
Determination of Bupropion and Its Impurities via a Chaotropic Chromatography Method Following Analytical Quality-by-Design Principles for Method Development


Abstract
:1. Introduction
2. Results and Discussion
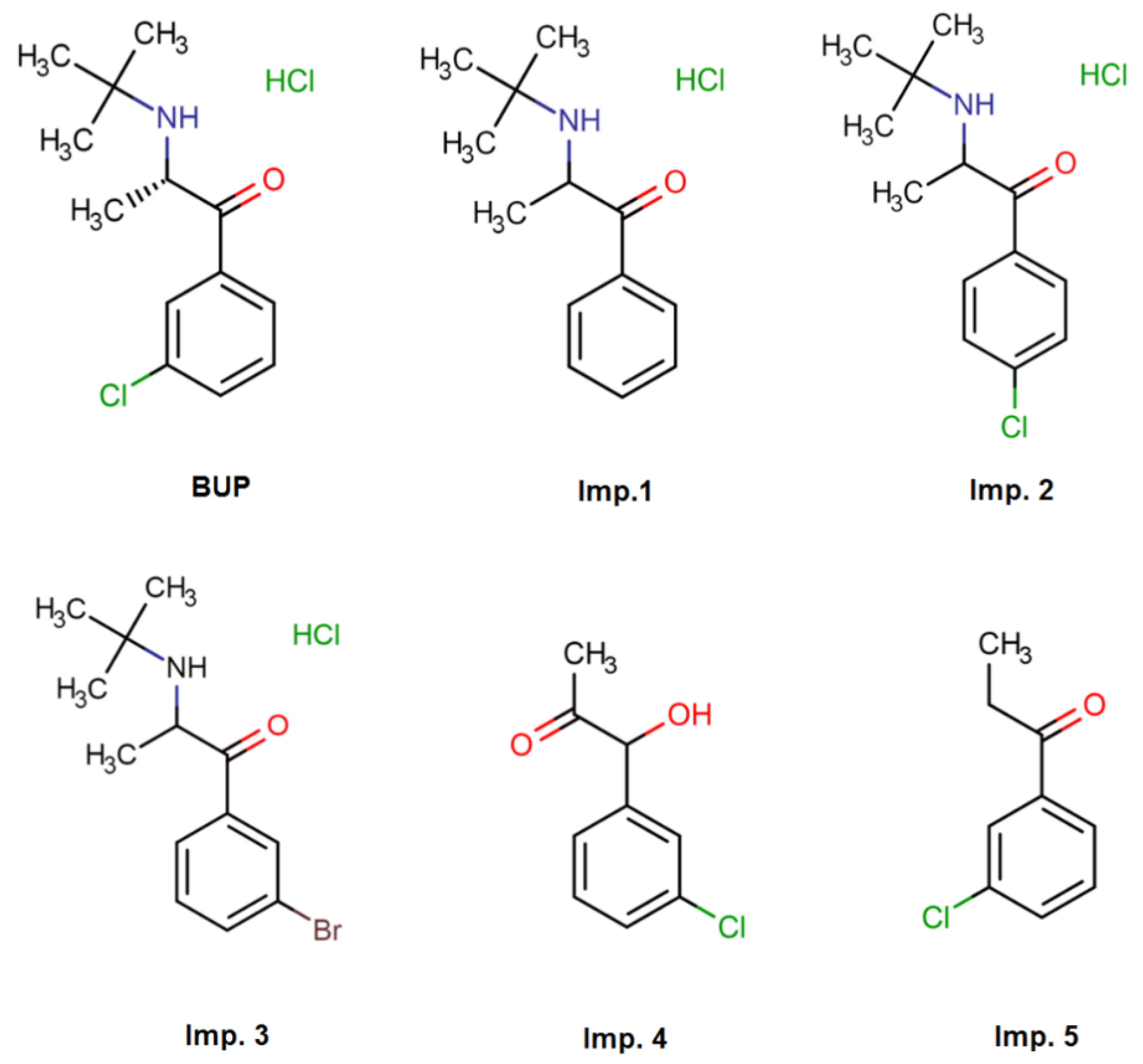
2.1. Preliminary Experiments and Screening Design
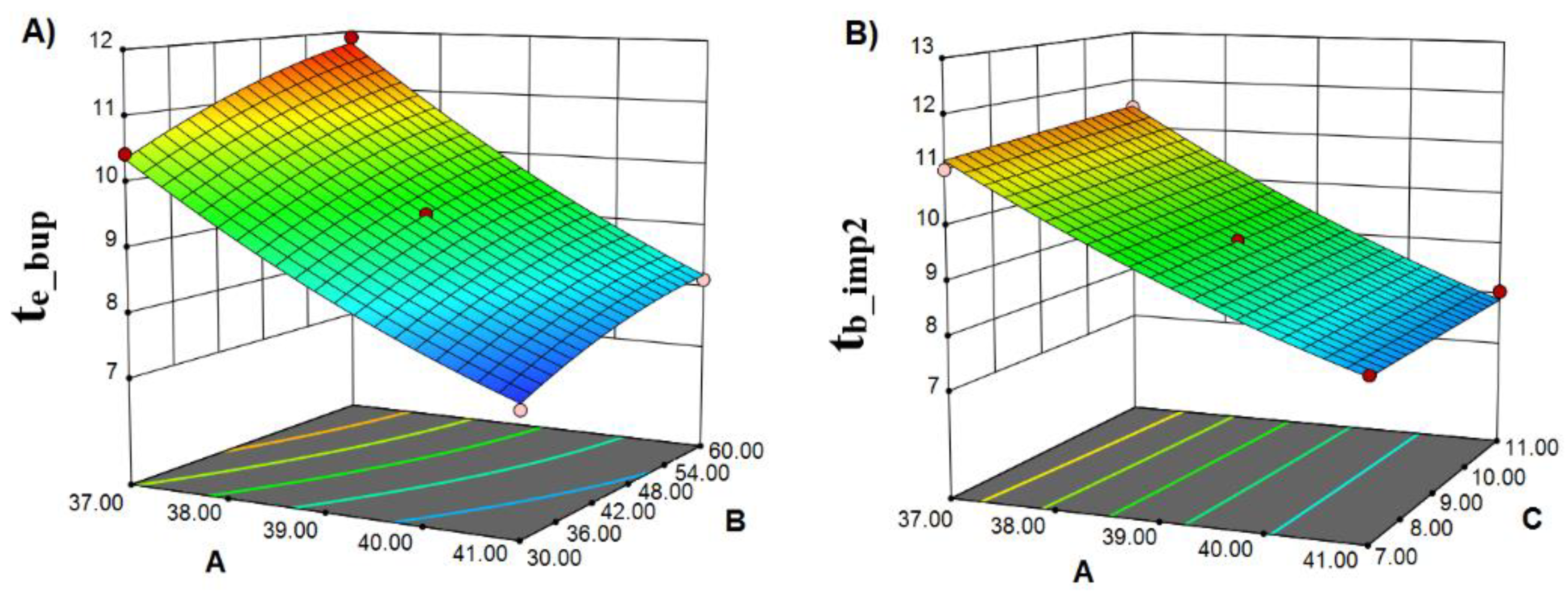
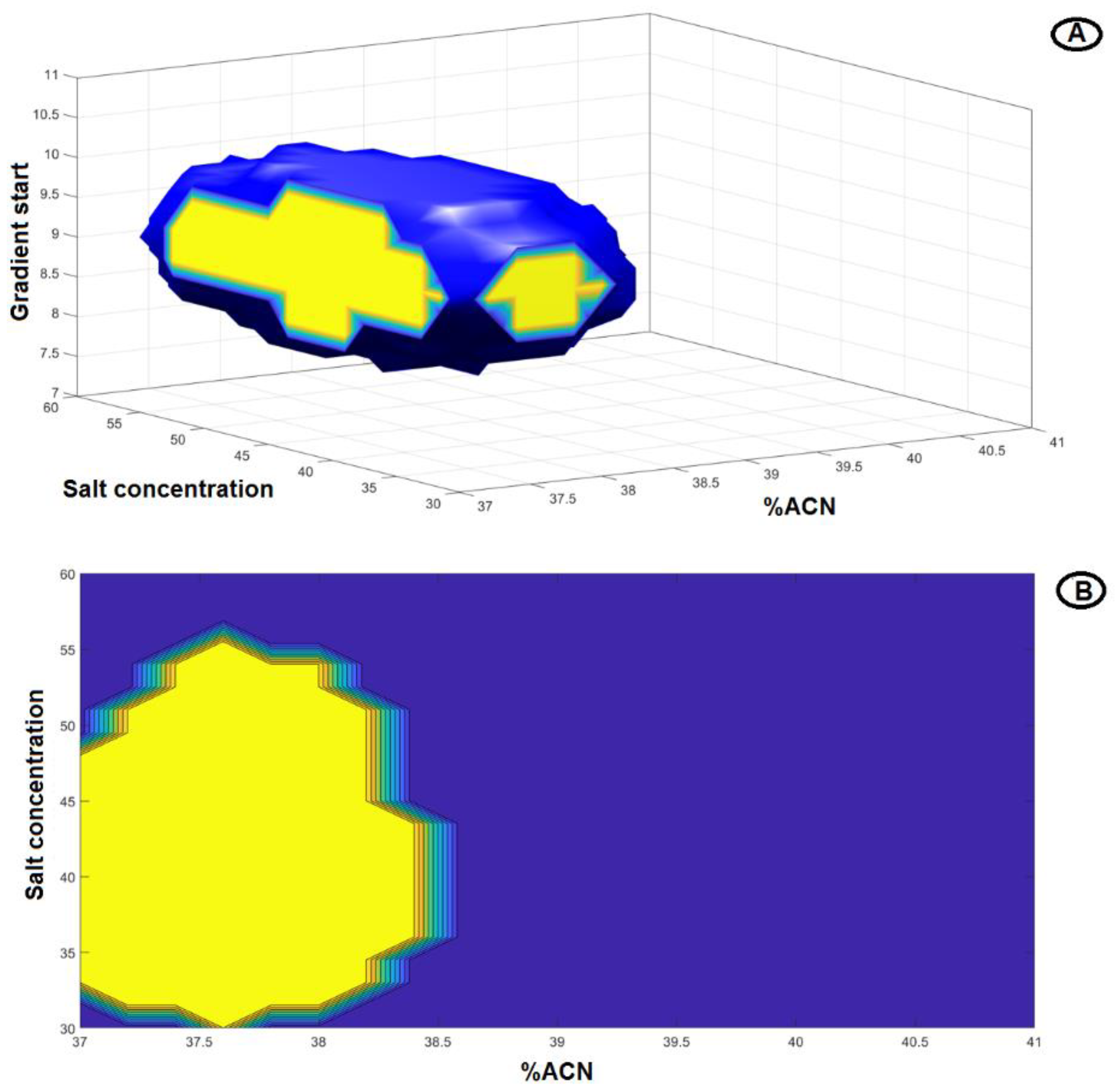
2.2. Robust Optimization of the Chromatographic Conditions Based on AQbD
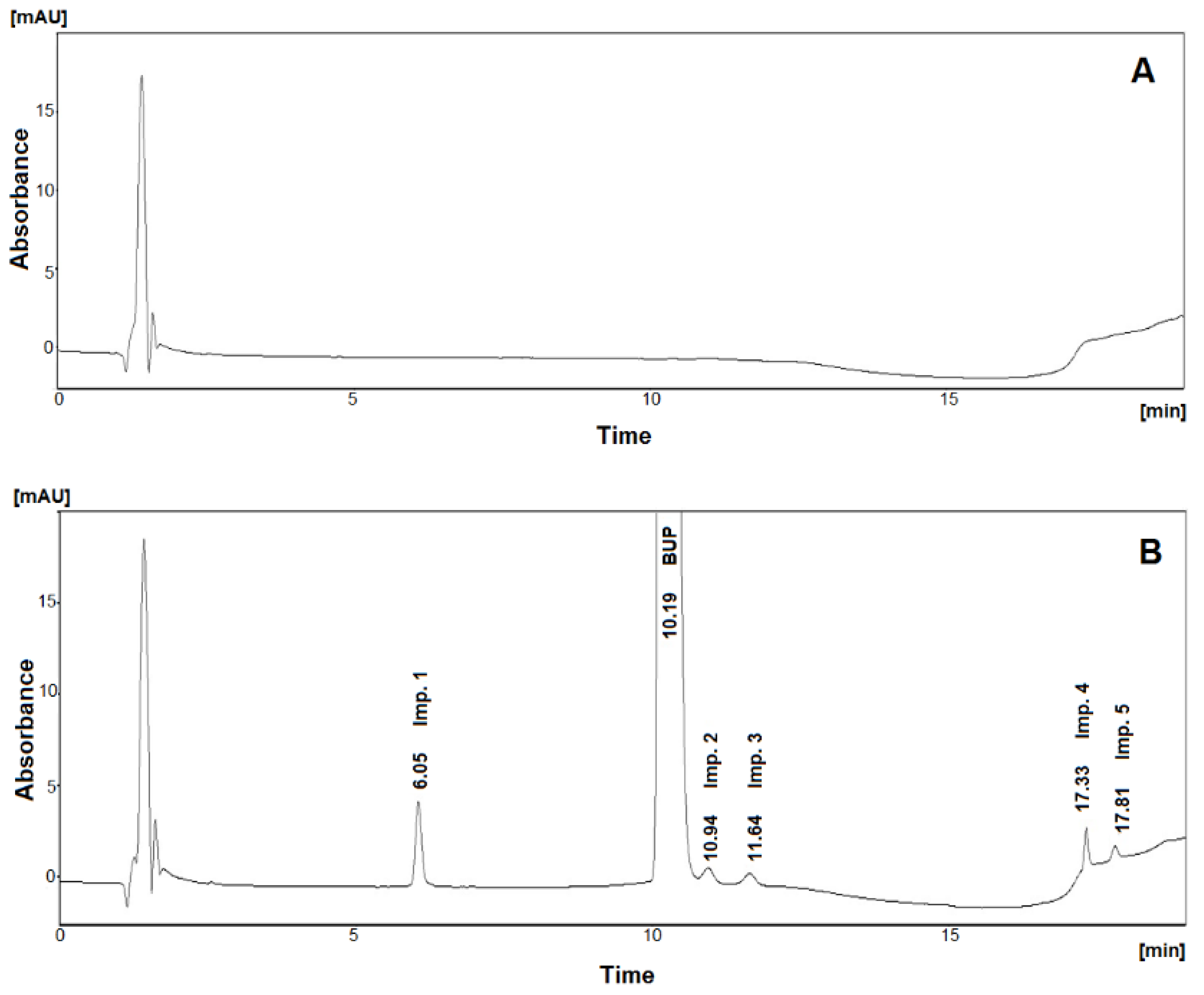
2.3. Method Validation
3. Materials and Methods
3.1. Reagents and Solvents
3.2. Instrumentation and Chromatographic Conditions
3.3. Solutions for Experimental Design
3.4. Solutions for Selectivity Estimation
3.5. Solutions for the Estimation of Linearity
3.6. Solutions for the Estimation of Accuracy
3.7. Solutions for the Estimation of Precision
3.8. Analysis of BUP Tablets
3.9. Software
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, S.R.; Berendt, R.T.; Ellison, C.D.; Ciavarella, A.B.; Asafu-Adjaye, E.; Khan, M.A.; Faustino, P.J. Profiles of Drug Substances, Excipients and Related Methodology; Academic Press: New York, NY, USA, 2016; Volume 41, pp. 1–30. [Google Scholar]
