
Identification of Potential Treatments for Acute Lymphoblastic Leukemia through Integrated Genomic Network Analysis



Abstract
:1. Introduction
2. Results
2.1. Prioritizing Variants from the GWAS Catalog
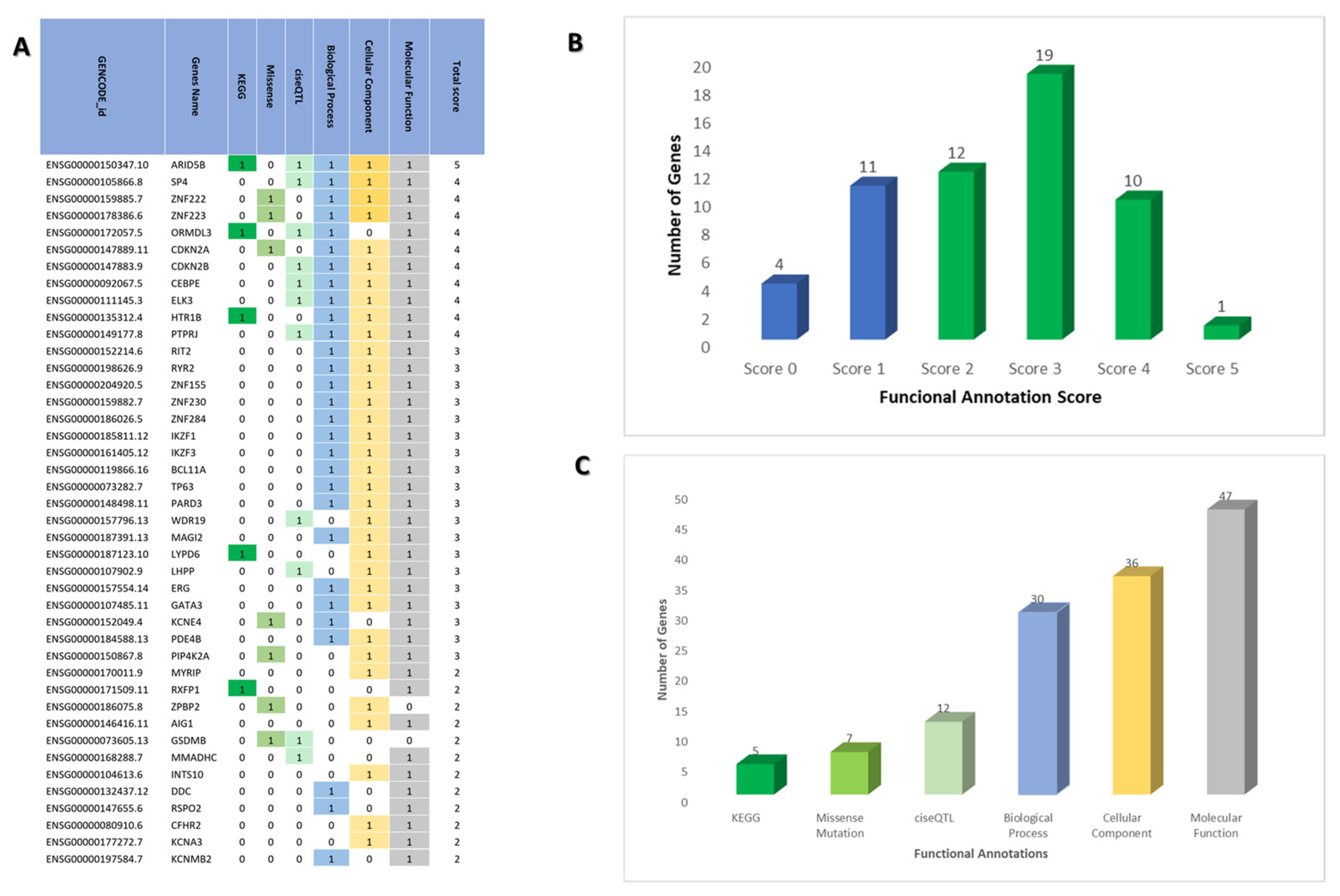
2.2. Prioritizing Biological Risk Gene for ALL
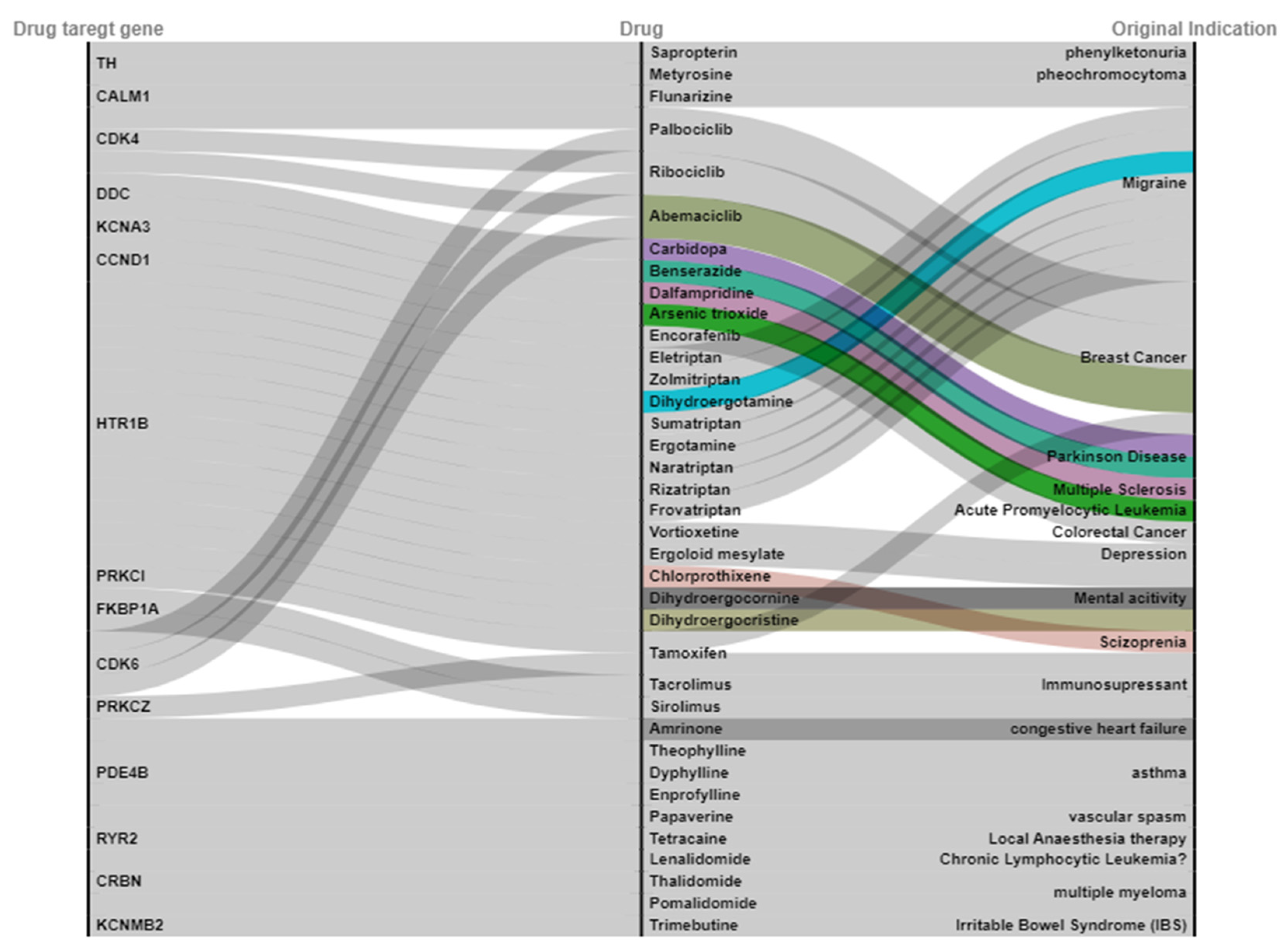
2.3. Drug Target Gene to Be Overlapped with a Drug Database
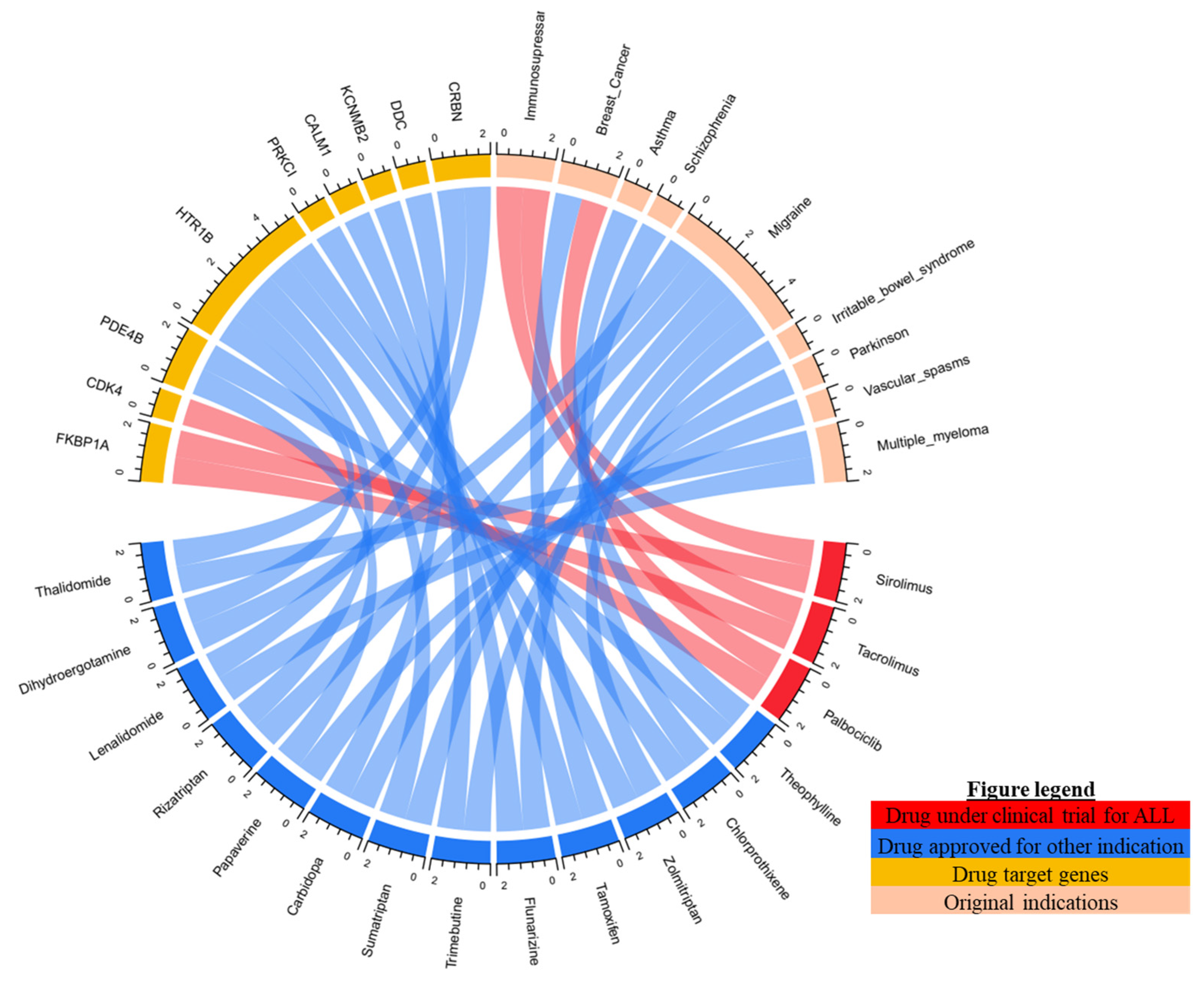
2.4. Candidate Drug for ALL Undergoing Clinical Trial
2.5. Candidate Drug for ALL according to CMap Analysis
3. Discussion
4. Materials and Methods
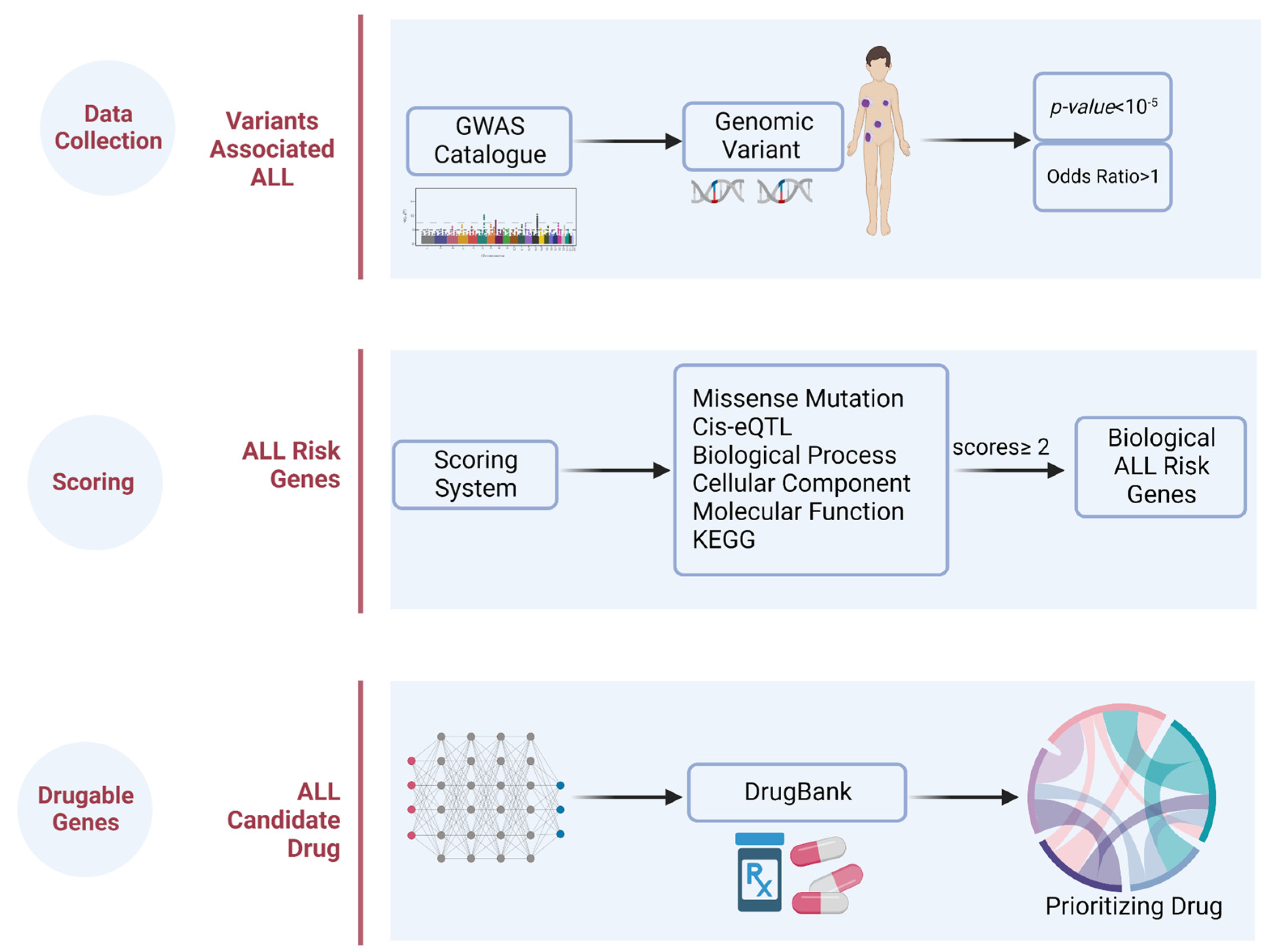
4.1. Design
4.2. Genetic Variants Associated with ALL
4.3. ALL Risk Genes
4.4. Prioritizing the Biological ALL-Risk Genes
4.5. Drug Identification
4.6. Connectivity Map (CMap) Analyses
4.7. Statistical and Integrated Genomic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaatsch, P. Epidemiology of childhood cancer. Cancer Treat. Rev. 2010, 36, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.A.; Shah, B.; Advani, A.; Aoun, P.; Boyer, M.W.; Burke, P.W.; DeAngelo, D.J.; Dinner, S.; Fathi, A.T.; Gauthier, J.; et al. Acute Lymphoblastic Leukemia, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 1079–1109. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Zhou, L.; Li, A.; Luo, S.; Wu, K. Global burden and trend of acute lymphoblastic leukemia from 1990 to 2017. Aging 2020, 12, 22869–22891. [Google Scholar] [CrossRef]
- Samra, B.; Jabbour, E.; Ravandi, F.; Kantarjian, H.; Short, N.J. Evolving therapy of adult acute lymphoblastic leukemia: State-of-the-art treatment and future directions. J. Hematol. Oncol. 