
Tunicamycin Protects against LPS-Induced Lung Injury


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
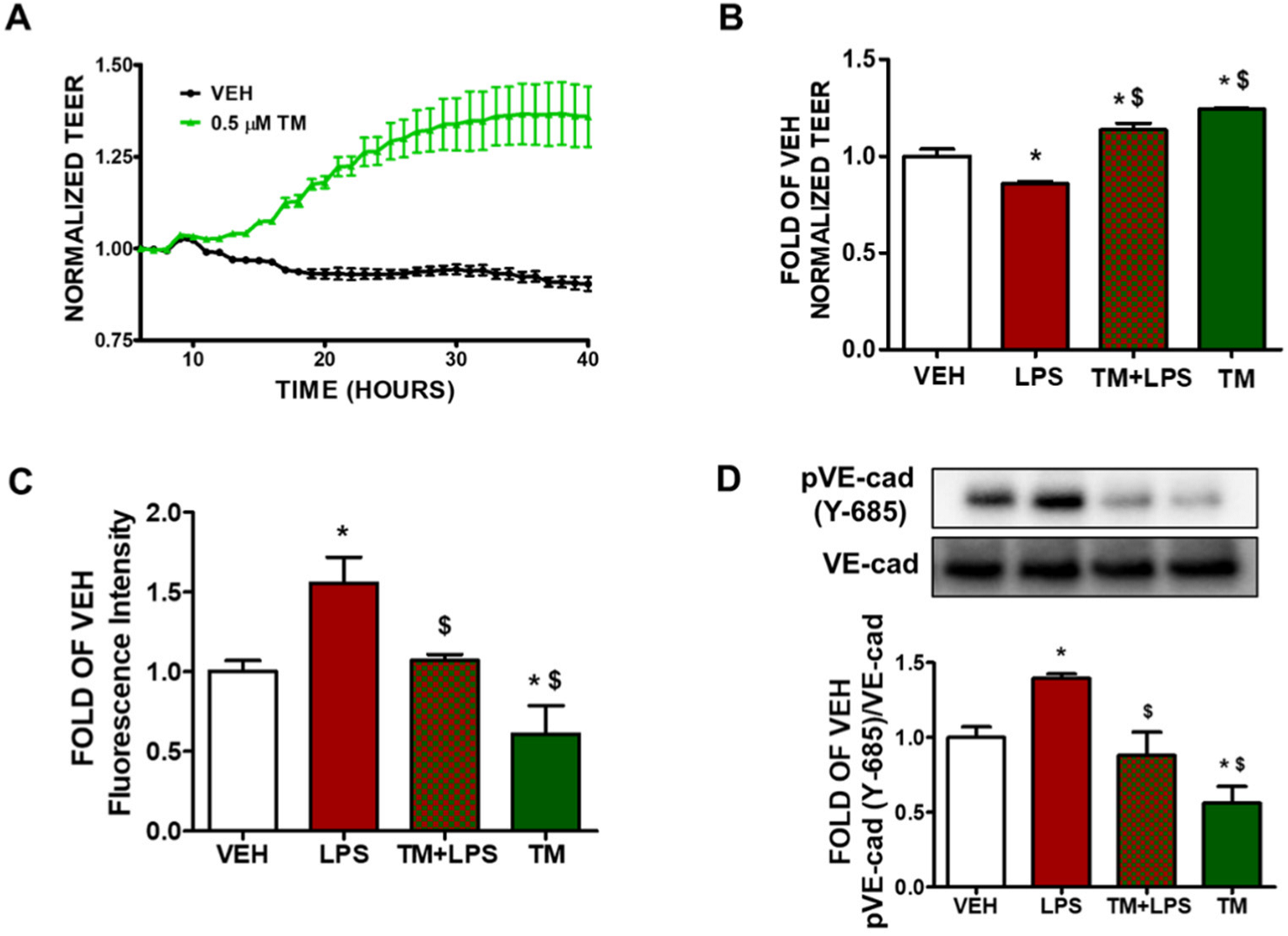
2.1. TM Enhances Lung Endothelial Integrity
2.2. TM Protects against LPS-Induced Increase in Lung Endothelial Permeability
2.3. TM Protects against LPS-Induced Induction of Paracellular Permeability in Lung Endothelial Cells
2.4. TM Suppresses LPS-Induced Phosphorylation of VE-Cadherin
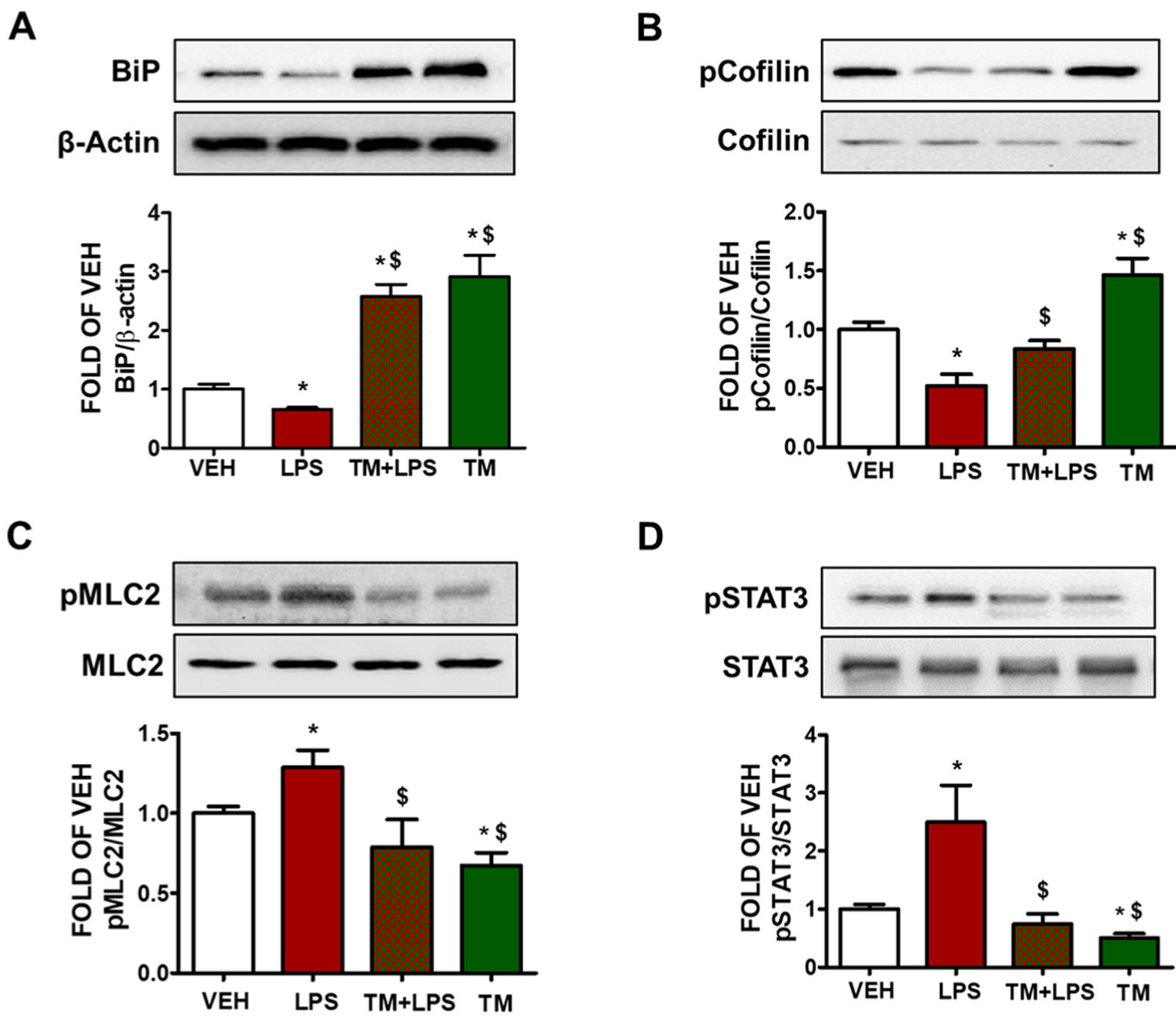
2.5. TM Counteracts LPS-Induced UPR Suppression
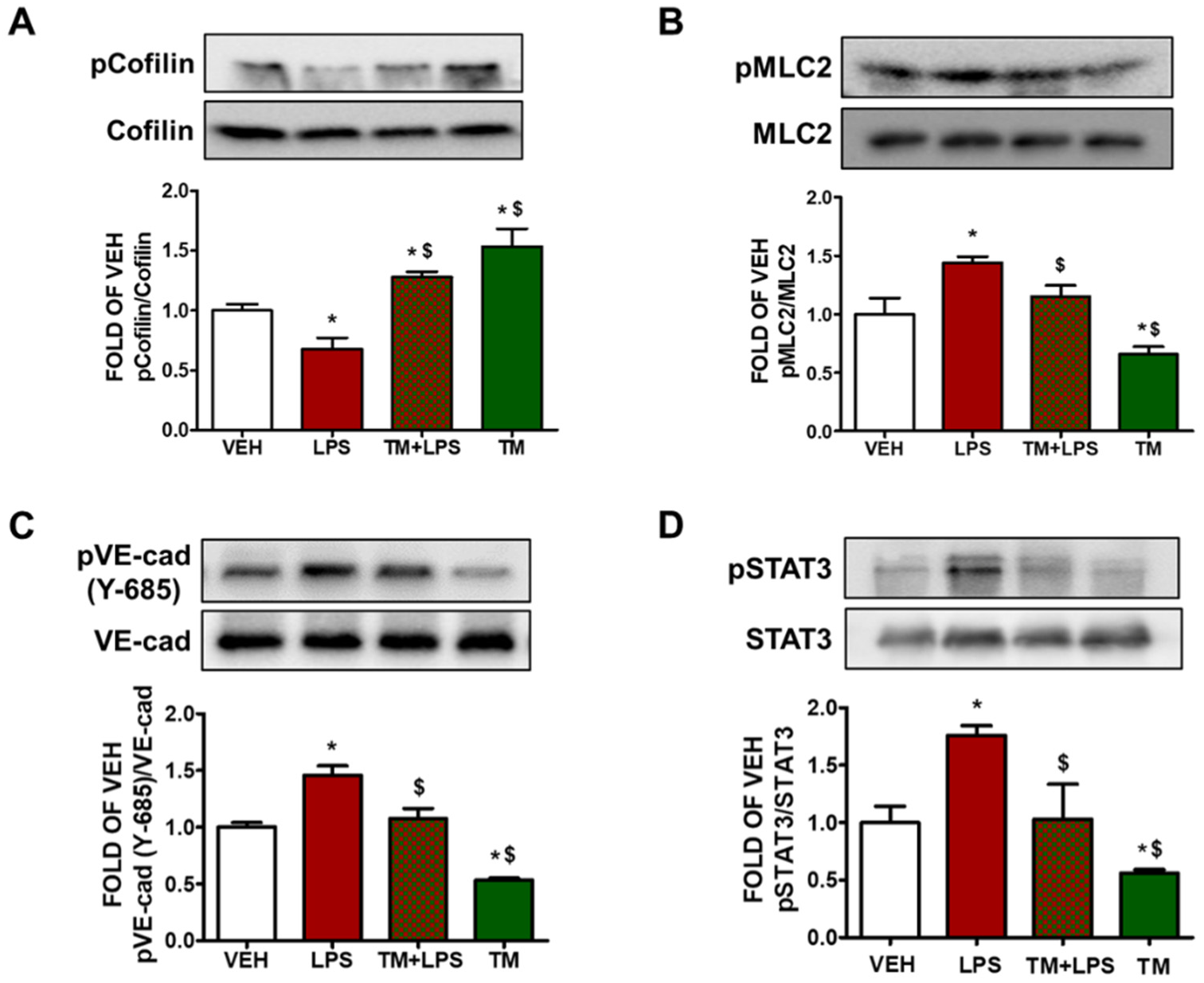
2.6. TM Suppresses LPS-Induced Activation of Cofilin in BPAEC
2.7. LPS-Induced Formation of Actin Stress Fibers Is Counteracted by TM in Lung Endothelial Cells
2.8. TM Suppresses LPS-Induced Activation of STAT3 in BPAEC
2.9. TM Protects against LPS-Induced Activation of Cofilin and MLC2 in Mice
2.10. TM Inhibits LPS-Induced Phosphorylation of VE-Cadherin in Mouse Lungs
2.11. TM Protects against LPS-Induced Activation of STAT3 Signaling in Mouse Lungs
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Animals
4.4. In Vivo Treatments
4.5. Western Blot Analysis
4.6. Fluorescein Isothiocyanate (FITC)-Dextran Permeability Assay
4.7. Measurement of Endothelial Barrier Function
4.8. Densitometry and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kubra, K.-T.; Barabutis, N. Brefeldin A and kifunensine modulate LPS-induced lung endothelial hyperpermeability in human and bovine cells. Am. J. Physiol. Cell Physiol. 2021, 321, C214–C220. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N. Growth Hormone Releasing Hormone in Endothelial Barrier Function. Trends Endocrinol. Metab. 2021, 32, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Millar, F.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Orsenigo, F.; Molendini, C.; Baluk, P.; McDonald, D.M. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009, 335, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannotta, M.; Trani, M.; Dejana, E. VE-Cadherin and Endothelial Adherens Junctions: Active Guardians of Vascular Integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal. Transduct. Target. Ther. 2017, 2, 17002. [Google Scholar] [CrossRef]
