
Design, Synthesis and Biological Evaluation of Biscarbamates as Potential Selective Butyrylcholinesterase Inhibitors for the Treatment of Alzheimer’s Disease
 , , ,
, , ,  and
and
Abstract
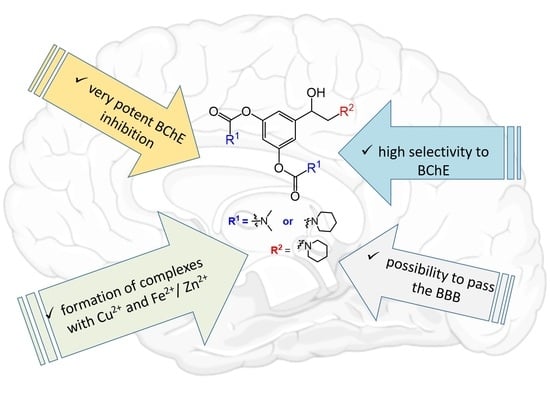
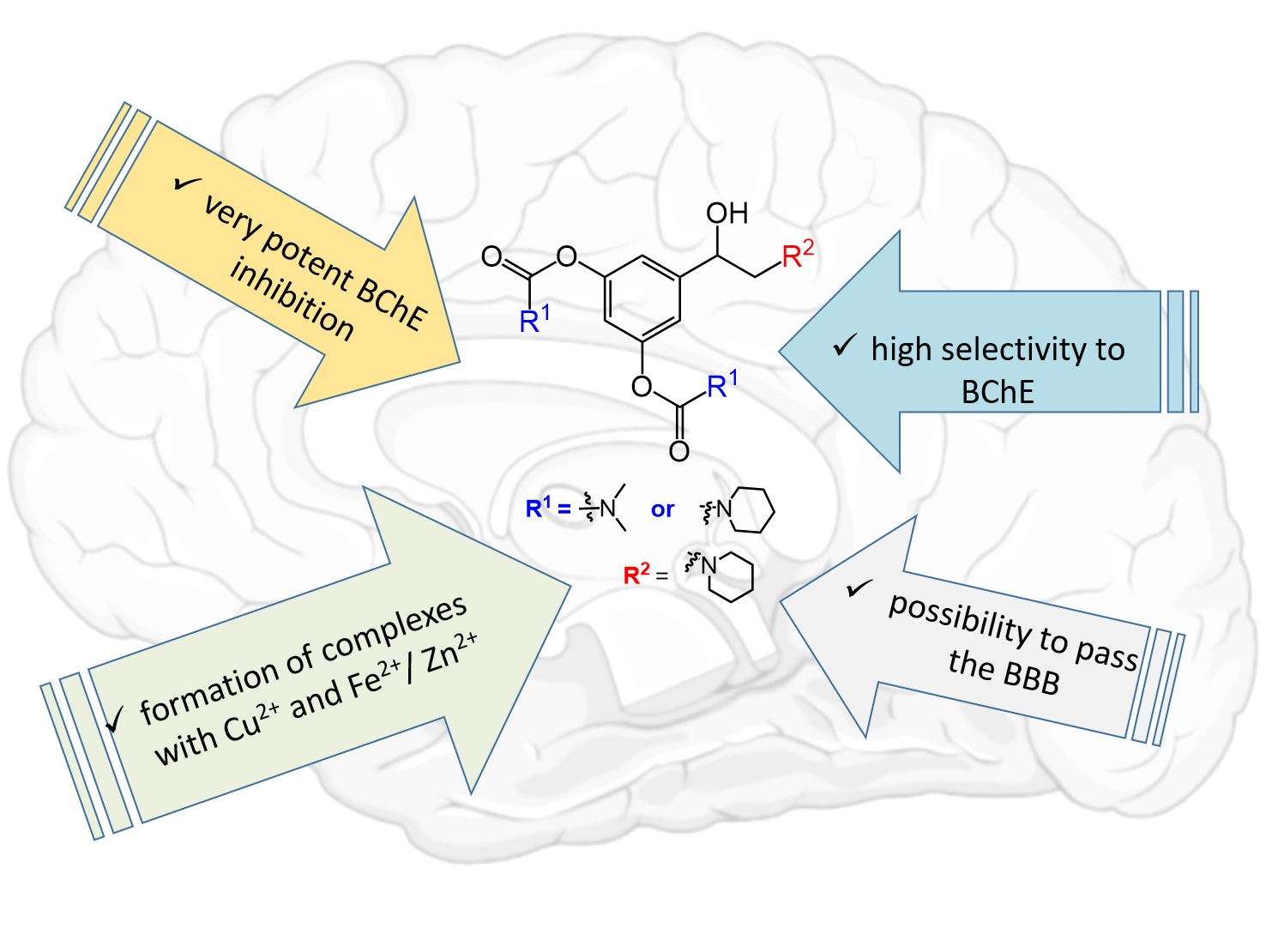
:
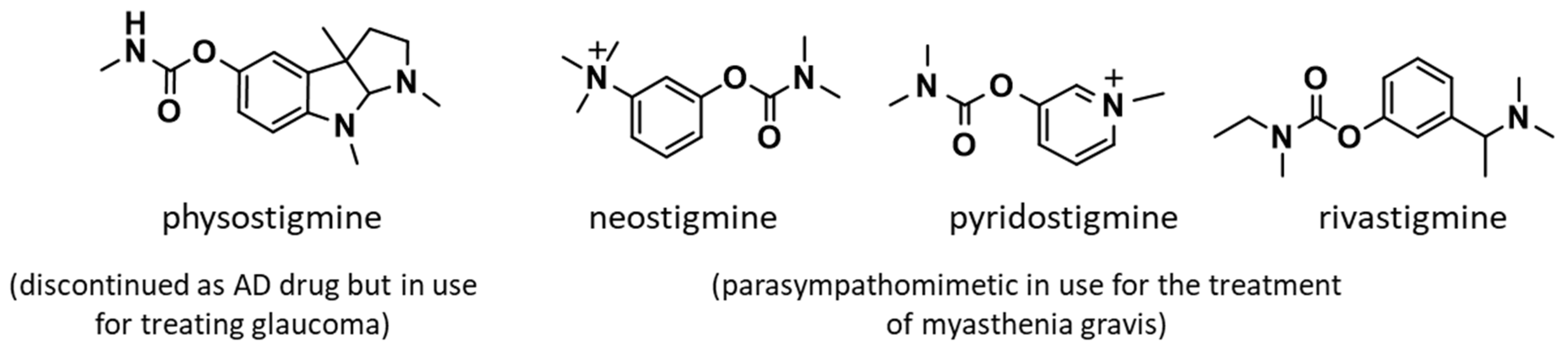
1. Introduction
2. Results and Discussion
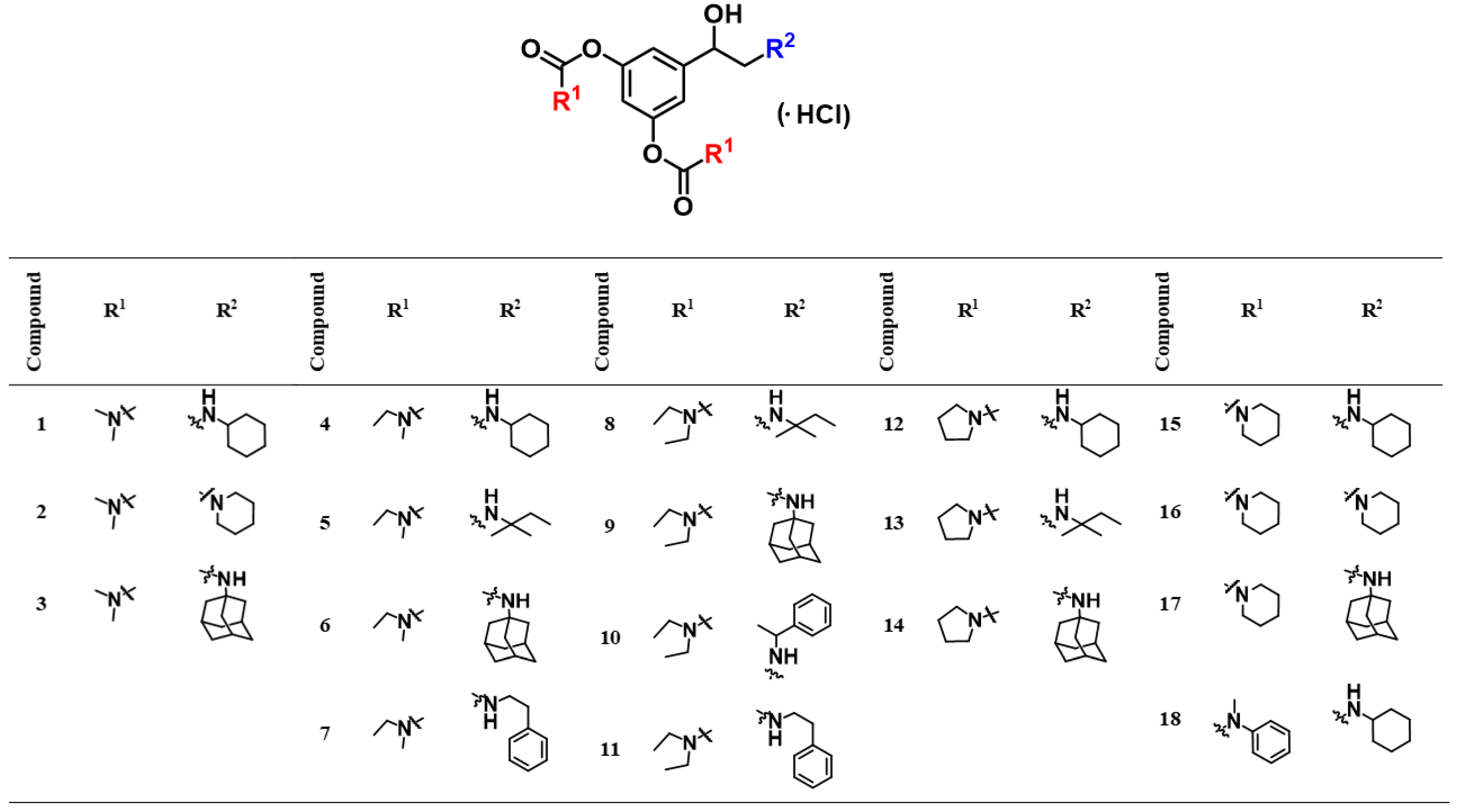
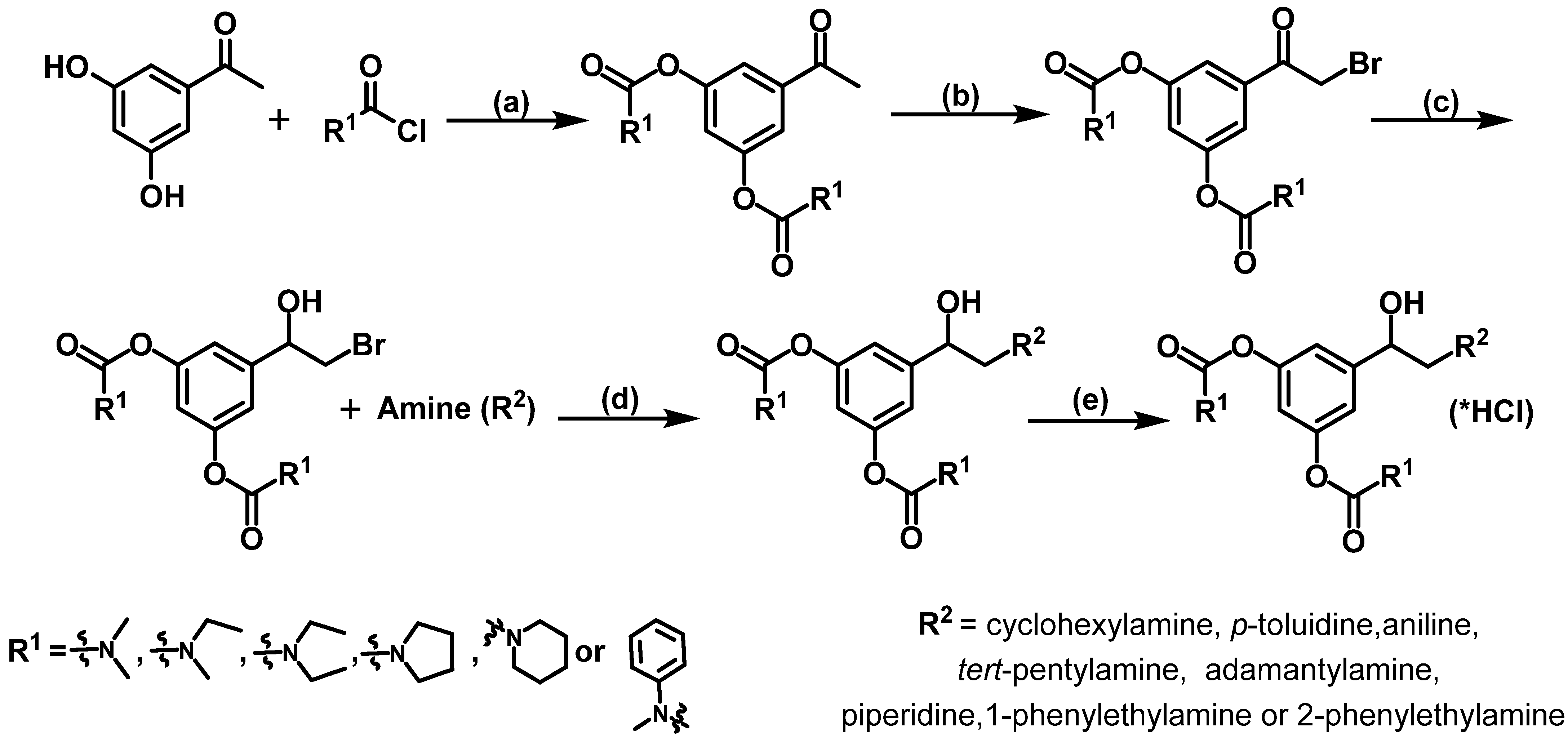
2.1. Design and Synthesis of Compounds
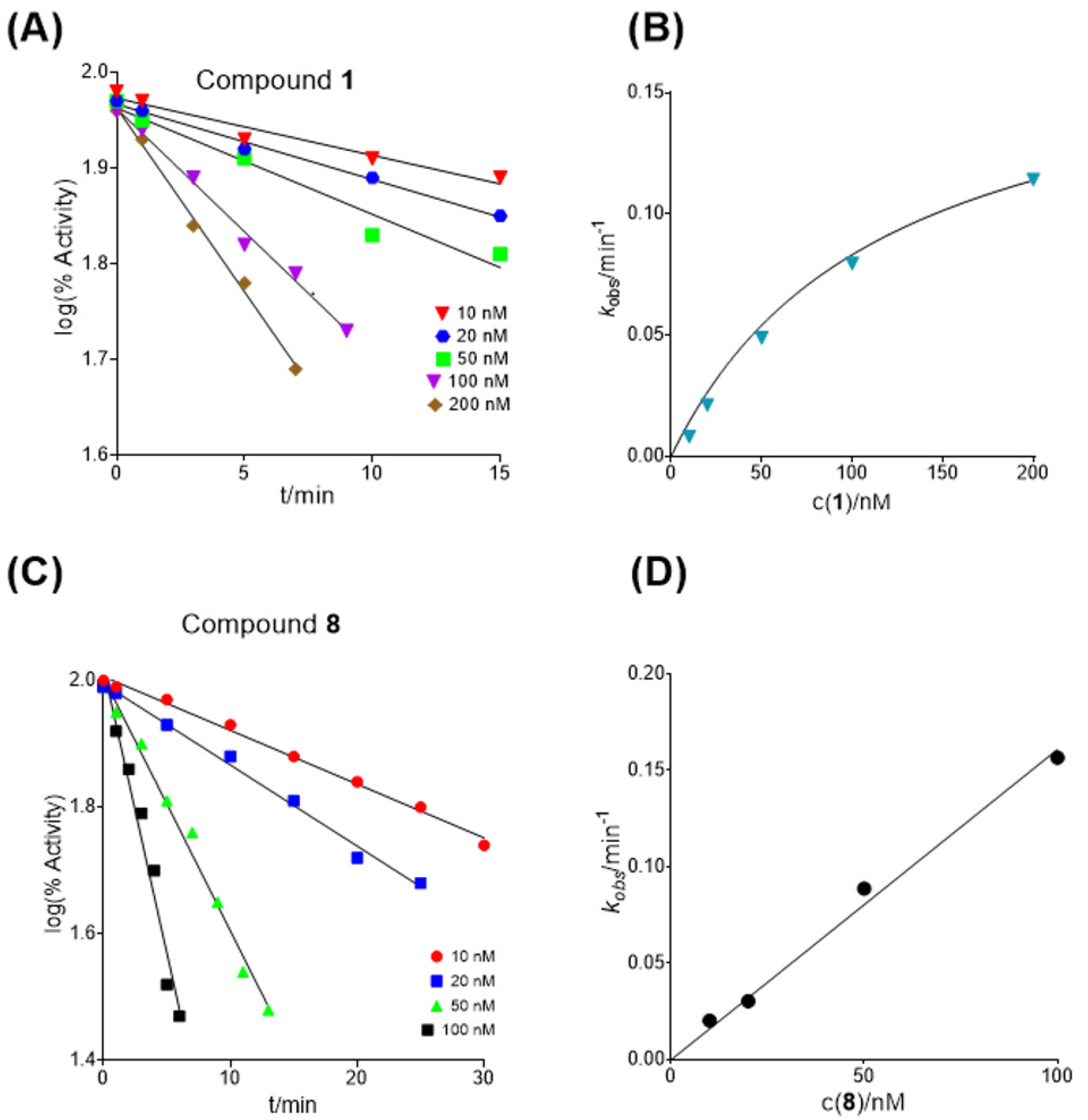
2.2. Kinetic Studies
2.2.1. Inhibition of Butyrylcholinesterase
2.2.2. Inhibition of Acetylcholinesterase
2.2.3. Selectivity of Inhibition
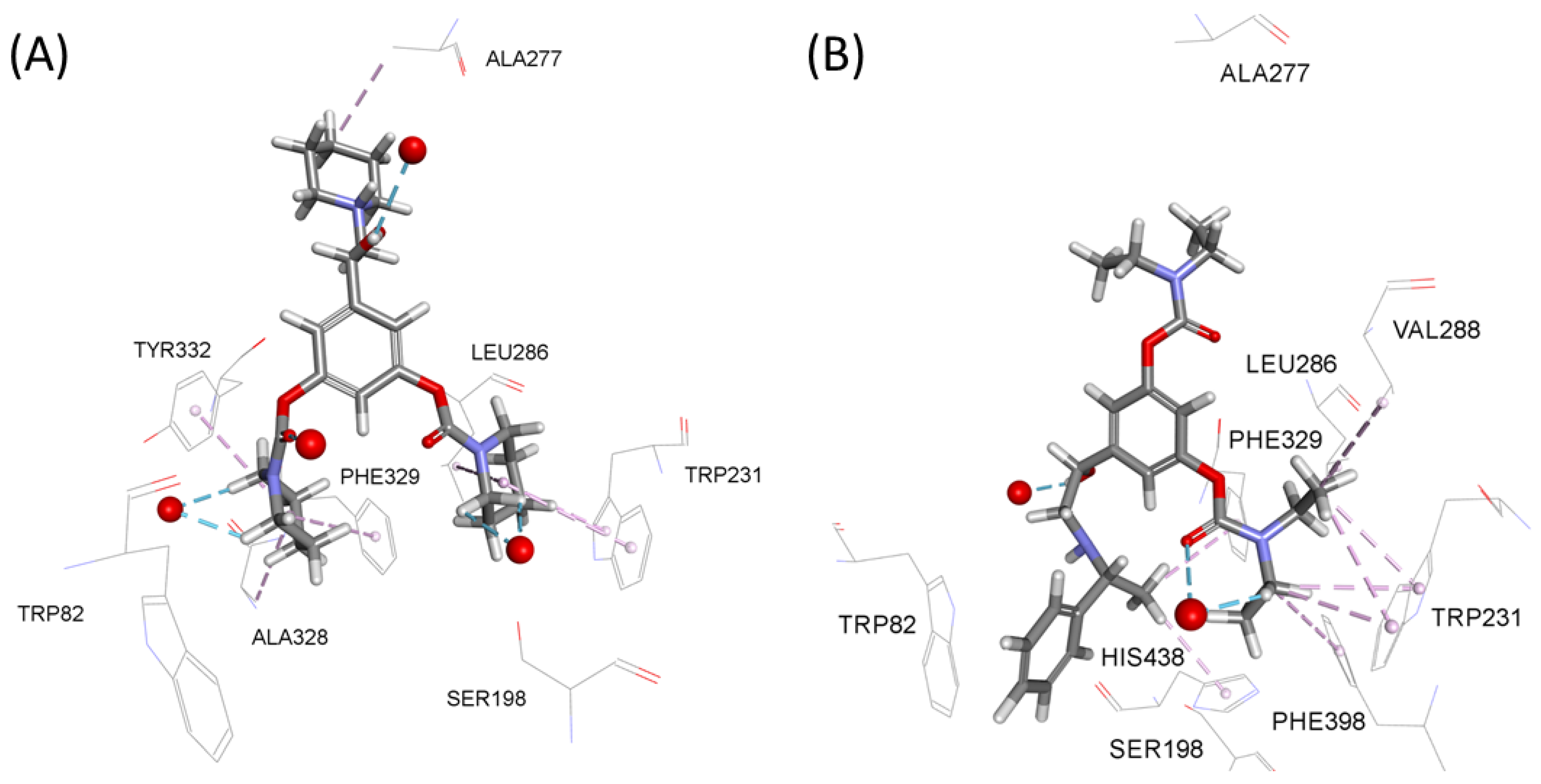
2.3. Docking Analysis
2.4. Decarbamylation Process
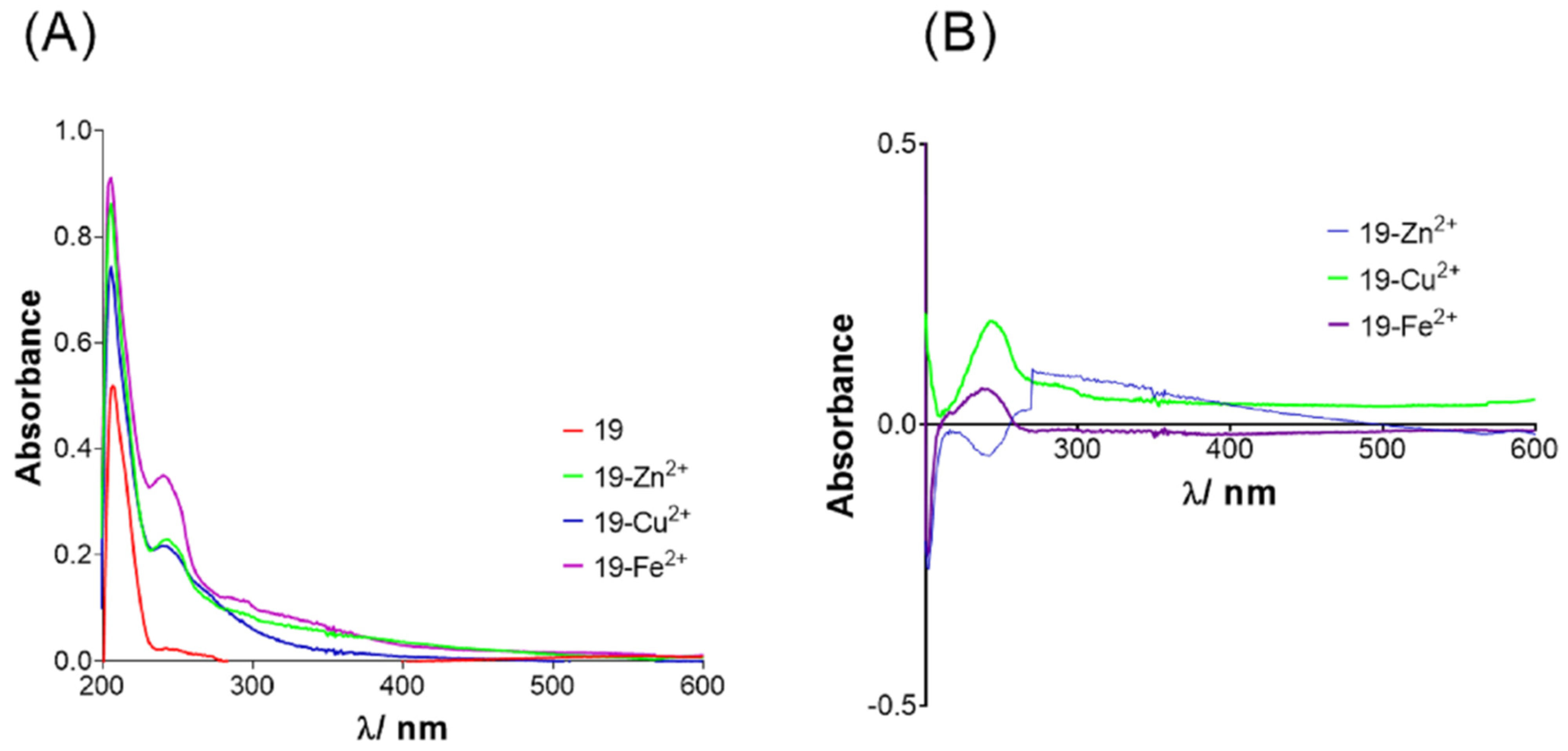
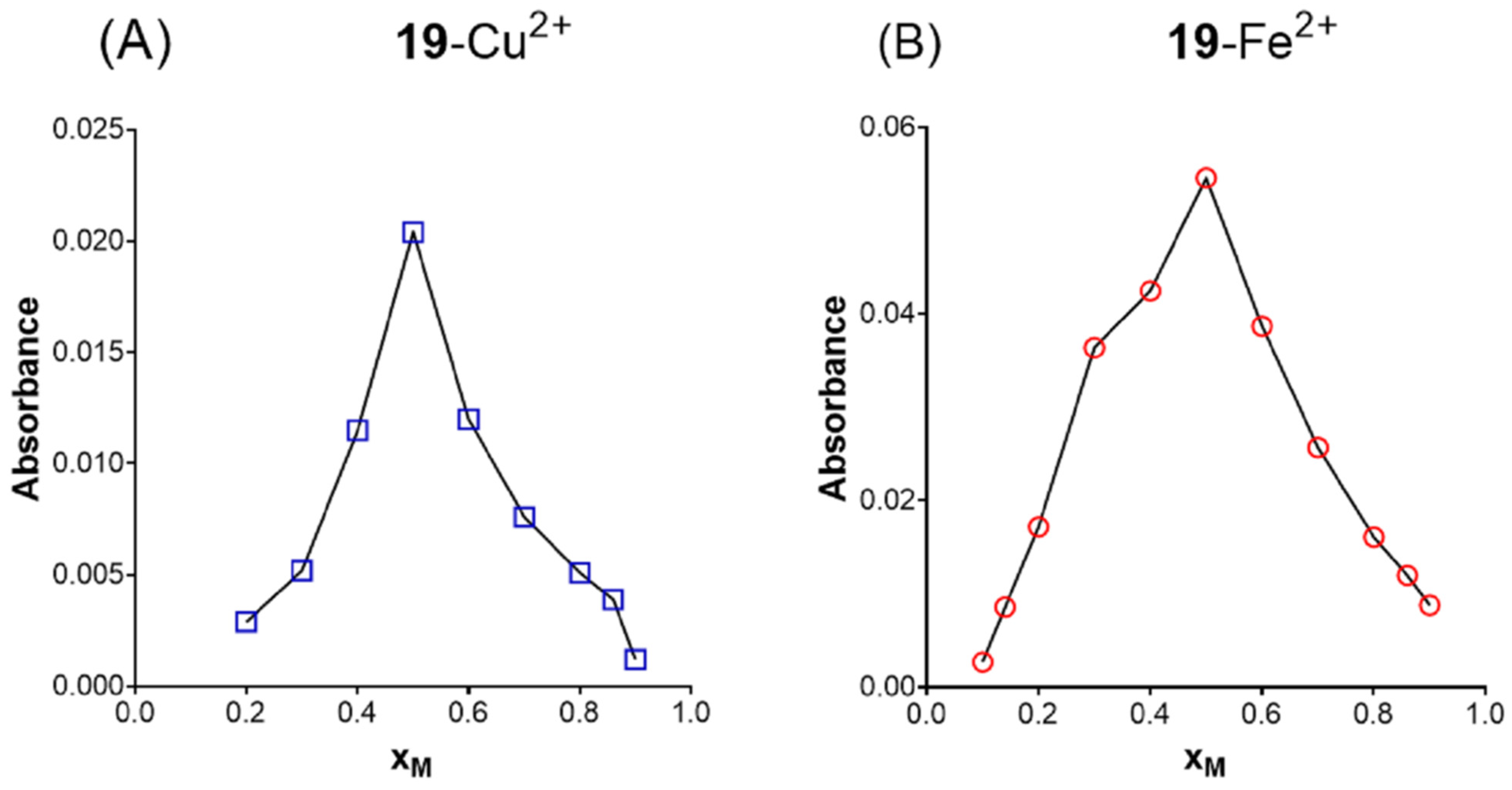
2.5. Metal Chelating Ability
2.6. The BBB Penetration Ability of Biscarbamates
2.7. Cytotoxicity
3. General Discussion
4. Materials and Methods
4.1. Synthesis of Compounds
4.2. Cholineseterase Inhibition
4.2.1. Enzyme Activity Measurements
4.2.2. Inhibition by Biscarbamates

4.3. Spontaneous Decarbamylation

4.4. Docking Studies
4.5. Metal Chelation Studies
4.6. In Silico Prediction of Blood–Brain Barrier (BBB) Penetration
4.7. Cytotoxicity of Biscarbamates
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 321–387. [Google Scholar]
- World Health Organization. Neurological Disorders: Public Health Challenges; WHO Library Cataloguing-in-Publication Data: Geneva, Switzerland, 2021. [Google Scholar]
- Catania, M.; Giaccone, G.; Salmona, M.; Tagaliavini, F.; Di Fede, G. Dreaming of a new world where Alzheimer‘s is a treatable disorder. Front. Aging Neurosci. 2019, 11, 317. [Google Scholar] [CrossRef]
- Gong, C.X.; Liu, F.; Iqbal, K. Multirfactorial hypothesis and multi-targets for Alzheimer‘s disease. J. Alzheimers Dis. 2018, 64, 107–117. [Google Scholar] [CrossRef]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. FDA-Approved Treatments for Alzheimer’s. Available online: https://www.alz.org/media/documents/fda-approved-treatments-alzheimers-ts.pdf (accessed on 15 July 2022).