2020, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Dull, J.; Yilmaz, M.; Khoury, J.D.; Ravandi, F.; Jain, N.; Einsele, H.; Garcia-Manero, G.; Konopleva, M.; Short, N.J.; et al. Outcome of patients with relapsed/refractory acute lymphoblastic leukemia after blinatumomab failure: No change in the level of CD19 expression. Am. J. Hematol. 2018, 93, 371–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, M.; Maciaszek, J.L.; Clark, M.E.; Pui, C.H.; Nichols, K.E. Recent advances in genetic predisposition to pediatric acute lymphoblastic leukemia. Expert Rev. Hematol. 2020, 13, 55–70. [Google Scholar] [CrossRef]
- Cerchione, C.; Locatelli, F.; Martinelli, G. Dasatinib in the Management of Pediatric Patients With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Front. Oncol. 2021, 11, 632231. [Google Scholar] [CrossRef]
- Brown, P.; Inaba, H.; Annesley, C.; Beck, J.; Colace, S.; Dallas, M.; DeSantes, K.; Kelly, K.; Kitko, C.; Lacayo, N.; et al. Pediatric Acute Lymphoblastic Leukemia, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 81–112. [Google Scholar] [CrossRef] [Green Version]
- Liu-Dumlao, T.; Kantarjian, H.; Thomas, D.A.; O’Brien, S.; Ravandi, F. Philadelphia-positive acute lymphoblastic leukemia: Current treatment options. Curr. Oncol. Rep. 2012, 14, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Schultz, K.R.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Devidas, M.; Wang, C.; Davies, S.M.; Gaynon, P.S.; Trigg, M.; et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: A children’s oncology group study. J. Clin. Oncol 2009, 27, 5175–5181. [Google Scholar] [CrossRef] [Green Version]
- Kort, E.; Jovinge, S. Drug Repurposing: Claiming the Full Benefit from Drug Development. Curr. Cardiol. Rep. 2021, 23, 62. [Google Scholar] [CrossRef] [PubMed]
- Reay, W.R.; Cairns, M.J. Advancing the use of genome-wide association studies for drug repurposing. Nat. Rev. Genet. 2021, 22, 658–671. [Google Scholar] [CrossRef]