- Arias, H.R. Is the inhibition of nicotinic acetylcholine receptors by bupropion involved in its clinical actions? Int. J. Biochem. Cell Biol. 2009, 41, 2098–2108. [Google Scholar] [CrossRef]
- Al-Khamis, K.I. Rapid determination of bupropion in human plasma by high performance liquid chromatography. J. Liq. Chromatogr. 1989, 12, 645–655. [Google Scholar] [CrossRef]
- Cooper, T.B.; Suckow, R.F.; Glassman, A. Determination of bupropion and its major basic metabolites in plasma by liquid chromatography with dual-wavelength ultraviolet detection. J. Pharm. Sci. 1984, 73, 1104–1107. [Google Scholar] [CrossRef]
- Loboz, K.K.; Gross, A.S.; Ray, J.; McLachlan, A.J. HPLC assay for bupropion and its major metabolites in human plasma. J. Chromatogr. B 2005, 823, 115–121. [Google Scholar] [CrossRef]
- Zhang, D.; Yuan, B.; Qiao, M.; Li, F. HPLC determination and pharmacokinetics of sustained-release bupropion tablets in dogs. J. Pharm. Biomed. Anal. 2003, 33, 287–293. [Google Scholar] [CrossRef]
- Coles, R.; Kharasch, E.D. Stereoselective analysis of bupropion and hydroxybupropion in human plasma and urine by LC/MS/MS. J. Chromatogr. B 2007, 857, 67–75. [Google Scholar] [CrossRef]
