
Two New Fatty Acid Derivatives, Omphalotols A and B and Anti-Helicobacter pylori Fatty Acid Derivatives from Poisonous Mushroom Omphalotus japonicus

 , and
, and 
Abstract
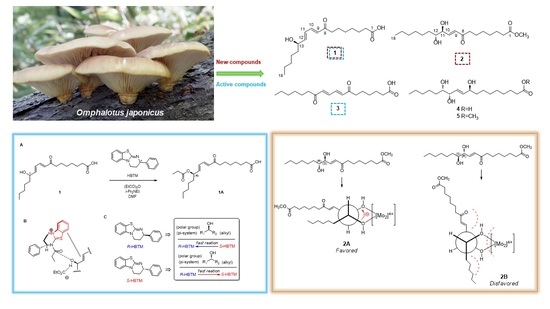
:
1. Introduction
2. Results and Discussion
2.1. Extraction of O. japonicus and Isolation of Compounds
2.2. Structural Elucidation of the Isolated Compounds 1–5
2.3. Antibacterial Activity Evaluation of Isolated Compounds against H. pylori
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Mushroom Material
3.3. Extraction of O. japonicus and Isolation of Compounds
3.3.1. Omphalotol A (1)
3.3.2. Omphalotol B (2)
3.4. MS/MS Analysis of Compounds 1 and 2
3.5. Experimental Procedures to Determine the Absolute Configuration of Compound 1
3.5.1. CEA Reaction
3.5.2. LC/MS Analysis
3.6. Absolute Configuration of the 1,2-diol Functionalities in Compound 2
3.7. Anti-Helicobacter pylori Activity
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, S.; Goyal, A. Recent Developments in Mushrooms as Anti-Cancer Therapeutics: A Review. 3 Biotech 2012, 2, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasser, S.P. Current Findings, Future Trends, and Unsolved Problems in Studies of Medicinal Mushrooms. Appl. Microbiol. Biotech. 2011, 89, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.R.M.; Lima, N. Biomedical Effects of Mushrooms with Emphasis on Pure Compounds. Biomed. J. 2014, 37, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ryoo, R.; Choi, J.H.; Kim, J.-H.; Kim, S.-H.; Kim, K.H. Trichothecene and Tremulane Sesquiterpenes from a Hallucinogenic Mushroom Gymnopilus junonius and Their Cytotoxicity. Arch. Pharm. Res. 2020, 43, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Yi, S.A.; Nam, K.H.; Ryoo, R.; Lee, J.; Kim, K.H. Pantheric Acids a–C from a Poisonous Mushroom, Amanita pantherina, Promote Lipid Accumulation in Adipocytes. J. Nat. Prod. 2019, 82, 3489–3493. [Google Scholar] [CrossRef]
- Lee, S.; Lee, D.; Ryoo, R.; Kim, J.-C.; Park, H.B.; Kang, K.S.; Kim, K.H. Calvatianone, a Sterol Possessing a 6/5/6/5-Fused Ring System with a Contracted Tetrahydrofuran B-Ring, from the Fruiting Bodies of Calvatia nipponica. J. Nat. Prod. 2020, 83, 2737–2742. [Google Scholar] [CrossRef]
- Aoki, S.; Aboshi, T.; Onodera, T.; Kimura, K.-i.; Arai, D.; Iizuka, Y.; Murayama, T. Omphaloprenol A: A New Bioactive Polyisoprenepolyol Isolated from the Mycelium of Poisonous Mushroom Omphalotus japonicus. Biosci. Biotechnol. Biochem. 2021, 85, 1364–1370. [Google Scholar] [CrossRef]
- Uto, Y.; Sasaki, K.; Takahashi, M.; Morimoto, K.; Inoue, K. Application of High-Speed Countercurrent Chromatography for the Purification of High-Purity Illudin S from Omphalotus japonicus. Anal. Sci. 2019, 35, 789–792. [Google Scholar] [CrossRef] [Green Version]
- Kelner, M.J.; McMorris, T.C.; Estes, L.; Rutherford, M.; Montoya, M.; Goldstein, J.; Samson, K.; Starr, R.; Taetle, R. Characterization of Illudin S Sensitivity in DNA Repair-Deficient Chinese Hamster Cells: Unusually High Sensitivity of Ercc2 and Ercc3 DNA Helicase-Deficient Mutants in Comparison to Other Chemotherapeutic Agents. Biochem. Pharmacol. 1994, 48, 403–409. [Google Scholar] [CrossRef]