- Sanseau, P.; Agarwal, P.; Barnes, M.R.; Pastinen, T.; Richards, J.B.; Cardon, L.R.; Mooser, V. Use of genome-wide association studies for drug repositioning. Nat. Biotechnol. 2012, 30, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Liu, Y.N.; Bi, Y.Y.; Wang, H. ARID5B gene polymorphisms and the risk of childhood acute lymphoblastic leukemia: A meta-analysis. Int. J. Hematol. 2019, 110, 272–284. [Google Scholar] [CrossRef]
- Zeng, H.; Wang, X.B.; Cui, N.H.; Nam, S.; Zeng, T.; Long, X. Associations between AT-rich interactive domain 5B gene polymorphisms and risk of childhood acute lymphoblastic leukemia: A meta-analysis. Asian Pac. J. Cancer Prev. 2014, 15, 6211–6217. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.M.; Xi, J.S.; Ma, Y.; Shao, L.; Nie, C.L.; Wang, G.J. ARID5B gene rs10821936 polymorphism is associated with childhood acute lymphoblastic leukemia: A meta-analysis based on 39,116 subjects. Tumour Biol. 2014, 35, 709–713. [Google Scholar] [CrossRef]
- Wang, P.; Deng, Y.; Yan, X.; Zhu, J.; Yin, Y.; Shu, Y.; Bai, D.; Zhang, S.; Xu, H.; Lu, X. The Role of ARID5B in Acute Lymphoblastic Leukemia and Beyond. Front. Genet. 2020, 11, 598. [Google Scholar] [CrossRef] [PubMed]
- Wilsker, D.; Patsialou, A.; Dallas, P.B.; Moran, E. ARID proteins: A diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ. 2002, 13, 95–106. [Google Scholar] [PubMed]
- Webb, C.F.; Bryant, J.; Popowski, M.; Allred, L.; Kim, D.; Harriss, J.; Schmidt, C.; Miner, C.A.; Rose, K.; Cheng, H.L.; et al. The ARID family transcription factor bright is required for both hematopoietic stem cell and B lineage development. Mol. Cell Biol. 2011, 31, 1041–1053. [Google Scholar] [CrossRef] [Green Version]