- Shah, P.A.; Parekh, J.M.; Shrivastav, P.S. Assessment of Critical Extraction and Chromatographic Parameters for the Determination of Bupropion and Its Three Primary Metabolites in Human Plasma by LC-MS/MS. Microchem. J. 2017, 135, 81–90. [Google Scholar] [CrossRef]
- Teitelbaum, A.M.; Flaker, A.M.; Kharasch, E.D. Development, validation and application of a comprehensive stereoselective LC/MS-MS assay for bupropion and oxidative, reductive, and glucuronide metabolites in human urine. J. Chromatogr. B 2016, 1027, 239–253. [Google Scholar] [CrossRef]
- Yeniceli, D.; Şener, E.; Korkmaz, O.T.; Doǧrukol-Ak, D.; Tuncel, N. A simple and sensitive LC-ESI-MS (ion trap) method for the determination of bupropion and its major metabolite, hydroxybupropion in rat plasma and brain microdialysates. Talanta 2011, 84, 19–26. [Google Scholar] [CrossRef]
- O’Byrne, P.M.; Williams, R.; Walsh, J.J.; Gilmer, J.F. The aqueous stability of bupropion. J. Pharm. Biomed. Anal. 2010, 53, 376–381. [Google Scholar] [CrossRef]
- Bansal, R.; Saini, B.; Bansal, Y.; Bansal, G. MSn, LC-MS-TOF and LC-PDA studies for identification of new degradation impurities of bupropion. Biomed. Chromatogr. 2013, 27, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.S.; Elghobashy, M.R.; Shokry, R.F.; Bebawy, L.I. Stability indicating HPLC and spectrophotometric methods for the determination of bupropion hydrochloride in the presence of its alkaline degradates and related impurity. Bull. Fac. Pharm. Cairo Univ. 2012, 50, 49–59. [Google Scholar] [CrossRef]
- United States Pharmacopeia and the National Formulary (USP 42-NF 37); The United States Pharmacopeial Convention: Rockville, MD, USA, 2019.
- LoBrutto, R.; Jones, A.; Kazakevich, Y.; McNair, H.M. Effect of the eluent pH and acidic modifiers on the HPLC retention of basic analytes. J. Chromatogr. A 2001, 913, 173–187. [Google Scholar] [CrossRef]
- LoBrutto, R.; Jones, A.; Kazakevich, Y. Effect of counter-anion concentration on retention in high-performance liquid chromatography of protonated basic analytes. J. Chromatogr. A 2001, 913, 189–196. [Google Scholar] [CrossRef]
- Pan, L.; LoBrutto, R.; Kazakevich, Y.V.; Thompson, R. Influence of inorganic mobile phase additives on the retention, efficiency and peak symmetry of protonated basic compounds in reversed phase liquid chromatography. J. Chromatogr. A 2004, 1049, 63–73. [Google Scholar] [CrossRef]
- Shulyak, N.; Piponski, M.; Kovalenko, S.; Stoimenova, T.B.; Drapak, I.; Piponska, M.; Rezk, M.R.; Abbeyquaye, A.D.; Oleshchuk, O.; Logoyda, L. Chaotropic salts impact in HPLC approaches for simultaneous analysis of hydrophilic and lipophilic drugs. J. Sep. Sci. 2021, 44, 2908–2916. [Google Scholar] [CrossRef]
- Čolović, J.; Rmandić, M.; Malenović, A. Characterization of bonded stationary phase performance as a function of qualitative and quantitative chromatographic factors in chaotropic chromatography with risperidone and its impurities as model substances. Anal. Bioanal. Chem. 2018, 410, 4855–4866. [Google Scholar] [CrossRef]
- Čolović, J.; Kalinić, M.; Vemić, A.; Erić, S.; Malenović, A. Investigation into the phenomena affecting the retention behavior of basic analytes in chaotropic chromatography: Joint effects of the most relevant chromatographic factors and analytes’ molecular properties. J. Chromatogr. A 2015, 1425, 150–157. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use, Topic Q8 (R2): Pharmaceutical Development; ICH: Geneva, Switzerland, 2009.