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Alzheimer Disease and Associated Disorders. In Butyrylcholinesterase: Its Role in Brain Function, 1st ed.; Giacobini, E., Ed.; Informa Healthcare: London, UK, 2003. [Google Scholar]
- Sharma, P.; Srivastava, P.; Seth, A.; Nath Tripathi, P.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53–89. [Google Scholar] [CrossRef]
- dos Santos Picançoa, L.C.; Ozelaa, P.F.; de Brito Britoa, M.F.; Pinheiroa, A.A.; Padilhab, E.C.; Bragac, F.S.; Tomich de Paula da Silvad, C.F.; Rodrigues dos Santos, C.B.; Rosad, J.M.C.; da Silva Hage-Melim, L.I. Alzheimer’s disease: A review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr. Med. Chem. 2018, 25, 3141–3159. [Google Scholar] [CrossRef]
- Greig, N.H.; Lahiri, D.K.; Sambamurti, K. Butyrylcholinesterase: An important new target in Alzheimer‘s disease therapy. Int. Psychogeriatr. 2002, 14, 77–91. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Zhou, S.; Huang, G. The biological activities of butyrylcholinesterase inhibitors. Biomed. Pharmacother. 2022, 146, 112556. [Google Scholar] [CrossRef]
- Li, S.; Li, J.L.; Travers, J.; Xu, T.; Sakamuru, S.; Klumpp-Thomas, C.; Huang, R.; Xia, M. Identification of compounds for butyrylcholinesterase inhibition. SLAS Discov. 2021, 26, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, H.; Chen, Y.; Sun, H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Decker, M. Homobivalent quinazolinimines as novel nanomolar inhibitors of cholinesterase with dirigible selectivity toward butyrylcholinesterase. J. Med. Chem 2006, 49, 5411–5413. [Google Scholar] [CrossRef]
- Karlsson, D.; Fallarero, A.; Gerda, B.; Mayer, C.; Prakash, O.; Mohan, G.; Vuorela, P.; Erker, T. The exploration of thienothiazines as selective butyrylcholinesterase inhibitors. Eur. J. Pharm. Sci. 2012, 47, 190–205. [Google Scholar] [CrossRef]