- Yokota, T.; Kanakura, Y. Role of tissue-specific AT-rich DNA sequence-binding proteins in lymphocyte differentiation. Int. J. Hematol. 2014, 100, 238–245. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, X.; Bhojwani, D.; E, S.; Goodings, C.; Zhang, H.; Seibel, N.L.; Yang, W.; Li, C.; Carroll, W.L.; et al. ARID5B Influences Antimetabolite Drug Sensitivity and Prognosis of Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020, 26, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Cheng, C.; Devidas, M.; Pei, D.; Fan, Y.; Yang, W.; Neale, G.; Scheet, P.; Burchard, E.G.; Torgerson, D.G.; et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2012, 30, 751–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Leon, A.; Ramirez-Martinez, M.; Fernandez-Garcia, D.; Amaro-Munoz, D.; Velazquez-Aragon, J.A.; Salas-Labadia, C.; Zapata-Tarres, M.; Velasco-Hidalgo, L.; Lopez-Santiago, N.; Lopez-Ruiz, M.I.; et al. Variants in ARID5B gene are associated with the development of acute lymphoblastic leukemia in Mexican children. Ann. Hematol. 2019, 98, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Han, Q.; Gu, Y.; Ge, Q.; Ma, J.; Sloane, J.; Gao, G.; Payne, K.J.; Szekely, L.; Song, C.; et al. Aberrant ARID5B expression and its association with Ikaros dysfunction in acute lymphoblastic leukemia. Oncogenesis 2018, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Leong, W.Z.; Tan, S.H.; Ngoc, P.C.T.; Amanda, S.; Yam, A.W.Y.; Liau, W.S.; Gong, Z.; Lawton, L.N.; Tenen, D.G.; Sanda, T. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017, 31, 2343–2360. [Google Scholar] [CrossRef]