- Kelner, M.J.; McMorris, T.C.; Montoya, M.A.; Estes, L.; Rutherford, M.; Samson, K.M.; Taetle, R. Characterization of Cellular Accumulation and Toxicity of Illudin S in Sensitive and Nonsensitive Tumor Cells. Cancer Chemother. Pharmacol. 1997, 40, 65–71. [Google Scholar] [CrossRef]
- Baekelandt, M. Irofulven (Mgi Pharma). Curr. Opin. Investig. Drugs 2002, 3, 1517–1526. [Google Scholar] [PubMed]
- Amato, R.J.; Perez, C.; Pagliaro, L. Irofulven, a Novel Inhibitor of DNA Synthesis, in Metastatic Renal Cell Cancer. Investig. New Drugs 2002, 20, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sturla, S.J. Profiling Patterns of Glutathione Reductase Inhibition by the Natural Product Illudin S and Its Acylfulvene Analogues. Mol. Biosyst. 2009, 5, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietsch, K.E.; Van Midwoud, P.M.; Villalta, P.W.; Sturla, S.J. Quantification of Acylfulvene–and Illudin S–DNA Adducts in Cells with Variable Bioactivation Capacities. Chem. Res. Toxicol. 2013, 26, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichihara, A.; Shirahama, H.; Matsumoto, T. Dihydroilludin S, a New Constituent from Lampteromyces japonicus. Tetrahedron Lett. 1969, 10, 3965–3968. [Google Scholar] [CrossRef]
- Kuramoto, M.; Tsukihara, T.; Ono, N. Neoilludins a and B, New Bioactive Components from Lampteromyces japonicus. Chem. Lett. 1999, 28, 1113–1114. [Google Scholar] [CrossRef]
- Uyakul, D.; Isobe, M.; Goto, T. Lampteromyces Bioluminescence: 3. Structure of Lampteroflavin, the Light Emitter in the Luminous Mushroom, L. japonicus. Bioorg. Chem. 1989, 17, 454–460. [Google Scholar] [CrossRef]
- Nakanishi, K.; Tada, M.; Yamada, Y.; Ohashi, M.; Komatsu, N.; Terakawa, H. Isolation of Lampterol, an Antitumour Substance from Lampteromyces japonicus. Nature 1963, 197, 292. [Google Scholar] [CrossRef]
- Fukuda, K.; Uematsu, T.; Hamada, A.; Akiya, S.; Komatsu, N.; Okubo, S. The Polysaccharide from Lampteromyces japonicus. Chem. Pharm. Bull. 1975, 23, 1955–1959. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-H.; Moon, E.-J.; Kim, S.-Y.; Lee, K.-R. Anti-Melanogenic Fatty Acid Derivatives from the Tuber-Barks of Colocasia antiquorum var. esculenta. Bull. Korean Chem. Soc. 2010, 31, 2051–2053. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Park, H.B.; Kim, K.H. Highly Sensitive, Simple, and Cost-and Time-Effective Method to Determine the Absolute Configuration of a Secondary Alcohol Using Competing Enantioselective Acylation Coupled with LC/MS. Anal. Chem. 2018, 90, 13212–13216. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, L.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of Absolute Configuration of Acyclic 1, 2-Diols with Mo2(Oac)4.1. Snatzke’s Method Revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [PubMed]