- Barabutis, N.; Akhter, M.S.; Kubra, K.-T.; Uddin, M.A. Restoring the endothelial barrier function in the elderly. Mech. Ageing Dev. 2021, 196, 111479. [Google Scholar] [CrossRef]
- Kubra, K.-T.; Akhter, M.S.; Uddin, M.A.; Barabutis, N. Unfolded protein response in cardiovascular disease. Cell. Signal. 2020, 73, 109699. [Google Scholar] [CrossRef]
- Kubra, K.-T.; Uddin, M.A.; Akhter, M.S.; Barabutis, N. Hsp90 inhibitors induce the unfolded protein response in bovine and mice lung cells. Cell. Signal. 2020, 67, 109500. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.A.; Kubra, K.-T.; Sonju, J.J.; Akhter, M.S.; Seetharama, J.; Barabutis, N. Effects of Heat Shock Protein 90 Inhibition In the Lungs. Med. Drug Discov. 2020, 6, 100046. [Google Scholar] [CrossRef] [PubMed]
- Naiel, S.; Tat, V.; Padwal, M.; Vierhout, M.; Mekhael, O.; Yousof, T.; Ayoub, A.; Abed, S.; Dvorkin-Gheva, A.; Ask, K. Protein Misfolding and Endoplasmic Reticulum Stress in Chronic Lung Disease: Will Cell-Specific Targeting Be the Key to the Cure? Chest 2020, 157, 1207–1220. [Google Scholar] [CrossRef]
- Hsu, J.-W.; Tang, P.-H.; Wang, I.-H.; Liu, C.-L.; Chen, W.-H.; Tsai, P.-C.; Chen, K.-Y.; Yu, C.-J.; Lee, F.-J.S. Unfolded protein response regulates yeast small GTPase Arl1p activation at late Golgi via phosphorylation of Arf GEF Syt1p. Proc. Natl. Acad. Sci. USA 2016, 113, E1683–E1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, N.P.; Hartman, T.M.; Li, J.; Velpula, K.K.; Naumann, T.A.; Guda, M.R.; Yu, B.; Bischoff, K.M. Modified tunicamycins with reduced eukaryotic toxicity that enhance the antibacterial activity of beta-lactams. J. Antibiot. 2017, 70, 1070–1077. [Google Scholar] [CrossRef]