- Zanatta, A.C.; Borges, M.S.; Mannochio-Russo, H.; Heredia-Vieira, S.C.; Campaner dos Santos, L.; Rinaldo, D.; Vilegas, W. Green chromatography as a novel alternative for the quality control of Serjania marginata Casar. Leaves. Microchem. J. 2022, 181, 107671. [Google Scholar] [CrossRef]
- Karmarkar, S.; Yang, X.; Garber, R.; Szajkovics, A.; Koberda, M. Quality by design (QbD) based development and validation of an HPLC method for amiodarone hydrochloride and its impurities in the drug substance. J. Pharm. Biomed. Anal. 2014, 100, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Iliou, K.; Malenović, A.; Loukas, Y.L.; Dotsikas, Y. Analysis of potential genotoxic impurities in rabeprazole active pharmaceutical ingredient via Liquid Chromatography-tandem Mass Spectrometry, following quality-by-design principles for method development. J. Pharm. Biomed. Anal. 2018, 149, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Kormány, R.; Rácz, N.; Fekete, S.; Horváth, K. Development of a fast and robust uhplc method for apixaban in-process control analysis. Molecules 2021, 26, 3505. [Google Scholar] [CrossRef] [PubMed]
- Malenović, A.; Dotsikas, Y.; Mašković, M.; Jančić-Stojanović, B.; Ivanović, D.; Medenica, M. Desirability-based optimization and its sensitivity analysis for the perindopril and its impurities analysis in a microemulsion LC system. Microchem. J. 2011, 99, 454–460. [Google Scholar] [CrossRef]
- Hanrahan, G.; Zhu, J.; Gibani, S.; Patil, D. Experimental Design. Chemom. Stat. Exp. Des. 2005, 8–13. [Google Scholar] [CrossRef]
- Flieger, J. The effect of chaotropic mobile phase additives on the separation of selected alkaloids in reversed-phase high-performance liquid chromatography. J. Chromatogr. A 2006, 1113, 37–44. [Google Scholar] [CrossRef]
- Crowther, J.B. Validation of pharmaceutical test methods. In Handbook of Modern Pharmaceutical Analysis; Ahuja, S., Scypinski, S., Eds.; Academic Press: New York, NY, USA, 2001; pp. 415–443. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Runs | Factors | Responses | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | te_bup | tb_imp2 | te_imp2 | tb_imp3 | tR_imp5 | |
| 1 | 2.4 | 37 | 80 | 30 | 8 | 13 | 12.53 | 12.45 | 12.66 | 12.71 | 15.51 |
| 2 | 2.4 | 43 | 20 | 40 | 8 | 9 | 5.89 | 5.31 | 5.62 | 5.55 | 15.20 |
| 3 | 3.2 | 40 | 50 | 35 | 6.5 | 11 | 9.05 | 9.03 | 9.41 | 9.51 | 13.29 |