- Bosak, A.; Ramić, A.; Šmidlehner, T.; Hrenar, T.; Primožič, I.; Koavrik, Z. Design and evaluation of selective butyrylcholinesterase inhibitors based on Cinchona alkaloid scaffold. PLoS ONE 2018, 13, 0205193. [Google Scholar] [CrossRef]
- Rani, A.; Singh, A.; Kaur, J.; Singh, G.; Bhatti, R.; Gumede, N.j.; Kisten, P.; Singh, P.; Sumanjit; Kumar, V. 1H-1,2,3-triazole grafted tacrine-chalcone conjugates as potential cholinesterase inhibitors with the evaluation of their behavioral tests and oxidative stress in mice brain cells. Bioorg. Chem. 2021, 114, 105053. [Google Scholar] [CrossRef]
- Singha, A.; Sahil Sharmaa, S.A.; Attrib, S.; Harmandeep Kaur Gulatia, P.K.; Bhagata, K.; Kumar, N.; Singha, H.; Singha, J.V.; Singh Bedia, P.M. New coumarin-benzotriazole based hybrid molecules as inhibitors of acetylcholinesterase and amyloid aggregation. Bioorg. Med. Chem. Lett 2020, 30, 127477. [Google Scholar] [CrossRef] [PubMed]
- Reiner, E.; Radić, Z. Mechanism of action of cholinesterase inhibitors. In Cholinesterase’s and Cholinesterase Inhibitors, 3rd ed.; Giaccobini, E., Dunitz, M., Eds.; Informa Healthcare: London, UK, 2000; pp. 103–144. [Google Scholar]
- Tunek, A.; Svensson, L.A. Bambuterol, a carbamate ester prodrug of terbutaline, as inhibitor of cholinesterases in human blood. Drug Metab. Dispos. 1988, 16, 759–764. [Google Scholar]
- Giacobini, E.; Pepeu, G. The Brain Cholinergic System in Health and Disease, 1st ed.; Giacobini, E., Ed.; Informa Healthcare: London, UK, 2006. [Google Scholar]
- Darvesh, S.; Walsh, R.; Kumar, R.; Caines, A.; Roberts, S.; Magee, D.; Rockwood, M.; Martin, E. Inhibition of human cholinesterases by drugs used to treat Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2003, 17, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Hoffman, H.; Chakkamparambil, B.; Grossberg, G.T. Evaluation of rivastigmine in Alzheimer’s disease. Neurodegener. Dis. Manag. 2020, 11, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.K.; Dandapat, J.; Dash, U.C.; Kanhar, S. Features and outcomes of drugs for combination therapy as multi-targets strategy to combat Alzheimer’s disease. J. Ethnopharmacol. 2018, 215, 42–73. [Google Scholar] [CrossRef]