- Hayakawa, K.; Hiramatsu, N.; Okamura, M.; Yamazaki, H.; Nakajima, S.; Yao, J.; Paton, A.W.; Paton, J.C.; Kitamura, M. Acquisition of Anergy to Proinflammatory Cytokines in Nonimmune Cells through Endoplasmic Reticulum Stress Response: A Mechanism for Subsidence of Inflammation. J. Immunol. 2009, 182, 1182–1191. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.A.; Akhter, M.S.; Siejka, A.; Catravas, J.D.; Barabutis, N. P53 supports endothelial barrier function via APE1/Ref1 suppression. Immunobiology 2019, 224, 532–538. [Google Scholar] [CrossRef]
- Citterio, C.; Vichi, A.; Pacheco-Rodriguez, G.; Aponte, A.M.; Moss, J.; Vaughan, M. Unfolded protein response and cell death after depletion of brefeldin A-inhibited guanine nucleotide-exchange protein GBF1. Proc. Natl. Acad. Sci. USA 2008, 105, 2877–2882. [Google Scholar] [CrossRef] [Green Version]
- Akhter, M.S.; Kubra, K.-T.; Uddin, M.A.; Barabutis, N. Kifunensine compromises lung endothelial barrier function. Microvasc. Res. 2020, 132, 104051. [Google Scholar] [CrossRef]
- Akhter, M.S.; Uddin, M.A.; Schally, A.V.; Kubra, K.-T.; Barabutis, N. Involvement of the unfolded protein response in the protective effects of growth hormone releasing hormone antagonists in the lungs. J. Cell Commun. Signal. 2020, 15, 125–129. [Google Scholar] [CrossRef]
- Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Paxman, R.J.; Plate, L.; Kelly, J.W.; Wiseman, R.L.; Glembotski, C.C. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 2019, 10, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yuan, Y.; Jiang, L.; Zhang, J.; Gao, J.; Shen, Z.; Zheng, Y.; Deng, T.; Yan, H.; Li, W.; et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014, 10, 1801–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chen, Y.; Zhou, Q.; Xu, J.; Qian, Q.; Ni, P.; Qian, Y. Mild Endoplasmic Reticulum Stress Protects Against Lipopolysaccharide-Induced Astrocytic Activation and Blood-Brain Barrier Hyperpermeability. Front. Cell. Neurosci. 2018, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.S.; Uddin, M.A.; Kubra, K.-T.; Barabutis, N. Autophagy, unfolded protein response and lung disease. Curr. Res. Cell Biol. 2020, 1, 100003. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N. Unfolded Protein Response in Lung Health and Disease. Front. Med. 2020, 7, 344. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N.; Verin, A.; Catravas, J.D. Regulation of pulmonary endothelial barrier function by kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L832–L845. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Lv, J.; Yang, W.; Xu, B.; Wang, Z.; Yu, Z.; Wu, J.; Yang, Y.; Han, Y. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis. Theranostics 2019, 9, 6424–6442. [Google Scholar] [CrossRef]
- Hayakawa, K.; Hiramatsu, N.; Okamura, M.; Yao, J.; Paton, A.W.; Paton, J.C.; Kitamura, M. Blunted activation of NF-kappaB and NF-kappaB-dependent gene expression by geranylgeranylacetone: Involvement of unfolded protein response. Biochem. Biophys. Res. Commun. 2008, 365, 47–53. [Google Scholar] [CrossRef]
- Engedal, N.; Torgersen, M.L.; Guldvik, I.J.; Barfeld, S.J.; Bakula, D.; Sætre, F.; Hagen, L.K.; Patterson, J.B.; Proikas-Cezanne, T.; Seglen, P.O.; et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 2013, 9, 1475–1490. [Google Scholar] [CrossRef] [Green Version]
- Doroghazi, J.R.; Ju, K.-S.; Brown, D.W.; Labeda, D.P.; Deng, Z.; Metcalf, W.W.; Chen, W.; Price, N.P.J. Genome Sequences of Three Tunicamycin-Producing Streptomyces Strains, S. chartreusis NRRL 12338, S. chartreusis NRRL 3882, and S. lysosuperificus ATCC 31396. J. Bacteriol. 2011, 193, 7021–7022. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Hwang, J.S.; Han, I.O. Tunicamycin inhibits Toll-like receptor-activated inflammation in RAW264.7 cells by suppression of NF-kappaB and c-Jun activity via a mechanism that is independent of ER-stress and N-glycosylation. Eur. J. Pharmacol. 2013, 721, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Maciel, M.; Hernández-Barrientos, D.; Herrera, I.; Selman, M.; Pardo, A.; Cabrera, S. Impaired autophagic activity and ATG4B deficiency are associated with increased endoplasmic reticulum stress-induced lung injury. Aging 2018, 10, 2098–2112. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Lee, M.D.; Wilson, C. The Endothelium Solves Problems That Endothelial Cells Do Not Know Exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef] [Green Version]