- Csordas, K.; Lautner-Csorba, O.; Semsei, A.F.; Harnos, A.; Hegyi, M.; Erdelyi, D.J.; Eipel, O.T.; Szalai, C.; Kovacs, G.T. Associations of novel genetic variations in the folate-related and ARID5B genes with the pharmacokinetics and toxicity of high-dose methotrexate in paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2014, 166, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Porazzi, P.; De Dominici, M.; Salvino, J.; Calabretta, B. Targeting the CDK6 Dependence of Ph+ Acute Lymphoblastic Leukemia. Genes 2021, 12, 1355. [Google Scholar] [CrossRef]
- Martinez-Cibrian, N.; Zeiser, R.; Perez-Simon, J.A. Graft-versus-host disease prophylaxis: Pathophysiology-based review on current approaches and future directions. Blood Rev. 2021, 48, 100792. [Google Scholar] [CrossRef]
- Paczesny, S.; Choi, S.W.; Ferrara, J.L. Acute graft-versus-host disease: New treatment strategies. Curr. Opin. Hematol. 2009, 16, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Blatt, J.; Rotenstein, D.; Dienes, S. Cytotoxicity of tamoxifen for acute lymphoblastic leukaemia in vitro. Br. J. Cancer 1984, 50, 837–839. [Google Scholar] [CrossRef]
- Adachi, K.; Honma, Y.; Miyake, T.; Kawakami, K.; Takahashi, T.; Suzumiya, J. Tamoxifen enhances the differentiation-inducing and growth-inhibitory effects of all-trans retinoic acid in acute promyelocytic leukemia cells. Int. J. Oncol. 2016, 48, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivas-Aguirre, M.; Torres-Lopez, L.; Gomez-Sandoval, Z.; Villatoro-Gomez, K.; Pottosin, I.; Dobrovinskaya, O. Tamoxifen Sensitizes Acute Lymphoblastic Leukemia Cells to Cannabidiol by Targeting Cyclophilin-D and Altering Mitochondrial Ca(2+) Homeostasis. Int. J. Mol. Sci. 2021, 22, 8688. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Tan, S.F.; Feith, D.J.; Kester, M.; Claxton, D.F.; Loughran, T.P., Jr.; Barth, B.M.; Fox, T.E.; Cabot, M.C. Modification of sphingolipid metabolism by tamoxifen and N-desmethyltamoxifen in acute myelogenous leukemia--Impact