

Design and Synthesis of Aminopyrimidinyl Pyrazole Analogs as PLK1 Inhibitors Using Hybrid 3D-QSAR and Molecular Docking
Abstract
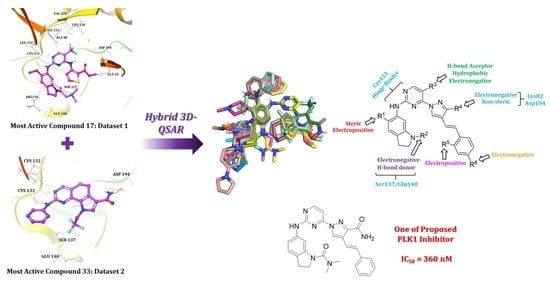
:
1. Introduction
2. Results and Discussion
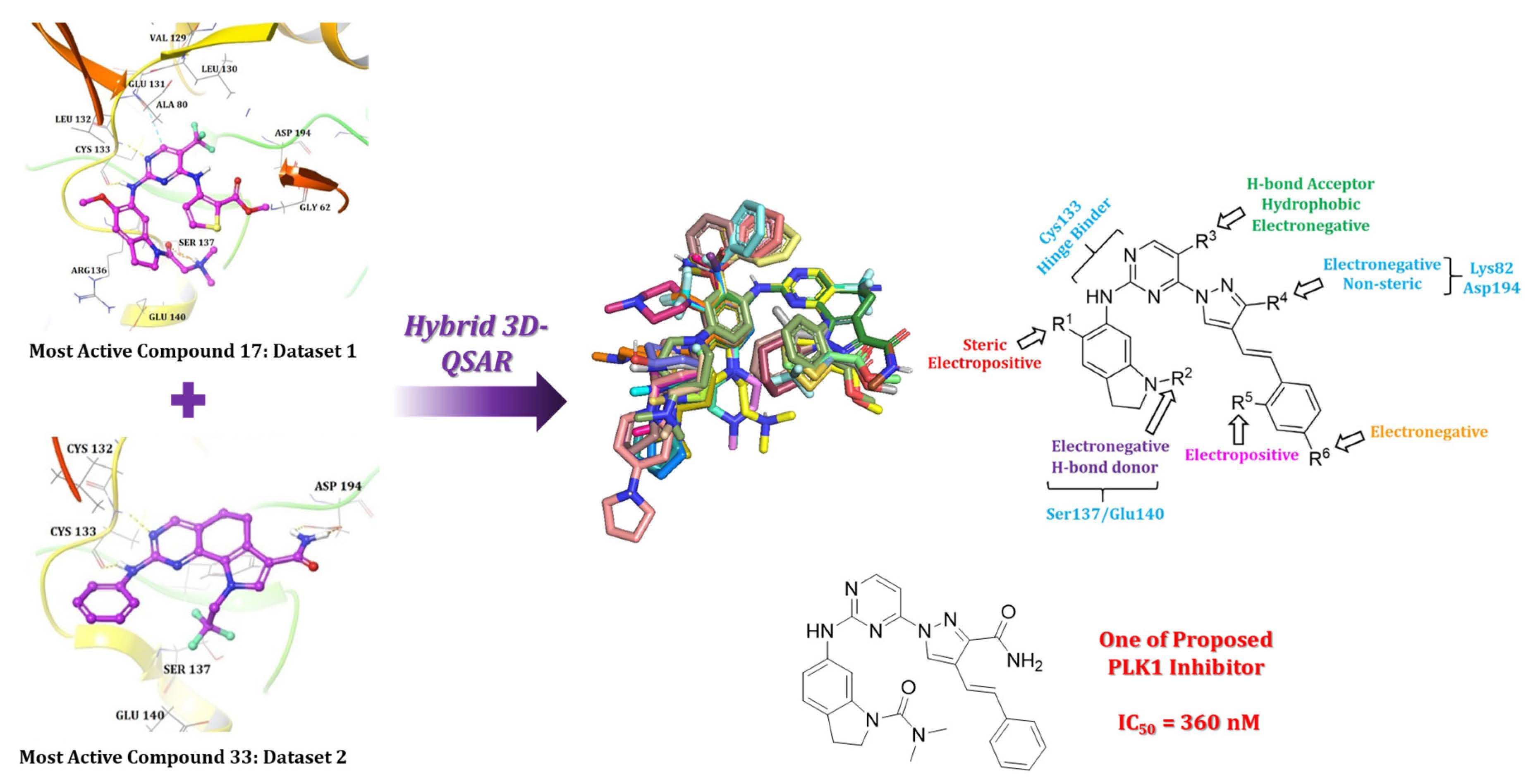
2.1. Molecular Docking
2.2. Hybrid 3D-QSAR Models
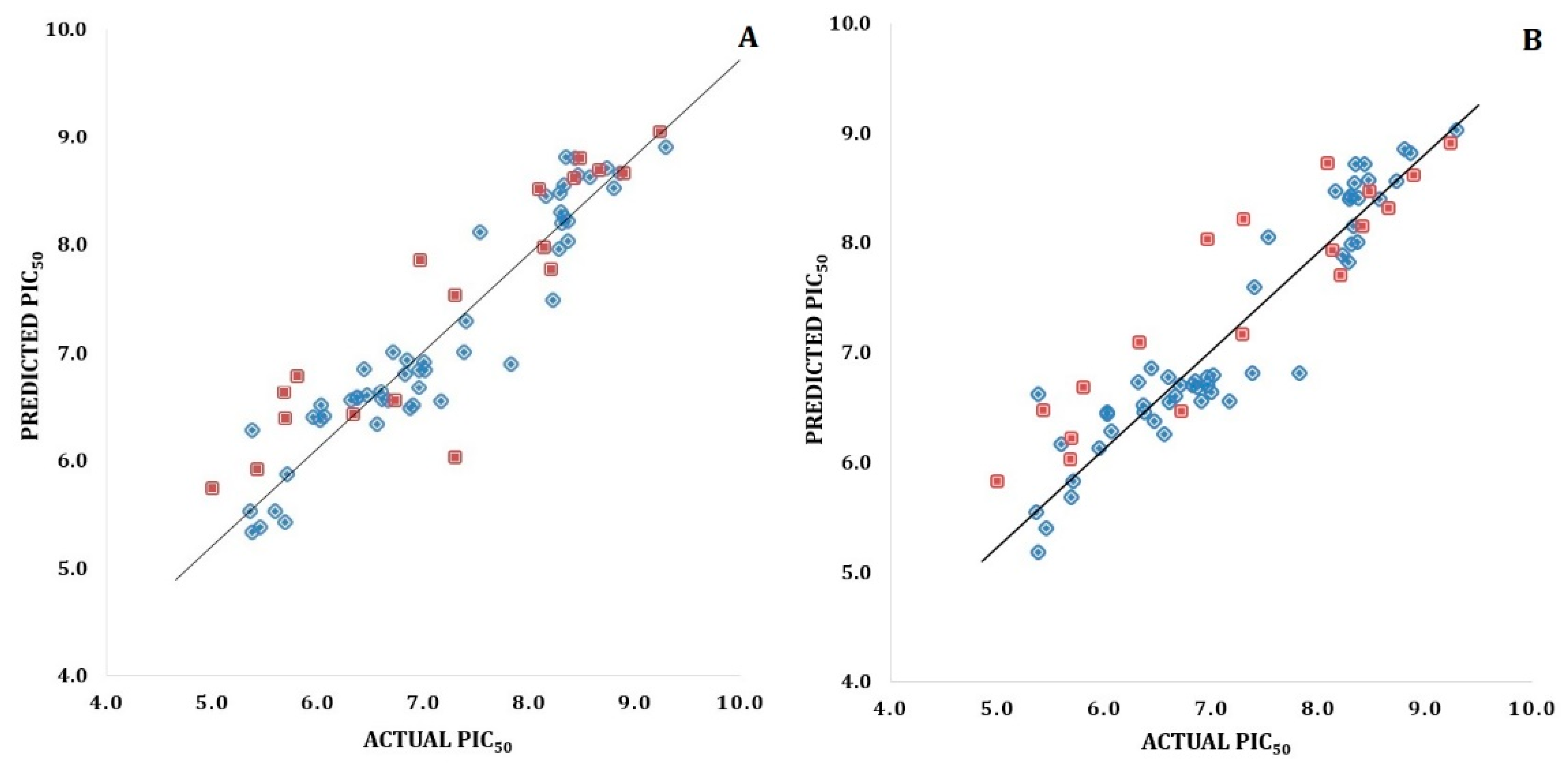
Validation of 3D-QSAR Models
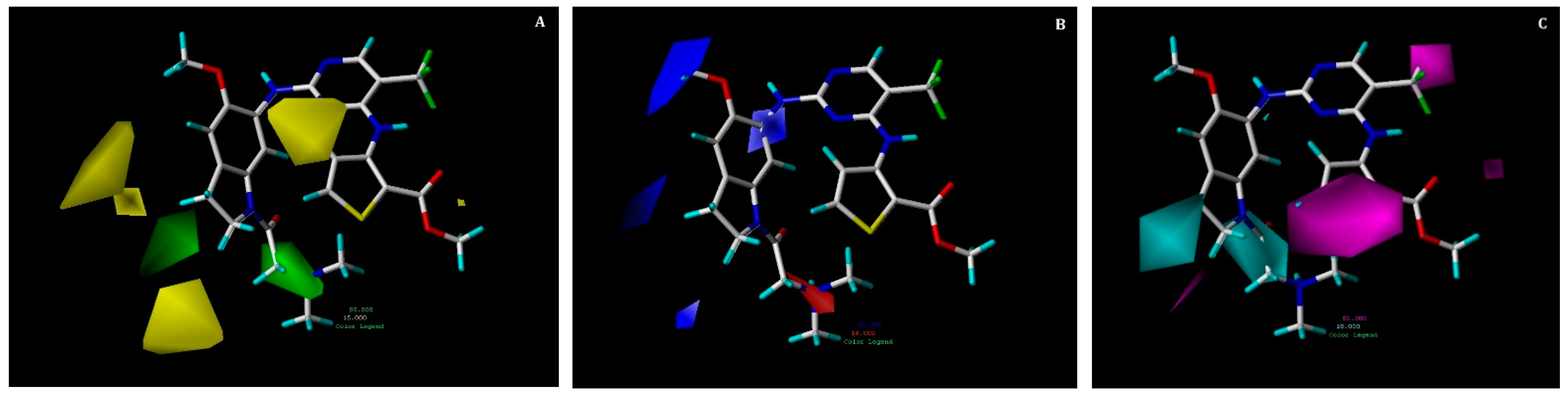
2.3. Contour Map Analysis
2.3.1. CoMFA Contour Maps
2.3.2. CoMSIA Contour Maps
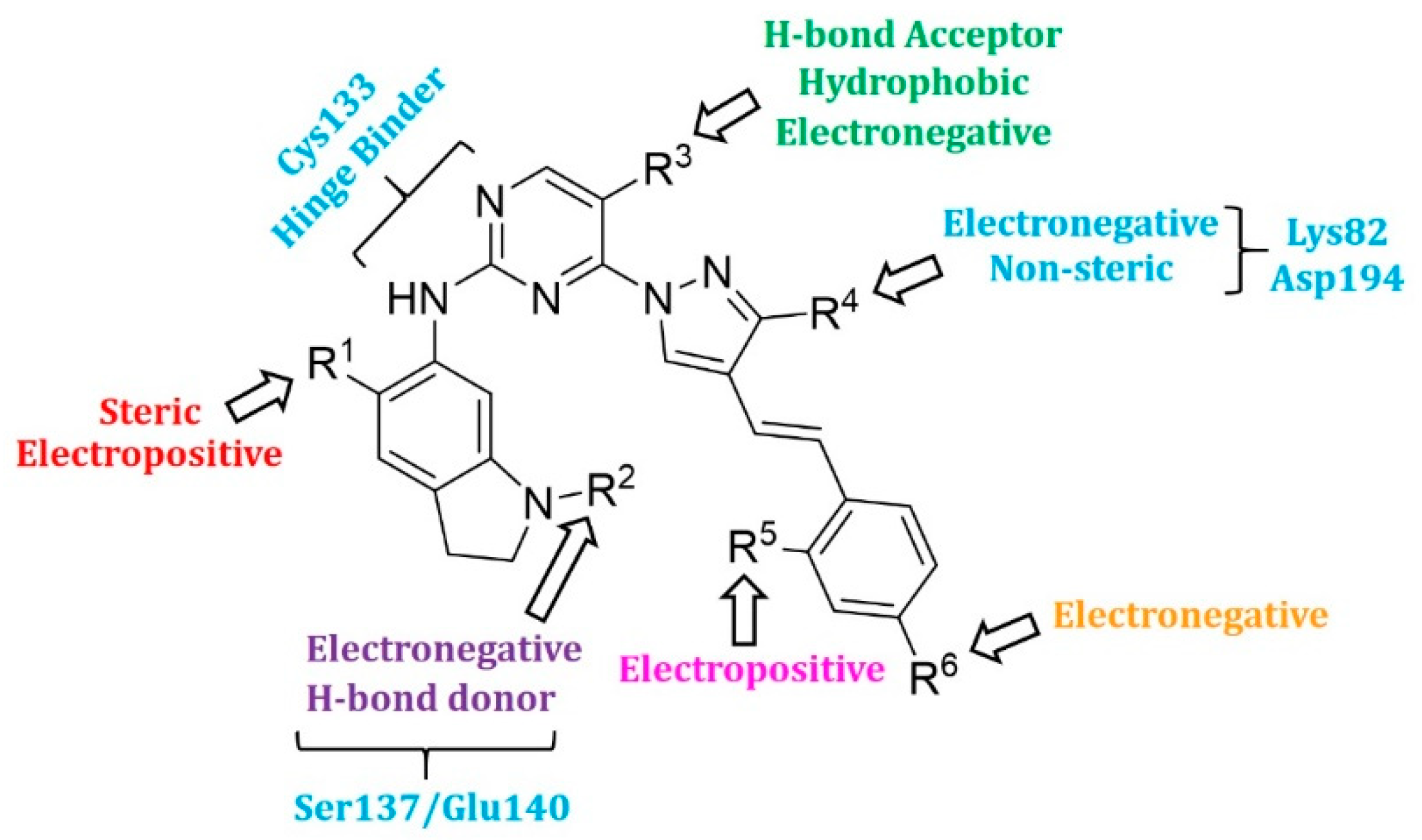
2.4. Designing New PLK1 Inhibitors
Synthesis of New PLK1 Inhibitors and Evaluation of IC50 Values
3. Materials and Methods
3.1. Training Set/Test Set Selection for CoMFA and CoMSIA
3.2. Molecular Docking
3.3. Receptor-Based Hybrid CoMFA and CoMSIA Models
3D-QSAR Model Validation
3.4. Synthesized PLK1 Inhibitors
3.5. Evaluation of IC50 Values
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kasahara, K.; Goto, H.; Izawa, I.; Kiyono, T.; Watanabe, N.; Elowe, S.; Nigg, E.A.; Inagaki, M. PI 3-kinase-dependent phosphorylation of Plk1–Ser99 promotes association with 14-3-3γ and is required for metaphase–anaphase transition. Nat. Commun. 2013, 4, 1882. [Google Scholar] [CrossRef]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Deng, Z.; Chen, G.; Liu, S.; Li, Y.; Zhong, J.; Zhang, B.; Li, L.; Huang, H.; Wang, Z.; Xu, Q.J.E. Discovery of methyl 3-((2-((1-(dimethylglycyl)-5-methoxyindolin-6-yl) amino)-5-(trifluoro-methyl) pyrimidin-4-yl) amino) thiophene-2-carboxylate as a potent and selective polo-like kinase 1 (PLK1) inhibitor for combating hepatocellular carcinoma. Eur. J. Med. Chem. 2020, 206, 112697. [Google Scholar] [CrossRef] [PubMed]
