
Development and Validation of Open-Source R Package HMCtdm for Therapeutic Drug Monitoring

 , , , and
, , , and
Abstract
:1. Introduction
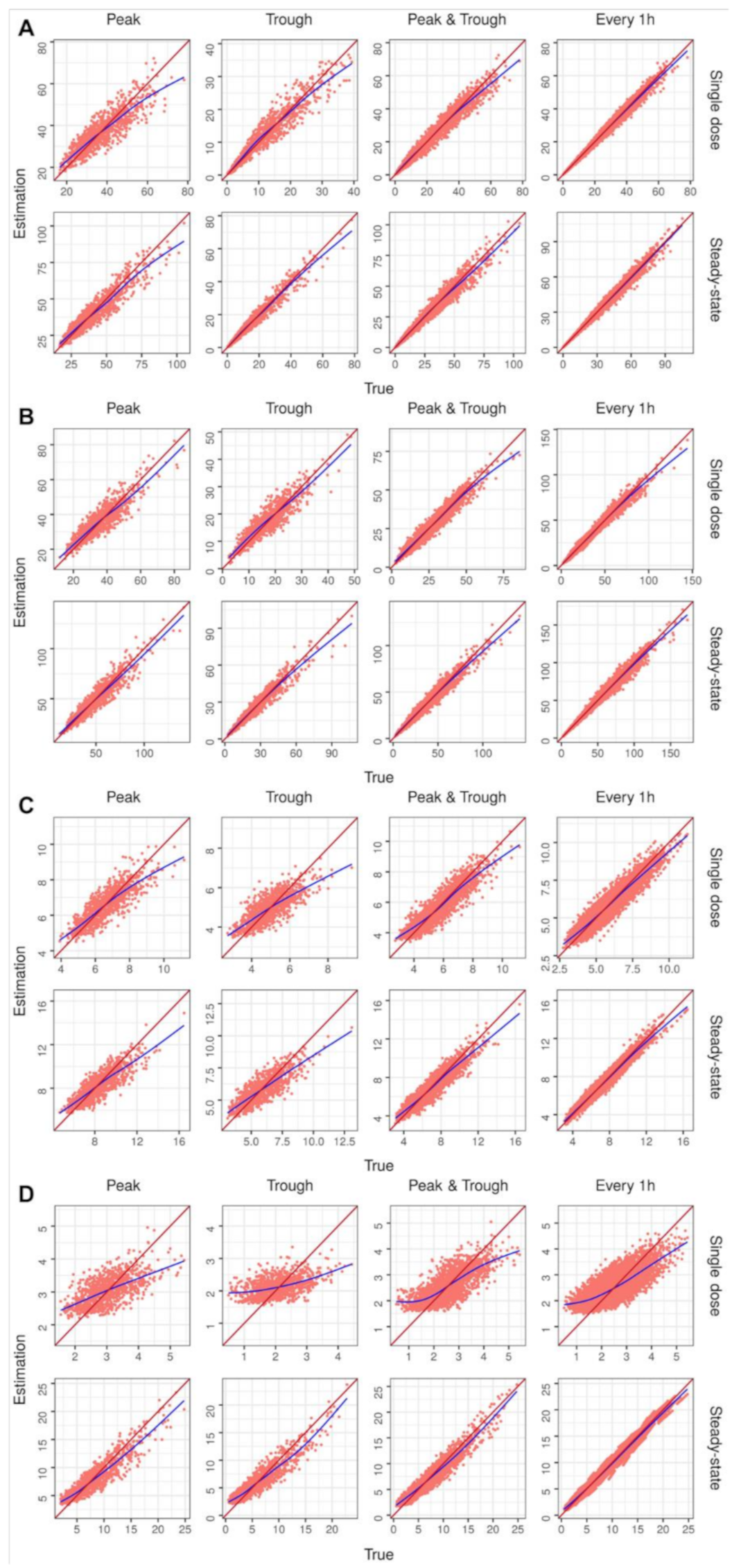
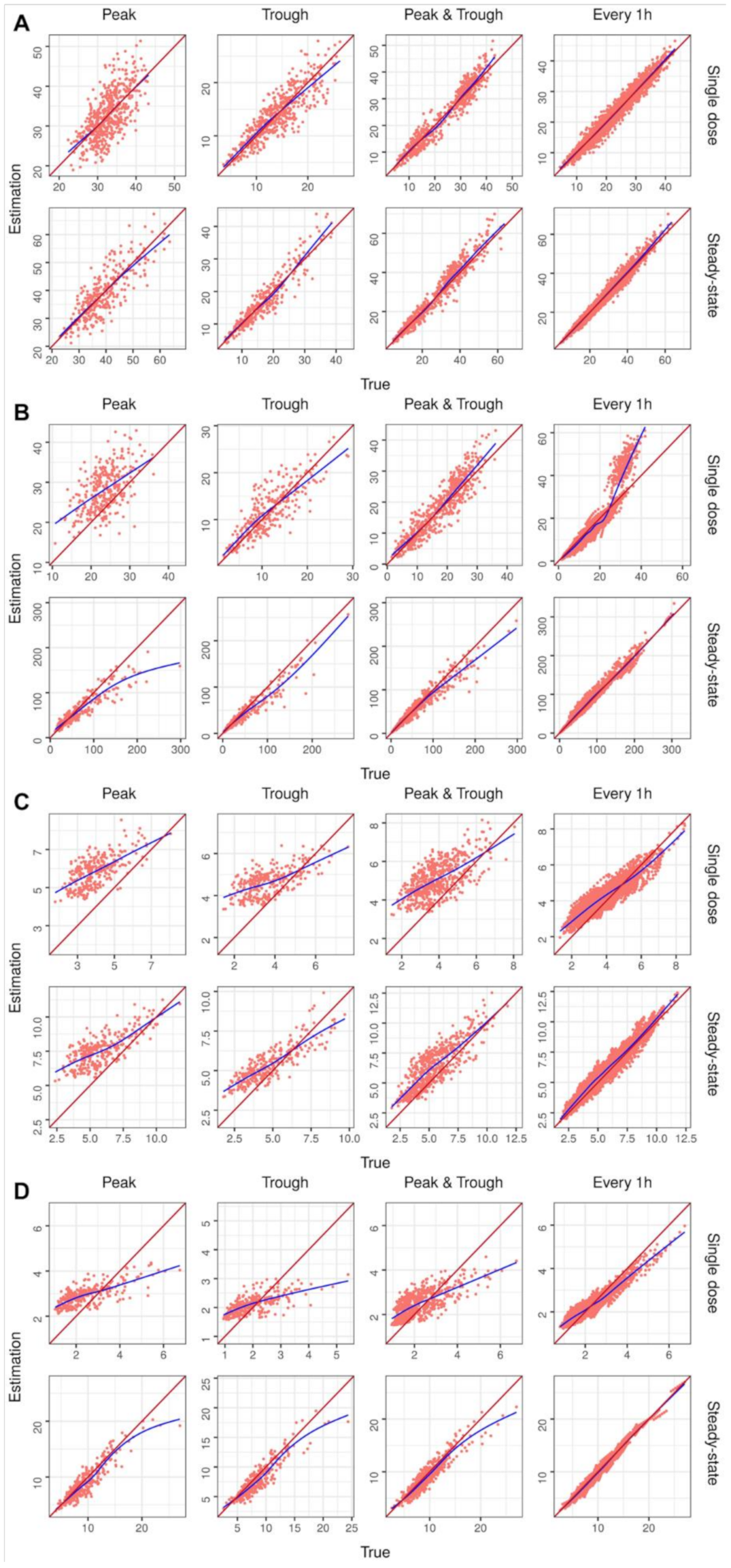
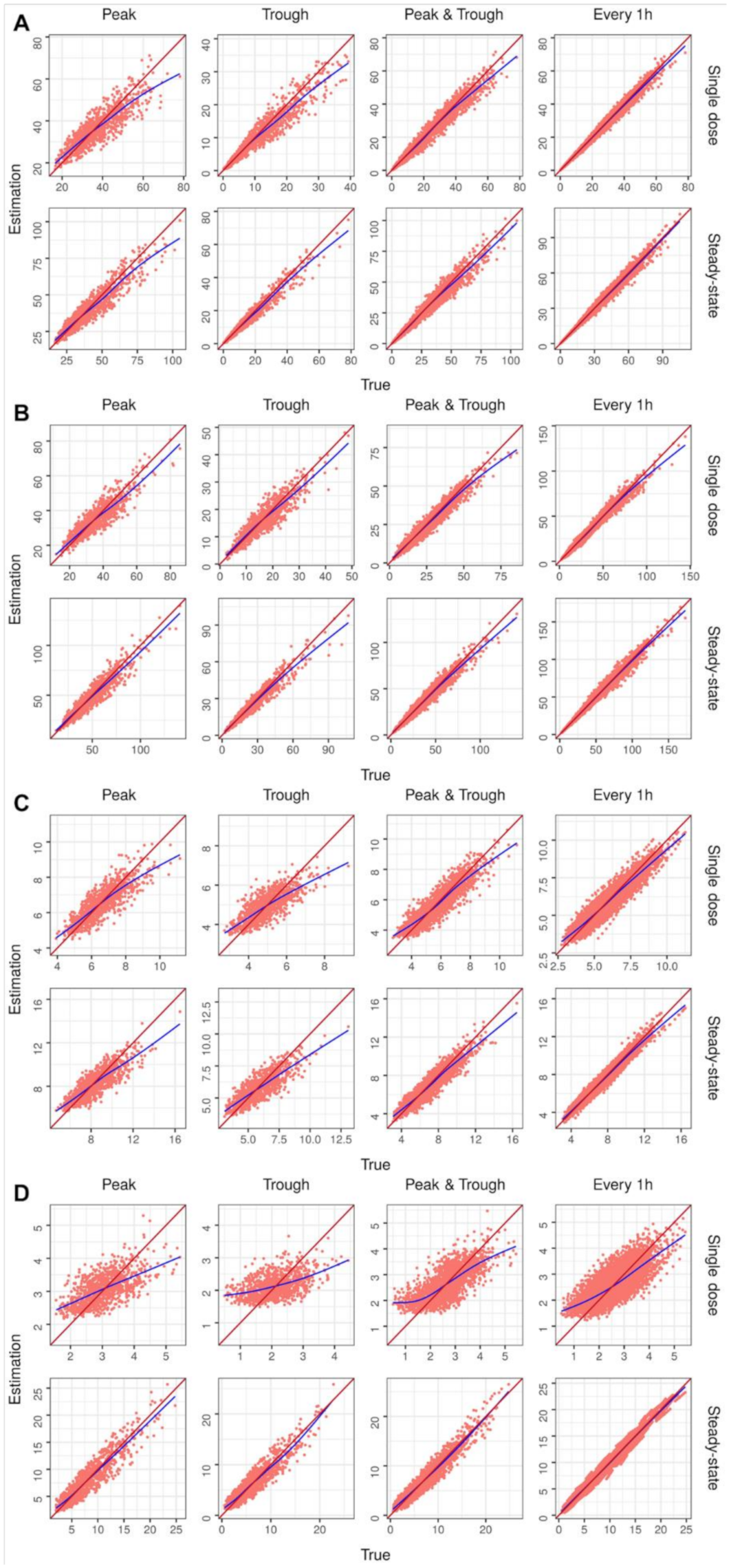
2. Results
3. Discussion
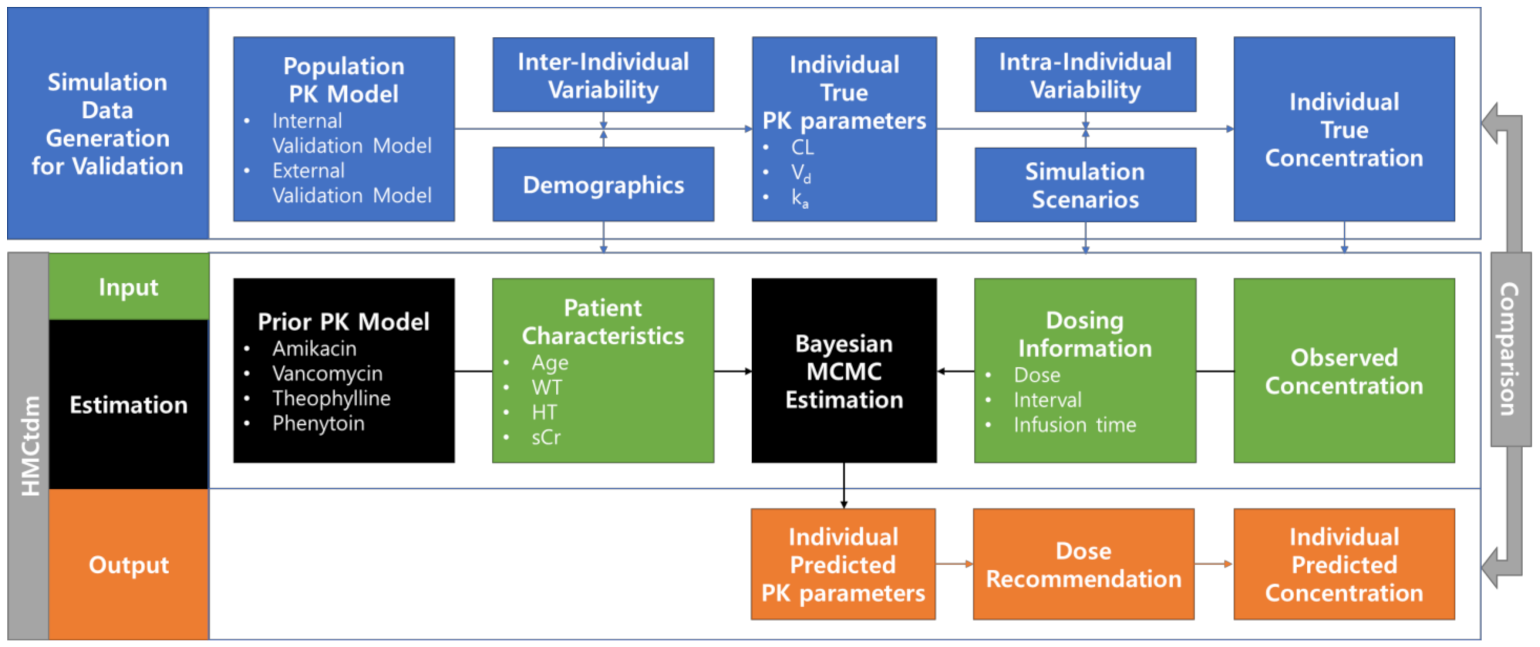
4. Materials and Methods
4.1. Development
4.1.1. Package Development
4.1.2. Pharmacokinetic Model
4.1.3. Estimation Method
4.1.4. Dose Target and Recommendation
4.2. Validation
4.2.1. Internal Validation PK model
4.2.2. External Validation of PK model
4.2.3. Performance Evaluation
4.3. MAP Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drennan, P.; Doogue, M.; van Hal, S.J.; Chin, P. Bayesian therapeutic drug monitoring software: Past, present and future. Int. J. Pharmacokinet. 2018, 3, 109. [Google Scholar] [CrossRef]
- Fuchs, A.; Csajka, C.; Thoma, Y.; Buclin, T.; Widmer, N. Benchmarking therapeutic drug monitoring software: A review of available computer tools. Clin. Pharmacokinet. 2013, 52, 9–22. [Google Scholar] [CrossRef]