| 4 | 2.4 | 37 | 20 | 40 | 5 | 13 | 8.71 | 8.67 | 8.95 | 9.01 | 14.07 |
| 5 | 4.0 | 37 | 80 | 30 | 5 | 13 | 10.99 | 10.89 | 11.11 | 11.13 | 14.27 |
| 6 | 2.4 | 43 | 80 | 40 | 5 | 13 | 6.78 | 6.71 | 7.00 | 7.05 | 12.38 |
| 7 | 2.4 | 43 | 20 | 30 | 8 | 13 | 5.41 | 5.32 | 5.56 | 5.56 | 13.84 |
| 8 | 4.0 | 37 | 20 | 40 | 8 | 13 | 9.80 | 9.83 | 10.23 | 10.63 | 15.24 |
| 9 | 4.0 | 37 | 80 | 40 | 5 | 9 | 9.71 | 9.55 | 9.75 | 9.81 | 31.50 |
| 10 | 3.2 | 40 | 50 | 35 | 6.5 | 11 | 9.07 | 9.05 | 9.34 | 9.52 | 13.32 |
| 11 | 2.4 | 37 | 80 | 40 | 8 | 9 | 11.18 | 10.68 | 12.52 | 10.55 | 33.00 |
| 12 | 2.4 | 37 | 20 | 30 | 5 | 9 | 9.28 | 9.11 | 9.29 | 10.49 | 32.00 |
| 13 | 2.4 | 43 | 80 | 30 | 5 | 9 | 7.71 | 7.87 | 7.97 | 8.09 | 32.50 |
| 14 | 4.0 | 43 | 20 | 30 | 5 | 13 | 6.88 | 6.77 | 7.01 | 7.11 | 12.99 |
| 15 | 4.0 | 43 | 20 | 40 | 5 | 9 | 6.03 | 5.85 | 6.11 | 6.14 | 11.29 |
| 16 | 3.2 | 40 | 50 | 35 | 6.5 | 11 | 9.01 | 8.99 | 9.36 | 9.51 | 13.31 |
| 17 | 4.0 | 37 | 20 | 30 | 8 | 9 | 11.11 | 10.98 | 11.17 | 11.17 | 31.00 |
| 18 | 4.0 | 43 | 80 | 40 | 8 | 13 | 7.10 | 6.93 | 7.20 | 7.25 | 13.67 |
| 19 | 4.0 | 43 | 80 | 30 | 8 | 9 | 7.86 | 7.86 | 8.13 | 8.32 | 24.36 |
| Runs | Factors | Responses | |||||
|---|---|---|---|---|---|---|---|
| A | B | C | te_bup | tb_imp2 | te_imp2 | tb_imp3 | |
| 1 | 39.00 | 45.00 | 9.00 | 9.35 | 9.39 | 9.65 | 9.93 |
| 2 | 41.00 | 45.00 | 7.00 | 7.93 | 7.93 | 7.93 | 8.00 |
| 3 | 39.00 | 60.00 | 11.00 | 9.79 | 9.83 | 10.13 | 10.41 |
| 4 | 41.00 | 45.00 | 11.00 | 8.05 | 8.05 | 8.05 | 8.15 |
| 5 | 37.00 | 30.00 | 9.00 | 10.43 | 10.56 | 10.85 | 11.12 |
| 6 | 41.00 | 30.00 | 9.00 | 7.19 | 7.19 | 7.47 | 7.56 |
| 7 | 39.00 | 60.00 | 7.00 | 9.75 | 9.83 | 10.03 | 10.31 |
| 8 | 37.00 | 45.00 | 11.00 | 11.34 | 11.43 | 11.81 | 12.23 |
| 9 | 37.00 | 45.00 | 7.00 | 10.98 | 11.01 | 11.21 | 11.39 |
| 10 | 39.00 | 45.00 | 9.00 | 9.33 | 9.38 | 9.65 | 9.95 |
| 11 | 37.00 | 60.00 | 9.00 | 11.90 | 12.02 | 12.37 | 12.59 |
| 12 | 39.00 | 45.00 | 9.00 | 9.35 | 9.41 | 9.61 | 9.96 |
| 13 | 39.00 | 45.00 | 9.00 | 9.35 | 9.41 | 9.68 | 9.99 |
| 14 | 39.00 | 30.00 | 7.00 | 8.63 | 8.68 | 8.93 | 9.14 |
| 15 | 41.00 | 60.00 | 9.00 | 8.11 | 8.11 | 8.39 | 8.53 |
| 16 | 39.00 | 30.00 | 11.00 | 8.62 | 8.67 | 8.92 | 9.19 |
| te_bup | tb_imp2 | te_imp2 | tb_imp3 | |
|---|---|---|---|---|
| bo | 9.35 | 9.4 | 9.65 | 9.90 |