- Beria, I.; Ballinari, D.; Bertrand, J.A.; Borghi, D.; Bossi, R.T.; Brasca, M.G.; Cappella, P.; Caruso, M.; Ceccarelli, W.; Ciavolella, A.J. Identification of 4,5-Dihydro-1 H-pyrazolo [4,3-H] quinazoline derivatives as a new class of orally and selective polo-like kinase 1 inhibitors. J. Med. Chem. 2010, 53, 3532–3551. [Google Scholar] [CrossRef] [PubMed]
- Iliaki, S.; Beyaert, R.; Afonina, I.S. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem. Pharmacol. 2021, 193, 114747. [Google Scholar] [CrossRef]
- Kothe, M.; Kohls, D.; Low, S.; Coli, R.; Cheng, A.C.; Jacques, S.L.; Johnson, T.L.; Lewis, C.; Loh, C.; Nonomiya, J. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry 2007, 46, 5960–5971. [Google Scholar] [CrossRef]
- Archambault, V.; Glover, D.M. Polo-like kinases: Conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 2009, 10, 265–275. [Google Scholar] [CrossRef]
- Bruinsma, W.; Raaijmakers, J.A.; Medema, R.H. Switching Polo-like kinase-1 on and off in time and space. Trends Biochem. Sci. 2012, 37, 534–542. [Google Scholar] [CrossRef]
- Ruan, H.; Kiselar, J.; Zhang, W.; Li, S.; Xiong, R.; Liu, Y.; Yang, S.; Lai, L. Integrative structural modeling of a multidomain polo-like kinase. Phys. Chem. Chem. Phys. 2020, 22, 27581–27589. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, a potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Guan, R.; Tapang, P.; Leverson, J.D.; Albert, D.; Giranda, V.L.; Luo, Y. Small interfering RNA–mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Caner Res. 2005, 65, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Elez, R.; Piiper, A.; Kronenberger, B.; Kock, M.; Brendel, M.; Hermann, E.; Pliquett, U.; Neumann, E.; Zeuzem, S. Tumor regression by combination antisense therapy against Plk1 and Bcl-2. Oncogene 2003, 22, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Kneisel, L.; Strebhardt, K.; Bernd, A.; Wolter, M.; Binder, A.; Kaufmann, R.J. Expression of polo-like kinase (PLK1) in thin melanomas: A novel marker of metastatic disease. J. Cutan. Pathol. 2002, 29, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Xie, Z.; Liao, C. Developing polo-like kinase 1 inhibitors. Future Med. Chem. 2020, 12, 869–871. [Google Scholar] [CrossRef]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef]
- Rudolph, D.; Impagnatiello, M.A.; Blaukopf, C.; Sommer, C.; Gerlich, D.W.; Roth, M.; Tontsch-Grunt, U.; Wernitznig, A.; Savarese, F.; Hofmann, M.H.; et al. Efficacy and mechanism of action of volasertib, a potent and selective inhibitor of Polo-like kinases, in preclinical models of acute myeloid leukemia. J. Pharmacol. Exp. Ther. 2015, 352, 579–589. [Google Scholar] [CrossRef]
- Sero, V.; Tavanti, E.; Vella, S.; Hattinger, C.M.; Fanelli, M.; Michelacci, F.; Versteeg, R.; Valsasina, B.; Gudeman, B.; Picci, P.J. Targeting polo-like kinase 1 by NMS-P937 in osteosarcoma cell lines inhibits tumor cell growth and partially overcomes drug resistance. Investig. New Drugs 2014, 32, 1167–1180. [Google Scholar] [CrossRef]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R.J. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Aided Mol. Des. 2000, 5, 231–243. [Google Scholar]