on enzyme activity and response to cytotoxics. Biochim. Biophys. Acta 2015, 1851, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Lopez, L.; Maycotte, P.; Linan-Rico, A.; Linan-Rico, L.; Donis-Maturano, L.; Delgado-Enciso, I.; Meza-Robles, C.; Vasquez-Jimenez, C.; Hernandez-Cruz, A.; Dobrovinskaya, O. Tamoxifen induces toxicity, causes autophagy, and partially reverses dexamethasone resistance in Jurkat T cells. J. Leukoc. Biol. 2019, 105, 983–998. [Google Scholar] [CrossRef]
- Berman, E.; McBride, M.; Lin, S.; Menedez-Botet, C.; Tong, W. Phase I trial of high-dose tamoxifen as a modulator of drug resistance in combination with daunorubicin in patients with relapsed or refractory acute leukemia. Leukemia 1995, 9, 1631–1637. [Google Scholar]
- National Center for Biotechnology Information. PRKCI Protein Kinase C Iota [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/5584 (accessed on 30 August 2022).
- Du, Y.; Li, K.; Wang, X.; Kaushik, A.C.; Junaid, M.; Wei, D. Identification of chlorprothixene as a potential drug that induces apoptosis and autophagic cell death in acute myeloid leukemia cells. FEBS J. 2020, 287, 1645–1665. [Google Scholar] [CrossRef]
- Bai, L.; Li, X.; Ma, X.; Zhao, R.; Wu, D. In Vitro Effect and Mechanism of Action of Ergot Alkaloid Dihydroergocristine in Chemoresistant Prostate Cancer Cells. Anticancer Res. 2020, 40, 6051–6062. [Google Scholar] [CrossRef]
- Galan-Diez, M.; Borot, F.; Ali, A.M.; Zhao, J.; Gil-Iturbe, E.; Shan, X.; Luo, N.; Liu, Y.; Huang, X.P.; Bisikirska, B.; et al. Subversion of Serotonin Receptor Signaling in Osteoblasts by Kynurenine Drives Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 1106–1127. [Google Scholar] [CrossRef]