- Pan, L.; Acuña, U.; Li, J.; Jena, N.; Ninh, T.; Pannell, C.; Chai, H.; Fuchs, J.; Carcache De Blanco, E.J.; Soejarto, D.D.; et al. Bioactive Flavaglines and Other Constituents Isolated from Aglaia perviridis. J. Nat. Prod. 2013, 76, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagle, D.G.; Gerwick, W.H. Structure and Stereochemistry of Constanolactones Ag, Lactonized Cyclopropyl Oxylipins from the Red Marine Alga Constantinea simplex. J. Org. Chem. 1994, 59, 7227–7237. [Google Scholar] [CrossRef]
- Liangyan, L.; Tao, F.; Zhenghui, L.; Jikai, L. Four New 2, 5-Disubstituted Furan Vicinal Diols from the Fungus Ceriporia alachuana. Chin. J. Org. Chem. 2017, 37, 1577–1581. [Google Scholar]
- Hamburger, M.; Handa, S.S.; Cordell, G.A.; Kinghorn, A.D.; Farnsworth, N.R. Plant Anticancer Agents, Xliii.(E, E)-7, 12-Dioxo-Octadeca-8, 10-Dien-1-Oic Acid (Ostopanic Acid), a Cytotoxic Fatty Acid from Ostodes paniculata. J. Nat. Prod. 1987, 50, 281–283. [Google Scholar] [CrossRef]
- Naidu, S.V.; Gupta, P.; Kumar, P. Enantioselective Syntheses of (−)-Pinellic Acid, A-and Β-Dimorphecolic Acid. Tetrahedron 2007, 63, 7624–7633. [Google Scholar] [CrossRef]
- Miura, A.; Kuwahara, S. A Concise Synthesis of Pinellic Acid Using a Cross-Metathesis Approach. Tetrahedron 2009, 65, 3364–3368. [Google Scholar] [CrossRef]
- McGee, D.J.; George, A.E.; Trainor, E.A.; Horton, K.E.; Hildebrandt, E.; Testerman, T.L. Cholesterol Enhances Helicobacter Pylori Resistance to Antibiotics and Ll-37. Antimicrob. Agents Chemother. 2011, 55, 2897–2904. [Google Scholar] [CrossRef] [Green Version]
- Chey, W.D.; Wong, B.C. Practice Parameters Committee of the American College of Gastroenterology. American College of Gastroenterology Guideline on the Management of Helicobacter Pylori Infection. Off. J. Am. Coll. Gastroenterol.|ACG 2007, 102, 1808–1825. [Google Scholar] [CrossRef]
- Tankovic, J.; Lamarque, D.; Lascols, C.; Soussy, C.J.; Delchier, J.C. Clarithromycin resistance of Helicobacter pylori has a major impact on the efficacy of the omeprazole-amoxicillin-clarithromycin therapy. Pathol. Biol. 2001, 49, 528–533. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Li, W.-Y.; Wu, D.-C.; Wang, J.-J.; Wu, C.-H.; Liao, J.-J.; Lin, C.-K. In vitro activity of 2-methoxy-1,4-naphthoquinone and stigmasta-7,22-diene-3b-ol from Impatiens balsamina L. against multiple antibiotic-resistant Helicobacter pylori. Evid. Based. Complement. Altern. Med. 2011, 2011, 704721. [Google Scholar]
- An, B.G.; Moon, B.S.; Kim, H.J.; Lim, H.C.; Lee, Y.C.; Lee, G.; Kim, S.H.; Park, M.; Kim, J.B. Antibiotic resistance in Helicobacter pylori strains and its effect on H. pylori eradication rates in a single center in Korea. Ann. Lab. Med. 2013, 33, 415–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Dunbar, K.B.; Mitui, M.; Arnold, C.A.; Lam-Himlin, D.M.; Valasek, M.A.; Thung, I.; Okwara, C.; Coss, E.; Cryer, B.; et al. Helicobacter pylori clarithromycin resistance and treatment failure are common in the USA. Dig. Dis. Sci. 2016, 61, 2373–2380. [Google Scholar] [CrossRef]
- Tanaka, T.; Kawase, M.; Tani, S. Urease inhibitory activity of simple α,β-unsaturated ketones. Life Sci. 2003, 73, 2985–2990. [Google Scholar] [CrossRef]