- Lenin, R.; Nagy, P.G.; Jha, K.A.; Gangaraju, R. GRP78 translocation to the cell surface and O-GlcNAcylation of VE-Cadherin contribute to ER stress-mediated endothelial permeability. Sci. Rep. 2019, 9, 10783. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Zhang, G.; Wang, X.; Nguyen, C.Y.; May, H.; Li, X.; Al-Hashimi, A.A.; Austin, R.C.; Gillette, T.G.; Fu, G.S.; et al. Endoplasmic Reticulum Chaperone GRP78 Protects Heart From Ischemia/Reperfusion Injury Through Akt Activation. Circ. Res. 2018, 122, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef]
- Vogel, S.M.; Malik, A.B. Cytoskeletal Dynamics and Lung Fluid Balance. Compr. Physiol. 2012, 2, 449–478. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Shimizu, J.; Miyawaki-Shimizu, K.; Vogel, S.M.; Bair, A.M.; Minshall, R.D.; Predescu, D.; Malik, A.B. Role of NF-kappaB-dependent caveolin-1 expression in the mechanism of increased endothelial permeability induced by lipopolysaccharide. J. Biol. Chem. 2008, 283, 4210–4218. [Google Scholar] [CrossRef] [Green Version]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 2008, 1778, 794–809. [Google Scholar] [CrossRef] [Green Version]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef]
- Liu, Z.; Tan, J.L.; Cohen, D.M.; Yang, M.T.; Sniadecki, N.J.; Ruiz, S.A.; Nelson, C.M.; Chen, C.S. Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. USA 2010, 107, 9944–9949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallez, Y.; Cand, F.; Cruzalegui, F.; Wernstedt, C.; Souchelnytskyi, S.; Vilgrain, I.; Huber, P. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: Identification of tyrosine 685 as the unique target site. Oncogene 2006, 26, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Tian, Y.; Dubrovskyi, O.; Zebda, N.; Sarich, N.; Tian, X.; Wang, Y.; Birukov, K.G. VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J. Cell. Physiol. 