- Banus-Mulet, A.; Etxabe, A.; Cornet-Masana, J.M.; Torrente, M.A.; Lara-Castillo, M.C.; Palomo, L.; Nomdedeu, M.; Diaz-Beya, M.; Sole, F.; Nomdedeu, B.; et al. Serotonin receptor type 1B constitutes a therapeutic target for MDS and CMML. Sci. Rep. 2018, 8, 13883. [Google Scholar] [CrossRef] [Green Version]
- Parcha, P.K.; Sarvagalla, S.; Ashok, C.; Sudharshan, S.J.; Dyavaiah, M.; Coumar, M.S.; Rajasekaran, B. Repositioning antispasmodic drug Papaverine for the treatment of chronic myeloid leukemia. Pharmacol. Rep. 2021, 73, 615–628. [Google Scholar] [CrossRef]
- Nam, J.; Kim, D.U.; Kim, E.; Kwak, B.; Ko, M.J.; Oh, A.Y.; Park, B.J.; Kim, Y.W.; Kim, A.; Sun, H.; et al. Disruption of the Myc-PDE4B regulatory circuitry impairs B-cell lymphoma survival. Leukemia 2019, 33, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- Lim, N.; Pavlidis, P. Evaluation of connectivity map shows limited reproducibility in drug repositioning. Sci. Rep. 2021, 11, 17624. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irham, L.M.; Wong, H.S.; Chou, W.H.; Adikusuma, W.; Mugiyanto, E.; Huang, W.C.; Chang, W.C. Integration of genetic variants and gene network for drug repurposing in colorectal cancer. Pharmacol. Res. 2020, 161, 105203. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.D.; Kellis, M. HaploReg v4: Systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016, 44, D877–D881. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G.T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Mauri, M.; Elli, T.; Caviglia, G.; Uboldi, G.; Azzi, M. RAWGraphs: A Visualisation Platform to Create Open Outputs. In Proceedings of the 12th Biannual Conference on Italian SIGCHI Chapter, Cagliari, Italy, 18–20 September 2017; pp. 1–5. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | No. of SNPs | No. of Hits in GWAS Catalog |
|---|---|---|
| IKZF1 | 3 | 13 |