- Klein, J. Phenserine. Expert. Opin. Investig. Drugs 2007, 16, 1087–1097. [Google Scholar] [CrossRef]
- Becker, R.E.; Greig, N.H. Was Phenserine a failure or were investigators mislead by methods? Curr. Alzheimer Res. 2012, 9, 1174–1181. [Google Scholar] [CrossRef]
- Greig, N.H.; Sambamurti, K.; Yu, Q.; Brossi, A.; Brunisma, G.B.; Lahiri, D.K. An overview of Phenserine tartrate, a novel acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 281–290. [Google Scholar] [CrossRef]
- Kamal, M.A.; Al-Jafari, A.A.; Yu, Q.S.; Greig, N.H. Kinetic analysis of the inhibition of human butyrylcholinesterase with cymserine. Biochim. Biophys. Acta -Gen. Subj. 2006, 1760, 200–206. [Google Scholar] [CrossRef]
- Kamal, M.A.; Qu, X.; Yu, Q.; Tweedie, D.; Holloway, H.W.; Yazhou, L.; Tan, Y.; Greig, N.H. Tetrahydrofurobenzofuran cymserine, a potent butyrylcholinesterase inhibitor and experimental Alzheimer drug candidate, enzyme kinetic analysis. J. Neural. Transm. 2008, 115, 889–898. [Google Scholar] [CrossRef]
- Somani, S.M.; Kutty, R.K.; Krishna, G. Eseroline, a metabolite of physostigmine, induces neuronal cell death. Toxicol. Appl. Pharmacol. 1990, 106, 28–37. [Google Scholar] [CrossRef]
- Alzheimer’s Drug Discovery Fondation. 2018 Alzheimer’s Clinical Trials Report. Available online: https://www.alzdiscovery.org/research-and-grants/clinical-trials-report/2018-report (accessed on 20 June 2022).
- Gazić, I.; Bosak, A.; Šinko, G.; Vinković, V.; Kovarik, Z. Preparative HPLC separation of bambuterol enantiomers and stereoselective inhibition of human cholinesterases. Anal. Bioanal. Chem. 2006, 385, 1513–1519. [Google Scholar] [CrossRef]
- Rosberg, B.; Schroder, C.; Nyberg, L.; Rosenborg, J.; Wiren, J.E. Bambuterol and terbutaline in human cerebrospinal fluid and plasma. Eur. J. Clin. Pharmacol. 1993, 45, 147–150. [Google Scholar] [CrossRef]
