% b | 6.74 ± 0.38 μM a | 30.1 ± 5.0% b |
| 12 | −OH | 6 | 9.66 ± 1.0 μM a | 0.258 ± 0.057 μM a | 30.1 ± 0.87% b |
| 13 | −OCH3 | 6 | 25.6 ± 9.7% b | 39.7± 6.3% b | 23.7 ± 7.5% b |
| 14 | H | 6 | 3.81 ± 1.4 μM a | 50.3 ± 3.4% b | 65.2 ± 3.4% b |
| 17 | −OH | 2.01 ± 0.65 μM a | 0.070 ± 0.009 μM a | 0.154 ± 0.007 μM a | |
| 21 | 8.92 ± 6.1% b | 26.3 ± 6.4% b | 5.64 ± 4.5% b | ||
| 22 | 1.95 ± 0.06% b | 17.9 ± 7.5% b | 5.56 ± 0.07% b | ||
| 25 | 17.6 ± 8.9% b | 11.3 ± 2.5% b | 21.0 ± 2.6% b | ||
| 26 | 23.6 ± 12% b | 2.18 ± 0.37 μM a | 7.07 ± 4.1% b | ||
| 27 | −CN | 4.98 ± 2.1 μM a | 0.462 ± 0.034 μM a | 63.6 ± 4.4% b | |
| Compounds | EC50 (nM) a or % Stimulation b at 10 μM | |
|---|---|---|
| OPRK1 Agonism against DynorphinA (370 nM) | OPRM1 Agonism against DAMGO (3.9 μM) | |
| Tianeptine | <10% b | <10% b |
| 12 | <10% b | 1200 ± 244 a |
| 14 | <10% b | 62.3 ± 2.9% b |
| 17 | <10% b | 384 ± 25 a |
| 21 | <10% b | <10% b |
| 22 | <10% b | 11.8 ± 1.9% b |
| 25 | <10% b | 2410 ± 144 a |
| 26 | <10% b | <10% b |
| 27 | <10% b | 65.9 ± 2.8% b |
| Compounds | EC50 (nM) a |
|---|---|
| OPRM1 | |
| Tianeptine | >5000 a |
| 12 | 134 ± 12 a |
| 17 | 1556 ± 195 a |
| 14 | 113 ± 3 a |
| 21 | >10,000 a |
| 22 | >10,000 a |
| 25 | >10,000 a |
| 26 | >10,000 a |
| 27 | 3285 ± 642 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, Y.-H.; Kim, Y.O.; Kang, K.M.; Lee, H.G.; Son, B.; Han, X.; Oh, E.; Kim, S.; Seo, S.H.; Park, J.-H.; et al. Development of Dibenzothiazepine Derivatives as Multifunctional Compounds for Neuropathic Pain. Pharmaceuticals 2022, 15, 407. https://doi.org/10.3390/ph15040407
Jung Y-H, Kim YO, Kang KM, Lee HG, Son B, Han X, Oh E, Kim S, Seo SH, Park J-H, et al. Development of Dibenzothiazepine Derivatives as Multifunctional Compounds for Neuropathic Pain. Pharmaceuticals. 2022; 15(4):407. https://doi.org/10.3390/ph15040407
Chicago/Turabian StyleJung, Young-Hwan, Yeo Ok Kim, Koon Mook Kang, Hyung Gon Lee, Borum Son, Xuehao Han, Eunseok Oh, Siwon Kim, Seon Hee Seo, Jong-Hyun Park, and et al. 2022. "Development of Dibenzothiazepine Derivatives as Multifunctional Compounds for Neuropathic Pain" Pharmaceuticals 15, no. 4: 407. https://doi.org/10.3390/ph15040407