| bA | −1.67 *** | −1.72 *** | −1.80 *** | −1.89 *** |
| bB | 0.59 *** | 0.59 *** | 0.59 *** | 0.61 *** |
| bC | 0.063 *** | 0.065 | 0.099 * | 0.14 * |
| bAB | −0.14 | −0.14 | −0.15 * | − |
| bAC | −0.061 | −0.076 | −0.12 | − |
| bBC | 0.012 | 0.00325 | 0.027 | − |
| bA2 | 0.22 ** | 0.21 * | 0.19 * | − |
| bB2 | 0.16 * | −0.14 | −0.063 | − |
| bC2 | 0.00075 | −0.00425 | −0.081 | − |
| R2 | 0.99677 | 0.9955 | 0.9977 | 0.9880 |
| adj. R2 | 0.9918 | 0.9888 | 0.9942 | 0.9851 |
| pred. R2 | 0.9476 | 0.9288 | 0.9644 | 0.9751 |
| Compound | LOD (μg/mL) | Linearity | Accuracy (Precision) | ||||
|---|---|---|---|---|---|---|---|
| Concentration Range (μg/mL) | a | b | r | Concentration Level (μg/mL) | % Recovery (% RSD) * | ||
| BUP | - | 200–400 | 12.318 | −43.145 | 0.9991 | 320 (80%) 400 (100%) 480 (120%) | 99.7 99.7 (0.17) 99.4 |
| Imp. 1 | 0.07 | 0.4–2.4 | 15.418 | 0.0397 | 0.9999 | 0.4 (LOQ) 2.0 (100%) 2.4 (120%) | 99.4 (1.7) 104.1 (0.3) 98.9 (1.8) |
| Imp. 2 | 0.06 | 0.2–1.0 | 11.558 | −0.576 | 0.9995 | 0.2 (LOQ) 0.8 (100%) 1.0 (120%) | 99.4 (6.2) 104.1 (1.3) 98.9 (0.9) |
| Imp. 3 | 0.04 | 0.2–1.0 | 9.291 | −0.472 | 0.9998 | 0.2 (LOQ) 0.8 (100%) 1.0 (120%) | 90.3 (3.6) 101 (1.5) 104.5 (1.3) |
| Imp. 4 | 0.6 | 1.8–12 | 0.847 | −0.458 | 0.9967 | 1.8 (LOQ) 9.0 (100%) 12 (120%) | 99.7 (2.4) 102.1 (1.5) 99.3 (4.6) |
| Imp. 5 | 0.06 | 0.2–0.8 | 10.511 | −0.0792 | 0.9993 | 0.2 (LOQ) 0.4 (100%) 0.8 (120%) | 102.8 (2.9) 103.4 (0.1) 103.3 (2.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkountanas, K.; Malenović, A.; Dotsikas, Y. Determination of Bupropion and Its Impurities via a Chaotropic Chromatography Method Following Analytical Quality-by-Design Principles for Method Development. Pharmaceuticals 2022, 15, 1196. https://doi.org/10.3390/ph15101196
Gkountanas K, Malenović A, Dotsikas Y. Determination of Bupropion and Its Impurities via a Chaotropic Chromatography Method Following Analytical Quality-by-Design Principles for Method Development. Pharmaceuticals. 2022; 15(10):1196. https://doi.org/10.3390/ph15101196
Chicago/Turabian StyleGkountanas, Kostas, Anđelija Malenović, and Yannis Dotsikas. 2022. "Determination of Bupropion and Its Impurities via a Chaotropic Chromatography Method Following Analytical Quality-by-Design Principles for Method Development" Pharmaceuticals 15, no. 10: 1196. https://doi.org/10.3390/ph15101196