- Wu, J.; Tian, Y.; Wang, S.; Pistolozzi, M.; Jin, Y.; Zhou, T.; Roy, G.; Xu, L.; Tan, W. Design, synthesis and biological evaluation of bambuterol analogues as novel inhibitors of butyrylcholinesterase. Eur. J. Med. Chem. 2016, 126, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Pistolozzi, M.; Liu, S.; Tan, W. Design, synthesis and biological evaluation of novel carbamates as potential inhibitors of acetylcholinesterase and butyrylcholinesterase. Biorg. Med. Chem. 2020, 28, 115324. [Google Scholar] [CrossRef]
- Bosak, A.; Smilovic, I.G.; Šinko, G.; Vinkovi’c, V.; Kovarik, Z. Metaproterenol, isoproterenol and their bisdimethyl-carbamate derivates as human cholinesterase inhibitors. J. Med. Chem. 2012, 55, 6716–6723. [Google Scholar] [CrossRef]
- Prester, L.; Simeon, V. Kinetics of the inhibition of human serum cholinesterase phenotypes with the dimethylcarbamate of (2-hydroxy-5-phenylbenzyl)-trimethylammonium bromide (Ro 02-0683). Biochemical. Pharmacol. 1991, 42, 2313–2316. [Google Scholar] [CrossRef]
- Simeon, V.; Reiner, E. Comparison between inhibition of acetylcolinesterase an cholinesterase by some N-methyl- and N, N-dimethyl carbamates. Arh. Hig. Rada Toksikol. 1973, 24, 199–206. [Google Scholar]
- Bartels, C.F.; Jensen, F.S.; Lockridge, O.; Van der Spek, A.F.L.; Rubinstein, H.M.; Lubrano, T.; La Du, B.N. DNA mutation associated with the human butyrylcholinesterase K-variant and its linkage to the atypical variant mutation and other polymorphic sites. Am. J. Hum. Genet. 1992, 50, 1086–1103. [Google Scholar]
- Kovarik, Z.; Simeon-Rudolf, V. Interaction of human butyrylcholinesterase variants with bambuterol and terbutaline. J. Enzym. Inhib. Med. Chem. 2004, 19, 113–117. [Google Scholar] [CrossRef] [PubMed]
- al-Jafari, A.A.; Kamal, M.A.; Greig, N.H.; Alhomida, A.S.; Perry, E.R. Kinetics of human erythrocyte acetylcholinesterase inhibition by a novel derivative of physostigmine: Phenserine. Biochem. Biophys. Res. Commun. 1998, 248, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sawada, Y.; Iga, T. Pharmacodynamic analysis of contractile potentiation by cholinesterase inhibitors in rats. J. Pharmacokinet. Biopharm. 