- Chirico, N.; Gramatica, P.J. Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some case studies on application of “rm2” metrics for judging quality of quantitative structure–activity relationship predictions: Emphasis on scaling of response data. J. Comp. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Thibaut, U.; Folkers, G.; Klebe, G.; Kubinyi, H.; Merz, A.; Rognan, D. Recommendations for CoMFA studies and 3D QSAR publications. Quant. Struct.-Act. Relat. 1994, 13, 1–3. [Google Scholar] [CrossRef]
- Poso, A.; von Wright, A.; Gynther, J. An empirical and theoretical study on mechanisms of mutagenic activity of hydrazine compounds. Mutat. Res. 1995, 332, 63–71. [Google Scholar] [CrossRef]
- Dagan-Wiener, A.; Nissim, I.; Ben Abu, N.; Borgonovo, G.; Bassoli, A.; Niv, M.Y. Bitter or not? BitterPredict, a tool for predicting taste from chemical structure. Sci. Rep. 2017, 7, 1–13. [Google Scholar]
- Cousins, K.R. Computer review of ChemDraw ultra 12.0. J. Am. Chem. Soc. 2011, 133, 8388. [Google Scholar] [CrossRef]
- Brooks, W.H.; Daniel, K.G.; Sung, S.-S.; Guida, W.C. Computational validation of the importance of absolute stereochemistry in virtual screening. J. Chem. Inf. Model. 2008, 48, 639–645. [Google Scholar] [CrossRef]