- Broeker, A.; Nardecchia, M.; Klinker, K.; Derendorf, H.; Day, R.; Marriott, D.; Carland, J.; Stocker, S.; Wicha, S. Towards precision dosing of vancomycin: A systematic evaluation of pharmacometric models for Bayesian forecasting. Clin. Microbiol. Infect. 2019, 25, 1286.e1–1286.e7. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.H.; Keizer, R.J. A hybrid machine learning/pharmacokinetic approach outperforms maximum a posteriori Bayesian estimation by selectively flattening model priors. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Uster, D.W.; Stocker, S.L.; Carland, J.E.; Brett, J.; Marriott, D.J.; Day, R.O.; Wicha, S.G. A model averaging/selection approach improves the predictive performance of model-informed precision dosing: Vancomycin as a case study. Clin. Pharmacol. Ther. 2021, 109, 175–183. [Google Scholar] [CrossRef]
- Sheiner, L.B.; Beal, S.; Rosenberg, B.; Marathe, V.V. Forecasting individual pharmacokinetics. Clin. Pharmacol. Ther. 1979, 26, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Lunn, D.J.; Thomas, A.; Best, N.; Spiegelhalter, D. WinBUGS-a Bayesian modelling framework: Concepts, structure, and extensibility. Stat. Comput. 2000, 10, 325–337. [Google Scholar] [CrossRef]