1996, 24, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Komatović, K.; Matošević, A.; Terzić-Jovanović, N.; Žunec, S.; Šegan, S.; Zlatović, M.; Maraković, N.; Bosak, A.; Opsenica, D.M. 4-Aminoquinoline-Based Adamantanes as Potential Anticholinesterase Agents in Symptomatic Treatment of Alzheimer’s Disease. Pharmaceutics 2022, 14, 1305. [Google Scholar] [CrossRef]
- Aldrige, W.N.; Reiner, E. Enzyme Inhibitors as Substrates, 1st ed.; Northoland Publishing Company: Amsterdam, The Netherlands, 1972. [Google Scholar]
- Groner, E.; Ashani, Y.; Schorer-Apelbaum, D.; Sterling, J.; Herzig, Y.; Weinstock, M. The kinetics of inhibition of human acetylcholinesterase and butyrylcholinesterase by two series of novel carbamates. Mol. Pharmacol. 2007, 71, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Rotenberg, M.; Almong, S. Evaluation of the decarbamylation process of cholinesterases during assay of enzyme activity. Clin. Chim. Acta 1995, 240, 107–116. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhou, H.; Wei, H.; Du, H.; Tan, W.; Zhan, Y.; Pistolozzi, M. A new method to characterize the kinetics of cholinesterases inhibited by carbamates. J. Pharm. Biomed. 2017, 144, 175–182. [Google Scholar] [CrossRef]
- Pal, T.; Patil, P.; Shrama, A. Synthesis, molecular docking and spectroscopic studies of pyridoxine carbamates as metal chelator. J. Mol. Struct. 2021, 1223, 128837. [Google Scholar] [CrossRef]
- Baum, L.; Ng, A. Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J. Alzheimer’s Dis. 2004, 6, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central nervous system multiparameter optimization desirability. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Hendey, B.; Monahan, A.J. The blood-brain barrier in neurodegenerative disease: A rhetorical perspective. J. Neurochem. 2009, 111, 291–314. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; de Lima Barros, P.; Levy, D.; Bydlowski, S.P. Ferroptosis mechanisms involved in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. New and rapid colorimetric determination of acetylcho-linesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Weinstock, M.; Groner, E. Rational design of a drug for Alzheimer’s disease with cholinesterase inhibitory and neuroprotective activity. Chem. Biol. Interact. 2008, 175, 216–221. [Google Scholar] [CrossRef]