- Lee, S.B.; Taylor, J.W. Isolation of DNA from fungal mycelia and single spores. In PCR Protocols: A Guide to Methods and Applications; Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 282–287. [Google Scholar]
- Gardes, M.; Bruns, T.D. Its Primers with Enhanced Specificity for Basidiomycetes-Application to the Identification of Mycorrhizae and Rusts. Molec. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- Khalil, A.A.K.; Park, W.S.; Lee, J.; Kim, H.J.; Akter, K.M.; Goo, Y.M.; Bae, J.Y.; Chun, M.S.; Kim, J.H.; Ahn, M.J. A new anti-Helicobacter pylori juglone from Reynoutria japonica. Arch. Pharmacal Res. 2019, 42, 505–511. [Google Scholar] [CrossRef]
- Wayne, P. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; approved standard; CLSI document M27-A3; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008; Volume 28, pp. 6–12. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 177.0 | 174.3 | ||
| 2 | 2.27 t (7.5) | 33.9 | 2.30 t (7.5) | 34.0 |
| 3 | 1.60 m b | 24.3 | 1.62 m b | 24.4 |
| 4 | 1.35 m b | 28.5 | 1.31 m b | 28.8 |
| 5 | 1.35 m b | 28.5 | 1.31 m b | 31.6 |
| 6 | 1.60 m b | 24.3 | 1.62 m b | 24.4 |
| 7 | 2.62 t (7.5) | 39.5 | 2.57 t (7.5) | 40.7 |
| 8 | 202.2 | 200.5 | ||
| 9 | 6.20 d (15.5) | 128.9 | 6.38 dd (16.0, 1.5) | 130.4 |
| 10 | 7.27 dd (15.5, 11.0) | 142.8 | 6.81 dd (16.0, 5.0) | 142.6 |
| 11 | 6.41 dd (15.0, 11.0) | 127.3 | 4.32 ddd (5.0, 3.5, 1.5) | 74.2 |
| 12 | 6.25 dd (15.0, 6.0) | 147.0 | 3.79 m | 74.0 |
| 13 | 4.17 q (6.0) | 71.1 | 1.43 m | 32.1 |
| 14 | 1.54 m | 36.5 | 1.31 m b | 28.8 |
| 15 | 1.35 m b | 24.8 | 1.31 m b | 25.2 |
| 16 | 1.33 m b | 22.2 | 1.31 m b | 22.5 |
| 17 | 1.33 m b | 31.7 | 1.31 m b | 31.6 |
| 18 | 0.91 t (7.0) | 13.0 | 0.90 t (7.0) | 13.9 |
| 1-OCH3 | 3.67 s | 51.3 | ||
| Compound | Concentration (μM) | Inhibition (%) | MIC (μM) | MIC50 (μM) | MIC90 (μM) |
|---|---|---|---|---|---|
| 1 | 100 | 27.4 ± 4.5 b | |||
| 2 | 11.1 ± 0.3 c | ||||
| 3 | 97.5 ± 0.8 a | 3.1 | 9 | 20 | |
| 4 | 2.5 ± 0.8 d | ||||
| 5 | 0.2 ± 0.1 d | ||||
| Quercetin * | 100 | 34.4 ± 0.6 b | 50 | ||
| Metronidazole * | 97.0 ± 0.1 a | 6.3 | 17 | 46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Kim, T.W.; Lee, Y.H.; Kang, D.-M.; Ryoo, R.; Ko, Y.-J.; Ahn, M.-J.; Kim, K.H. Two New Fatty Acid Derivatives, Omphalotols A and B and Anti-Helicobacter pylori Fatty Acid Derivatives from Poisonous Mushroom Omphalotus japonicus. Pharmaceuticals 2022, 15, 139. https://doi.org/10.3390/ph15020139
Lee S, Kim TW, Lee YH, Kang D-M, Ryoo R, Ko Y-J, Ahn M-J, Kim KH. Two New Fatty Acid Derivatives, Omphalotols A and B and Anti-Helicobacter pylori Fatty Acid Derivatives from Poisonous Mushroom Omphalotus japonicus. Pharmaceuticals. 2022; 15(2):139. https://doi.org/10.3390/ph15020139
Chicago/Turabian StyleLee, Seulah, Tae Wan Kim, Yong Hoon Lee, Dong-Min Kang, Rhim Ryoo, Yoon-Joo Ko, Mi-Jeong Ahn, and Ki Hyun Kim. 2022. "Two New Fatty Acid Derivatives, Omphalotols A and B and Anti-Helicobacter pylori Fatty Acid Derivatives from Poisonous Mushroom Omphalotus japonicus" Pharmaceuticals 15, no. 2: 139. https://doi.org/10.3390/ph15020139