| ARID5B | 4 | 12 |
| GATA3 | 1 | 11 |
| SLC7A8, CEBPE | 2 | 7 |
| CDKN2A | 3 | 4 |
| LHPP | 3 | 4 |
| PIP4K2A | 3 | 4 |
| CCDC26 | 3 | 3 |
| ELK3 | 1 | 3 |
| GPATCH2L | 1 | 3 |
| OR5AL1, OR5AL2P | 1 | 3 |
| PDE4B | 3 | 3 |
| RNU6-366P, CPSF2 | 1 | 3 |
| TP63 | 1 | 3 |
| CSGALNACT1, INTS10 | 1 | 2 |
| DDC, FIGNL1 | 1 | 2 |
| ERG | 1 | 2 |
| AGBL1 | 1 | 2 |
| PTPRJ | 1 | 2 |
| RN7SL361P, BCL11A | 1 | 2 |
| RNU6-1091P, IKZF1 | 2 | 2 |
| RPL6P5 | 1 | 2 |
| Other genes with 1 hit | 33 | 33 |
| Not known genes | 2 | 3 |
| TOTAL | 74 | 128 |
| Drugs | Original Indications | Mode of Actions | Drug Target Genes | CMap (Score) |
|---|---|---|---|---|
| Chlorprothixene | Schizophrenia | inhibitor | HTR1B | 88.76 |
| Sirolimus | Immunosuppressant | inhibitor | FKBP1A | 87.80 |
| Dihydroergocristine | Cerebrovascular Diseases | antagonist | HTR1B | 84.11 |
| Papaverine | Vascular spasm | Inhibitor | PDE4B | 83.98 |
| Tamoxifen | Breast cancer | inhibitor | PRKC, PRKCI | 80.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zazuli, Z.; Irham, L.M.; Adikusuma, W.; Sari, N.M. Identification of Potential Treatments for Acute Lymphoblastic Leukemia through Integrated Genomic Network Analysis. Pharmaceuticals 2022, 15, 1562. https://doi.org/10.3390/ph15121562
Zazuli Z, Irham LM, Adikusuma W, Sari NM. Identification of Potential Treatments for Acute Lymphoblastic Leukemia through Integrated Genomic Network Analysis. Pharmaceuticals. 2022; 15(12):1562. https://doi.org/10.3390/ph15121562
Chicago/Turabian StyleZazuli, Zulfan, Lalu Muhammad Irham, Wirawan Adikusuma, and Nur Melani Sari. 2022. "Identification of Potential Treatments for Acute Lymphoblastic Leukemia through Integrated Genomic Network Analysis" Pharmaceuticals 15, no. 12: 1562. https://doi.org/10.3390/ph15121562