- Van den Driessche, G.; Fourches, D. Adverse drug reactions triggered by the common HLA-B* 57: 01 variant: A molecular docking study. J. Cheminform. 2017, 9, 1–17. [Google Scholar] [CrossRef]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Giardina, S.F.; Werner, D.S.; Pingle, M.; Feinberg, P.B.; Foreman, K.W.; Bergstrom, D.E.; Arnold, L.D.; Barany, F. Novel, self-assembling dimeric inhibitors of human β tryptase. J. Med. Chem. 2020, 63, 3004–3027. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Ghosh, S.; Keretsu, S.; Cho, S.J. Designing of the N-ethyl-4-(pyridin-4-yl) benzamide based potent ROCK1 inhibitors using docking, molecular dynamics, and 3D-QSAR. PeerJ 2021, 9, e11951. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Jayatilleke, P.R.; Nair, A.C.; Zauhar, R.; Welsh, W. Computational Studies on HIV-1 Protease Inhibitors: Influence of Calculated Inhibitor−Enzyme Binding Affinities on the Statistical Quality of 3D-QSAR CoMFA Models. J. Med. Chem. 2000, 43, 4446–4451. [Google Scholar] [CrossRef] [PubMed]
- Gadhe, C.G.; Kothandan, G.; Cho, S.J. Large variation in electrostatic contours upon addition of steric parameters and the effect of charge calculation schemes in CoMFA on mutagenicity of MX analogues. Mol. Sim. 2012, 38, 861–871. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Full Model | Test Set 12 | ||
|---|---|---|---|---|
| CoMFA | CoMSIA (SEH) | CoMFA | CoMSIA (SEH) | |
| q2 | 0.517 | 0.540 | 0.628 | 0.580 |
| ONC | 6 | 6 | 6 | 6 |
| SEP | 0.844 | 0.824 | 0.717 | 0.762 |
| r2 | 0.847 | 0.855 | 0.905 | 0.895 |
| SEE | 0.475 | 0.462 | 0.363 | 0.381 |
| F value | 58.087 | 61.993 | 71.401 | 63.990 |
| LOF | - | - | 0.607 | 0.609 |
| BS-r2 | - | - | 0.929 | 0.936 |