- Plummer, M. JAGS: A program for analysis of Bayesian graphical models using Gibbs sampling. In Proceedings of the 3rd International Workshop on Distributed Statistical Computing, Vienna, Austria, 20–23 March 2003; pp. 1–10. [Google Scholar]
- Carpenter, B.; Gelman, A.; Hoffman, M.D.; Lee, D.; Goodrich, B.; Betancourt, M.; Brubaker, M.; Guo, J.; Li, P.; Riddell, A. Stan: A probabilistic programming language. J. Stat. Softw. 2017, 76, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, B.; Johnston, C. Introduction to Bayesian pharmacometric data analysis using NONMEM®. In Proceedings of the ACoP10, Orlando, FL, USA, 20–23 October 2019. [Google Scholar]
- Jayachandran, D.; Laínez-Aguirre, J.; Rundell, A.; Vik, T.; Hannemann, R.; Reklaitis, G.; Ramkrishna, D. Model-based individualized treatment of chemotherapeutics: Bayesian population modeling and dose optimization. PLoS ONE 2015, 10, e0133244. [Google Scholar] [CrossRef]
- Pananos, A.D.; Lizotte, D.J. Comparisons between Hamiltonian Monte Carlo and maximum a posteriori for a Bayesian model for Apixaban induction dose & dose personalization. In Proceedings of the Machine Learning for Healthcare Conference, Virtual Meeting, 7–8 August 2020; pp. 397–417. [Google Scholar]
- Maier, C.; Hartung, N.; de Wiljes, J.; Kloft, C.; Huisinga, W. Bayesian data assimilation to support informed decision making in individualized chemotherapy. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 153–164. [Google Scholar] [CrossRef]
- Wakefield, J. Bayesian individualization via sampling-based methods. J. Pharmacokinet. Biopharm. 1996, 24, 103–131. [Google Scholar] [CrossRef] [PubMed]
- Torsten. Torsten: Library of C++ Functions that Support Applications of Stan in Pharmacometrics; Metrum Research Group LLC: Tariffville, CT, USA, 2015. [Google Scholar]
- Baron, K.T.; Hindmarsh, A.; Petzold, L.; Gillespie, B.; Margossian, C.; Pastoor, D. Mrgsolve: Simulate from ODE-Based Population PK/PD and Systems Pharmacology Models; Metrum Research Group LLC: Tariffville, CT, USA, 2019. [Google Scholar]
- D’Argenio, D.Z. Optimal sampling times for pharmacokinetic experiments. J. Pharmacokinet. Biopharm. 1981, 9, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; van Hest, R.M.; Fleuren, L.M.; Roggeveen, L.F.; Bosman, R.J.; van der Voort, P.H.; Girbes, A.R.; Mathot, R.A.; van Hasselt, J.G.; Elbers, P.W. Why we should sample sparsely and aim for a higher target: Lessons from model-based therapeutic drug monitoring of vancomycin in intensive care patients. Br. J. Clin. Pharmacol. 2021, 87, 1234–1242. [Google Scholar] [CrossRef]
- Mould, D.; D’haens, G.; Upton, R. Clinical decision support tools: The evolution of a revolution. Clin. Pharmacol. Ther. 2016, 99, 405–418. [Google Scholar] [CrossRef] [PubMed]
- The R Development Core Team. R: A Language and Environment for Statistical Computing; Version 4.1.0.; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Betancourt, M. A conceptual introduction to Hamiltonian Monte Carlo. arXiv 2017, arXiv:1701.02434. Available online: https://arxiv.org/abs/1701.02434 (accessed on 1 November 2021).