2011, 227, 3405–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, A.G.; Smith, T.H.; Chen, B.; Bhattacharya, S.; Cordova, I.C.; Kenakin, T.; Vaidehi, N.; Trejo, J. N-linked glycosylation of protease-activated receptor-1 at extracellular loop 2 regulates G-protein signaling bias. Proc. Natl. Acad. Sci. USA 2015, 112, E3600–E3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Chen, Y.C.; Lan, T.; Qian, H.; Wang, Y.; Jiang, L. LPS-induced nuclear translocation of RhoA is dependent on NF-kappaB in the human lung cancer cell line A549. Oncol. Lett. 2012, 3, 1283–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Nelson, W.J. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell–cell adhesion. J. Cell Biol. 2007, 178, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Kubra, K.-T.; Uddin, M.A.; Akhter, M.S.; Barabutis, N. Luminespib counteracts the Kifunensine-induced lung endothelial barrier dysfunction. Curr. Res. Toxicol. 2020, 1, 111–115. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Barabutis, N.; Uddin, M.A.; Catravas, J.D. Hsp90 inhibitors suppress P53 phosphorylation in LPS-induced endothelial inflammation. Cytokine 2019, 113, 427–432. [Google Scholar] [CrossRef]
- Akhter, M.; Uddin, M.; Kubra, K.-T.; Barabutis, N. Elucidation of the Molecular Pathways Involved in the Protective Effects of AUY-922 in LPS-Induced Inflammation in Mouse Lungs. Pharmaceuticals 2021, 14, 522. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubra, K.-T.; Uddin, M.A.; Barabutis, N. Tunicamycin Protects against LPS-Induced Lung Injury. Pharmaceuticals 2022, 15, 134. https://doi.org/10.3390/ph15020134
Kubra K-T, Uddin MA, Barabutis N. Tunicamycin Protects against LPS-Induced Lung Injury. Pharmaceuticals. 2022; 15(2):134. https://doi.org/10.3390/ph15020134
Chicago/Turabian StyleKubra, Khadeja-Tul, Mohammad A. Uddin, and Nektarios Barabutis. 2022. "Tunicamycin Protects against LPS-Induced Lung Injury" Pharmaceuticals 15, no. 2: 134. https://doi.org/10.3390/ph15020134