| BS-SD | - | - | 0.020 | 0.020 |
| r2pred | - | - | 0.796 | 0.783 |
| rm2 | - | - | 0.665 | 0.581 |
| Delta rm2 | - | - | 0.181 | 0.214 |
| Compound Structure | Name | R1 | R2 | R3 | R4 | R5 | R6 | Predicted pIC50 |
|---|---|---|---|---|---|---|---|---|
 | D3 |  |  | CF3 | CONH2 | H | Cl | 10.217 |
 | D5 |  | 9.715 | |||||
 | D10 |  | 10.272 | |||||
 | D14 |  | 9.811 | |||||
 | D17 |  | 9.903 |
| Compound Structure | Name | R1 | R2 | R3 | R4 | R5 | R6 | IC50 (µM) | Predicted IC50 (nM) |
|---|---|---|---|---|---|---|---|---|---|
 | D39 | H |  | H |  | H | H | 1.43 | 0.35 |
 | D40 | H |  | H | CONH2 | H | H | 0.359 | 0.13 |
| Compound | Structure | NMR | HRMS |
|---|---|---|---|
| D39 |  | 1H NMR (400 MHz, DMSO-d6) δ 10.05–9.95 (m, 1H), 9.40 (s, 1H), 0.16 (d, J = 28.1 Hz, 1H), 8.62 (d, J = 5.3 Hz, 1H), 7.58 (d, J = 7.3 Hz, 1H), 7.48 (d, J = 1. 8 Hz, 1H), 7.41 (t, J = 7.6Hz, 1H), 7.33 (dd, J = 5.0, 3.6 Hz, 1H), 7.31–7.09 (m, 5H), 4.40 (q, J = 7.1 Hz, 2H), 4.31–4.23 (m, 3H), 4.10 (q, J = 5.2 Hz, 2H), 3.22 (s, 1H), 2.27 (d, J = 16.0 Hz, 1H), 2.11 (d, J = 23. 6 Hz, 6H), 1.38 (t, J = 7.1 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 168.4 (S), 161.7 (s), 161.2 (s), 159.6 (s), 156.5 (s), 143.3 (s), 142.9 (s), 138.9 (s), 137.2 (s), 131.6 (s), 128.8 (s), 127.9 (s), 126.5 (s), 125.9 (s), 125.1 (s), 124.5 (s), 124.3 (s), 117.5 (s), 114.5 (s), 107.1 (s), 99.1 (s), 63.9 (S), 61.0 (s), 47.9 (s), 45.2 (s), 27.1 (s), 14.2 (s) | HRMS (ESI+) calculated for [M + H]+ C30H31N7O3: 538.2561, found 538.2572. |