- Pistolozzi, M.; Du, H.; Wei, H.; Tan, W. Stereoselective inhibition of human butyrylcholinesterase by the enantiomers of bambuterol and their intermediates. Drug Metab. Dispos. 2015, 43, 344–352. [Google Scholar] [CrossRef]
- Perola, E.; Cellai, L.; Lamba, D.; Filocamo, L.; Brufani, M. Long chain analogs of physostigmine as potential drugs for Alzheimer’s disease: New insights into the mechanism of action in the inhibition of acetylcholinesterase. Biochim. Biophys. Acta Bioenerg. 1997, 1343, 41–50. [Google Scholar] [CrossRef]
- Kovarik, Z.; Čalić, M.; Šinko, G.; Bosak, A. Structure-activity approach in the reactivation of tabun-phosphorylatedhuman acetylcholinesterase with bispyridinium para-aldoximes. Arh. Hig. Rada. Toksikol. 2007, 58, 201–209. [Google Scholar] [CrossRef]
- Koska, J.; Spassov, V.Z.; Maynard, A.J.; Yan, L.; Austin, N.; Flook, P.K.; Venkatachalam, C.M. Fully automated molecular mechanics based induced fit protein-ligand docking method. J. Chem. Inf. Model. 2008, 48, 1965–1973. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Wang, X.B.; Xie, S.S.; Jiang, N.; Wang, K.D.G.; Yao, H.Q.; Sun, H.B.; Kong, L.Y. Multifunctional tacrine flavonoid hybrids with cholinergic, β-amyloid-reducing, and metal chelating properties for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2013, 69, 632–646. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Bergamini, C.; Farto, R.; Soukup, A.; Korabency, J.; Andrisano, V.; Bartolini, M.; Bolognesi, M.L. Novel 8-hydroxyquinoline derivatives as multitarget compounds for the treatment of Alzheimer’s disease. ChemMedChem 2016, 11, 1284–1295. [Google Scholar] [CrossRef]
- Chemicalize. Calculation Module. 2018. Available online: https://chemicalize.com/ (accessed on 22 May 2022).