- Neal, R.M. Monte Carlo Implementation. In Bayesian Learning for Neural Networks; Springer: New York, NY, USA, 1996; pp. 55–98. [Google Scholar]
- Lenert, L.; Peck, C.C.; Brown, W.D. One-Compartment Forecaster Reference Materials; Technical Report No. 10, Appendix 1, 114–115; Division of Clinical Pharmacology Uniformed Services of the Health Sciences: Bethesda, MD, USA, 1982. [Google Scholar]
- Cockcroft, D.W.; Gault, H. Prediction of creatinine clearance from serum creatinine. Nephron 1976, 16, 31–41. [Google Scholar] [CrossRef]
- Anonymous. Amikacin Inj.; package insert; Dongkwang Pharmaceutical Co., LTD.: Seoul, Korea, 2017. [Google Scholar]
- Anonymous. Vancomycin HCl Injection; package insert; HK Inno. N Co.: Seoul, Korea, 2020. [Google Scholar]
- Anonymous. TEHOLAN-B®; package insert; Alvogen Korea Co.: Seoul, Korea, 2017. [Google Scholar]
- Anonymous. Hydantoin Tab; package insert; Whan in Pharmaceutical Co., Ltd.: Seoul, Korea, 2019. [Google Scholar]
- Gilbert, D.N.; Chamber, H.F.; Saag, M.S.; Pavia, A.T. The Sanford Guide to Antimicrobial Therapy 2020, 50th ed.; Antimicrobial Therapy, Incorporated: Sperryville, VA, USA, 2020; pp. 114–130. [Google Scholar]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Rotschafer, J.C. Therapeutic monitoring of vancomycin for serious methicillin-resistant Staphylococcus aureus infections: A revised consensus guideline and review by the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists. Clin. Infect. Dis. 2020, 71, 1361–1364. [Google Scholar]
- Malson, G. Therapeutic Drug Monitoring—Medicines Formulary; Version 7; Wirral University Teaching Hospital: Birkenhead, UK, 2013. [Google Scholar]
- Jang, S.; Lee, Y.; Park, M.; Song, Y.; Kim, J.; Kim, H.; Ahn, B.; Park, K. Population pharmacokinetics of amikacin in a Korean clinical population. Int. J. Clin. Pharmacol. Ther. 2011, 49, 371–381. [Google Scholar] [CrossRef]
- Bae, S.H.; Yim, D.-S.; Lee, H.; Park, A.-R.; Kwon, J.-E.; Sumiko, H.; Han, S. Application of Pharmacometrics in Pharmacotherapy: Open-Source Software for Vancomycin Therapeutic Drug Management. Pharmaceutics 2019, 11, 224. [Google Scholar] [CrossRef] [Green Version]
- Tanigawara, Y.; Komada, F.; Shimizu, T.; Iwakawa, S.; Iwai, T.; Maekawa, H.; Hori, R.; Okumura, K. Population pharmacokinetics of theophylline. III. Premarketing study for a once-daily administered preparation. Biol. Pharm. Bull. 1995, 18, 1590–1598. [Google Scholar] [CrossRef] [Green Version]
- Odani, A.; Hashimoto, Y.; Takayanagi, K.; Otsuki, Y.; Koue, T.; Takano, M.; Yasuhara, M.; Hattori, H.; Furusho, K.; Inui, K. Population pharmacokinetics of phenytoin in Japanese patients with epilepsy: Analysis with a dose-dependent clearance model. Biol. Pharm. Bull. 1996, 19, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Le Louedec, F.; Puisset, F.; Thomas, F.; Chatelut, É.; White-Koning, M. Easy and reliable maximum a posteriori Bayesian estimation of pharmacokinetic parameters with the open-source R package mapbayr. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 1208–1220. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Time | Peak | Trough | Peak and Trough | Every 1 h | ||||