| D40 |  | 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 1H), 9.36 (s, 1H), 9.23 (s, 1H), 8.63 (d, J = 5.3 Hz, 1H), 7.97 (s, 1H), 7.63 (d, J = 16.7 Hz, 1H), 7.59 (s, 1H), 7.55 (d, J = 7.4 Hz, 2H), 7.48 (d, J = 2.8 Hz, 1H), 7.47 (d, J = 5.3 Hz, 1H), 7.40 (t, J = 7.6 Hz, 2H), 7.29 (t, J = 7.3 Hz, 1H), 7.17 (d, J = 8.1 Hz, 1H), 7.07 (d, J = 7.8 Hz, 1H), 4.27 (t, J = 8.4 Hz, 2H), 3.24 (s, 2H), 3.09 (t, J = 8.3 Hz, 2H), 2.16 (s. 6H); 13C NMR (101 MHz, DMSO-d6) δ 168.2 (s), 163.7 (s), 160.8 (s), 159.6 (s), 156.6 (s), 145.8 (s), 143.3 (s), 139.0 (s), 137.5 (s), 130.7 (s), 128.7 (s), 127.6 (s), 126.4 (s), 125.7 (s), 125.0 (s), 124.5 (s), 123.2 (s), 118.3 (s), 114.4 (s), 107.0 (s), 99.1 (s), 63.8 (s), 47.8 (s), 45.2 (s), 27.1 (s) | HRMS (ESI+) calculated for [M + H]+ C28H29N8O2: 509.2408, found 509.2403. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhujbal, S.P.; Kim, H.; Bae, H.; Hah, J.-M. Design and Synthesis of Aminopyrimidinyl Pyrazole Analogs as PLK1 Inhibitors Using Hybrid 3D-QSAR and Molecular Docking. Pharmaceuticals 2022, 15, 1170. https://doi.org/10.3390/ph15101170
Bhujbal SP, Kim H, Bae H, Hah J-M. Design and Synthesis of Aminopyrimidinyl Pyrazole Analogs as PLK1 Inhibitors Using Hybrid 3D-QSAR and Molecular Docking. Pharmaceuticals. 2022; 15(10):1170. https://doi.org/10.3390/ph15101170
Chicago/Turabian StyleBhujbal, Swapnil P., Hyejin Kim, Hyunah Bae, and Jung-Mi Hah. 2022. "Design and Synthesis of Aminopyrimidinyl Pyrazole Analogs as PLK1 Inhibitors Using Hybrid 3D-QSAR and Molecular Docking" Pharmaceuticals 15, no. 10: 1170. https://doi.org/10.3390/ph15101170