- Zandona, A.; Maraković, N.; Mišetić, P.; Madunić, J.; Miš, K.; Padovan, J.; Pirkemajer, S.; Katalinić, M. Activation of (un)regulated cell death as a new perspective for bispyridinium and imidazolium oximes. Arch. Toxicol. 2021, 95, 2737–2754. [Google Scholar] [CrossRef]
- ECACC. Fundamental Techniques in Cell Culture Laboratory Handbook, 4th ed.; Merck KGaA: Darmstadt, Germany, 2018. [Google Scholar]
- Zandona, A.; Katalinić, M.; Šinko, G.; Kastelic, A.R.; Primožiĉ, I.; Kovarik, Z. Targeting organophosphorus com-pounds poisoning by novel quinuclidine-3 oximes: Development of butyrylcholinesterase-based bioscavengers. Arch. Toxicol. 2020, 94, 3157–3171. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BChE | AChE | ki(BChE)/ki(AChE) | ||||
|---|---|---|---|---|---|---|---|
| ki∙106 (M−1 min−1) | Ki (µM) | kmax (min−1) | ki∙106 (M−1 min−1) | Ki (µM) | kmax (min−1) | ||
| 1 | 1.54 ± 0.40 | 0.116 ± 0.027 | 0.180 ± 0.021 | 0.0261 ± 0.0102 | 14.9 ± 4.8 | 0.389 ± 0.086 | 59 |
| 2 | 1.74 ± 0.48 | 0.0754 ± 0.0188 | 0.131 ± 0.015 | 0.0312 ± 0.0032 | 5.16 ± 1.11 | 0.161 ± 0.014 | 56 |
| 3 | 0.0192 ± 0.0021 | 27.0 ± 6.5 | 0.582 ± 0.091 | 0.00330 ± 0.0013 | 80.5 ± 22.7 | 0.269 ± 0.044 | 6 |
| 4 | 1.47 ± 0.193 | 0.485 ± 0.049 | 0.739 ± 0.057 | 1.73 ± 0.44 | 0.383 ± 0.081 | 0.661 ± 0.090 | 0.88 |
| 5 | 2.28 ± 0.42 | 0.0485 ± 0.0083 | 0.110 ± 0.007 | 0.0402 ± 0.0063 | 4.57 ± 0.66 | 0.184 ± 0.013 | 56 |
| 6 | 3.68 ± 1.23 | 0.115 ± 0.038 | 0.423 ± 0.293 | 2.00 ± 0.70 | - | - | 1.8 |
| 7 | 0.205 ± 0.052 | 1.42 ± 0.30 | 0.292 ± 0.034 | 0.0310 ± 0.0132 | 3.29 ± 0.52 | 0.102 ± 0.007 | 6.6 |
| 8 | 1.64 ± 0.37 | - | - | 4.56 ± 1.18 | 0.0439 ± 0.0191 | 0.200 ± 0.141 | 0.36 |
| 9 | 2.18 ± 0.52 | 0.236 ± 0.045 | 0.514 ± 0.061 | 2.02 ± 0.65 | 0.181 ± 0.053 | 0.364 ± 0.161 | 1 |
| 10 | 0.0145 ± 0.0049 | - | - | 0.00693 ± 0.00210 | - | - | 2 |
| 11 | 0.0144 ± 0.0053 | - | - | 0.00679 ± 0.000217 | - | - | 2 |
| 12 | 1.05 ± 0.45 | 0.212 ± 0.078 | 0.224 ± 0.050 | 0.0193 ± 0.0096 | 41.4 ± 35.6 | 0.801 ± 0.576 | 55 |
| 13 | 4.51 ± 152 | 0.127 ± 0.025 | 0.571 ± 0.057 | 0.00360 ± 0.000303 | - | - | 1288 |
| 14 | 1.80 ± 0.37 | 0.378 ± 0.049 | 0.679 ± 0.062 | 0.00841 ± 0.00053 | - | - | 209 |
| 15 | 2.62 ± 0.97 | 0.155 ± 0.052 | 0.399 ± 0.073 | 0.0136 ± 0.0050 | - | - | 184 |
| 16 | 38.0 ± 6.7 | 0.0119 ± 0.0030 | 0.453 ± 0.065 | 0.0382 ± 0.0131 | 6.93 ± 2.04 | 0.264 ± 0.040 | 1087 |
| 17 | 1.89 ± 0.59 | 0.0910 ± 0.0229 | 0.171 ± 0.019 | 0.165 ± 0.074 | 2.23 ± 0.95 | 0.369 ± 0.116 | 12 |
| 18 | 1.63 ± 0.57 | 0.0622 ± 0.0129 | 0.101 ± 0.008 | 0.0182 ± 0.0063 | 15.8 ± 3.6 | 0.288 ± 0.044 | 91 |
| Bambuterol | 4.4 ± 0.2 * | - | - | 0.000220 ± 0.000070 ** | 1900 ± 590 | 0.42 ± 0.007 | 19,600 |
| Rivastigmine | 0.0551 ± 0.0022 | 0.00222 ± 0.00046 | 25 | ||||
| Compound | HEK293 | HepG2 | SH-SY5Y |
|---|---|---|---|
| 1 | ≥800 | ≥800 | ≥800 |
| 2 | ≥800 | ≥800 | ≥800 |
| 3 | 240 ± 26 | 324 ± 30 | 197 ± 38 |
| 4 | 347 ± 8 | 398 ± 22 | 525 ± 32 |
| 5 | ≥800 | ≥800 | ≥800 |
| 6 | 112 ± 6 | 47.9 ± 1.3 | 89.1 ± 3.0 |
| 7 | 42.7 ± 9.2 | 83.2 ± 4.8 | 214 ± 15 |
| 8 | 234 ± 17 | 252 ± 10 | 240 ± 29 |
| 9 | 20.9 ± 2.6 | 14.8 ± 2.1 | 19.1 ± 1.1 |
| 10 | 11.5 ± 2.9 | 76.7 ± 13.0 | 17.0 ± 4.2 |
| 11 | 36.3 ± 1.2 | 33.7 ± 4.3 | 38.9 ± 3.6 |
| 12 | 251 ± 25 | 248 ± 5.7 | 178 ± 26 |
| 13 | 281 ± 9 | 457 ± 53 | ≥800 |
| 14 | 32.4 ± 6.5 | 41.7 ± 5.4 | 46.8 ± 4.3 |
| 15 | 38.0 ± 2.1 | 27.5 ± 1.7 | 30.2 ± 3.2 |
| 16 | 288 ± 32 | 295 ± 47 | 126 ± 25 |
| 17 | 6.76 ± 0.31 | 7.59 ± 0.26 | 8.19 ± 0.72 |
| 18 | 8.32 ± 0.95 | 15.3 ± 0.3 | 7.41 ± 0.09 |
| Bambuterol | ≥800 | ≥800 | ≥800 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matošević, A.; Knežević, A.; Zandona, A.; Maraković, N.; Kovarik, Z.; Bosak, A. Design, Synthesis and Biological Evaluation of Biscarbamates as Potential Selective Butyrylcholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. Pharmaceuticals 2022, 15, 1220. https://doi.org/10.3390/ph15101220
Matošević A, Knežević A, Zandona A, Maraković N, Kovarik Z, Bosak A. Design, Synthesis and Biological Evaluation of Biscarbamates as Potential Selective Butyrylcholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. Pharmaceuticals. 2022; 15(10):1220. https://doi.org/10.3390/ph15101220
Chicago/Turabian StyleMatošević, Ana, Anamarija Knežević, Antonio Zandona, Nikola Maraković, Zrinka Kovarik, and Anita Bosak. 2022. "Design, Synthesis and Biological Evaluation of Biscarbamates as Potential Selective Butyrylcholinesterase Inhibitors for the Treatment of Alzheimer’s Disease" Pharmaceuticals 15, no. 10: 1220. https://doi.org/10.3390/ph15101220