|---|---|---|---|---|---|---|---|---|
| MPE (%) | RMSE (mg/L) | MPE (%) | RMSE (mg/L) | MPE(%) | RMSE (mg/L) | MPE(%) | RMSE (mg/L) | |
| Amikacin | ||||||||
| Single dose | 0.91 | 5.11 | 4.92 | 2.48 | 1.37 | 3.22 | −0.32 | 1.35 |
| Steady state | −0.85 | 5.31 | 2.14 | 2.49 | 0.70 | 3.81 | −0.35 | 1.75 |
| Vancomycin | ||||||||
| Single dose | 2.32 | 4.51 | 6.72 | 2.85 | 2.27 | 3.06 | 0.23 | 2.04 |
| Steady state | 0.39 | 6.12 | 1.89 | 3.80 | 1.12 | 4.14 | −0.11 | 2.49 |
| Theophylline | ||||||||
| Single dose | −0.01 | 0.61 | 1.56 | 0.59 | 0.78 | 0.58 | −0.03 | 0.40 |
| Steady state | −0.25 | 0.85 | 0.90 | 0.77 | −0.18 | 0.68 | −0.45 | 0.35 |
| Phenytoin | ||||||||
| Single dose | 3.54 | 0.52 | 12.04 | 0.56 | 7.53 | 0.53 | 5.28 | 0.46 |
| Steady state | 5.28 | 1.60 | 13.91 | 1.52 | 7.24 | 1.18 | 2.27 | 0.58 |
| Sampling Time | Peak | Trough | Peak and Trough | Every 1 h | ||||
|---|---|---|---|---|---|---|---|---|
| MPE (%) | RMSE (mg/L) | MPE (%) | RMSE (mg/L) | MPE(%) | RMSE (mg/L) | MPE(%) | RMSE (mg/L) | |
| Amikacin | ||||||||
| Single dose | 0.25 | 4.44 | 1.15 | 2.34 | 0.75 | 2.86 | −0.07 | 1.62 |
| Steady state | −0.14 | 5.14 | −0.10 | 2.72 | 1.86 | 3.49 | 0.32 | 1.83 |
| Vancomycin | ||||||||
| Single dose | 21.91 | 6.66 | 6.99 | 2.95 | 5.49 | 3.45 | −5.25 | 4.47 |
| Steady state | −2.72 | 21.19 | −5.08 | 15.92 | −0.62 | 13.11 | 3.38 | 6.60 |
| Theophylline | ||||||||
| Single dose | 53.35 | 2.04 | 34.62 | 1.29 | 37.92 | 1.48 | 12.53 | 0.60 |
| Steady state | 37.15 | 2.09 | 19.43 | 1.07 | 21.26 | 1.37 | 6.93 | 0.58 |
| Phenytoin | ||||||||
| Single dose | 34.39 | 0.91 | 14.65 | 0.57 | 21.15 | 0.70 | 8.07 | 0.34 |
| Steady state | −5.13 | 1.43 | −8.01 | 1.44 | −4.36 | 1.16 | −1.96 | 0.54 |
| Sampling Time | Peak | Trough | Peak and Trough | Every 1 h | ||||
|---|---|---|---|---|---|---|---|---|
| MPE (%) | RMSE (mg/L) | MPE (%) | RMSE (mg/L) | MPE(%) | RMSE (mg/L) | MPE(%) | RMSE (mg/L) | |
| Amikacin | ||||||||
| Single dose | −0.33 | 5.16 | −4.65 | 2.72 | −2.28 | 3.34 | −1.16 | 1.38 |
| Steady state | −1.85 | 5.41 | −2.64 | 2.66 | −1.93 | 3.89 | −0.84 | 1.77 |
| Vancomycin | ||||||||
| Single dose | −0.15 | 4.53 | 0.22 | 2.82 | −1.20 | 3.14 | −1.16 | 2.06 |
| Steady state | −0.97 | 6.17 | −1.79 | 3.94 | −1.26 | 4.22 | −0.80 | 2.46 |
| Theophylline | ||||||||
| Single dose | −0.26 | 0.61 | 1.22 | 0.59 | 0.48 | 0.58 | −0.31 | 0.40 |
| Steady state | −0.42 | 0.85 | −0.11 | 0.78 | −0.81 | 0.69 | −0.71 | 0.36 |
| Phenytoin | ||||||||
| Single dose | 4.15 | 0.52 | 11.56 | 0.56 | 7.72 | 0.54 | 5.32 | 0.45 |
| Steady state | 2.07 | 1.53 | 7.06 | 1.41 | 3.86 | 1.11 | 1.21 | 0.56 |
| Pharmacokinetic Parameters | ||||||
|---|---|---|---|---|---|---|
| Drug (Model) | Amikacin (1 CMT IV) | Vancomycin (2 CMT IV) | ||||
| Parameters | Mean (CV) | Lower | Upper | Mean (CV) | Lower | Upper |
| CLslope | 0.815 (0.4) | 0.3 | 1.7 | 0.75 (0.33) | 0.3 | 1.7 |
| CLnr (mL/min/kg) | 0.0417 (0.25) | 0.0001 | 0.17 | 0.05 (0.2) | 0.01 | 0.2 |
| Vnr (L/kg) | 0.27 (0.3) | 0.15 | 0.65 | 0.21 (0.2) | 0.08 | 0.4 |
| k12 (1/h) | - | - | - | 1.12 (0.25) | 0.6 | 1.6 |
| k21 (1/h) | - | - | - | 0.48 (0.25) | 0.2 | 1.0 |
| Drug (Model) | Theophylline (1 CMT oral) | Phenytoin (1 CMT oral) | ||||
| Parameters | Mean (CV) | Lower | Upper | Mean (CV) | Lower | Upper |
| CLslope | - | - | - | 0.01 | - | - |
| CLnr (mL/h/kg) | 40.0 (0.5) | 15.0 | 90.0 | - | - | - |
| Vnr (L/kg) | 0.5 (0.2) | 0.35 | 0.65 | 0.8 (0.2) | 0.3 | 1.4 |
| ka | 0.27 | - | - | - | - | - |
| F | 1 | - | - | 0.92 | - | - |
| Vmax (mg/kg/d) | - | - | - | 500 (0.3) | 250.0 | 2000.0 |
| km (mcg/mL) | - | - | - | 5.0 (0.5) | 2.0 | 9.0 |
| Parameter Equations | ||||||
| Model | Linear Pharmacokinetics | Nonlinear Pharmacokinetics | ||||
| CL (L/h) | ||||||
| V (L) | ||||||
| Variability Equations | ||||||
| Parameters | ||||||
| Concentration | ||||||
(mg/L) | (mg/L) | |||||
| Drug | Amikacin | Vancomycin | Theophylline | Phenytoin | |
|---|---|---|---|---|---|
| Dose (mg) [25,26,27,28] | 500 | 1000 | 200 | 100 | |
| Infusion rate (mg/h) | 1000 | 500 | - | 50 * | |
| Dosing Interval (h) | 8 | 12 | 12 | 8 | |
| Sampling time (h) [29,30,31] | |||||
| Set 1 | Peak | 1 | 2 | 4 | 2 |
| Set 2 | Trough | 8 | 12 | 12 | 8 |
| Set 3 | Peak and trough | 1, 8 | 2, 12 | 4, 12 | 2, 8 |
| Set 4 | Every 1 h | 1 to 8 | 1 to 12 | 1 to 12 | 1 to 8 |
| Component | Equation |
|---|---|
| Amikacin [32] | |
| Pharmacokinetic Parameters | |
| Interindividual Variability | |
| Residual errors | |
| Vancomycin [33] | |
| Pharmacokinetic Parameters | |
| Interindividual Variability | |
| Residual errors | |
| Theophylline [34] | |
| Pharmacokinetic Parameters | * |
| Interindividual Variability | |
| Residual errors | |
| Phenytoin [35] | |
| Pharmacokinetic Parameters | |
| Interindividual Variability | |
| Residual errors | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Song, M.; Lim, W.; Song, E.; Han, J.; Kim, B.-H. Development and Validation of Open-Source R Package HMCtdm for Therapeutic Drug Monitoring. Pharmaceuticals 2022, 15, 127. https://doi.org/10.3390/ph15020127
Lee S, Song M, Lim W, Song E, Han J, Kim B-H. Development and Validation of Open-Source R Package HMCtdm for Therapeutic Drug Monitoring. Pharmaceuticals. 2022; 15(2):127. https://doi.org/10.3390/ph15020127
Chicago/Turabian StyleLee, Sooyoung, Moonsik Song, Woojae Lim, Eunjung Song, Jongdae Han, and Bo-Hyung Kim. 2022. "Development and Validation of Open-Source R Package HMCtdm for Therapeutic Drug Monitoring" Pharmaceuticals 15, no. 2: 127. https://doi.org/10.3390/ph15020127