
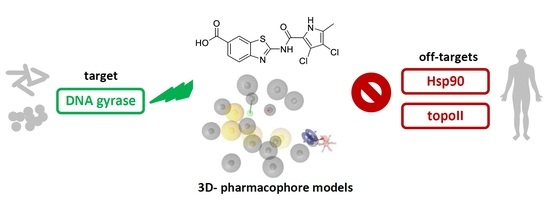
Selective DNA Gyrase Inhibitors: Multi-Target in Silico Profiling with 3D-Pharmacophores

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
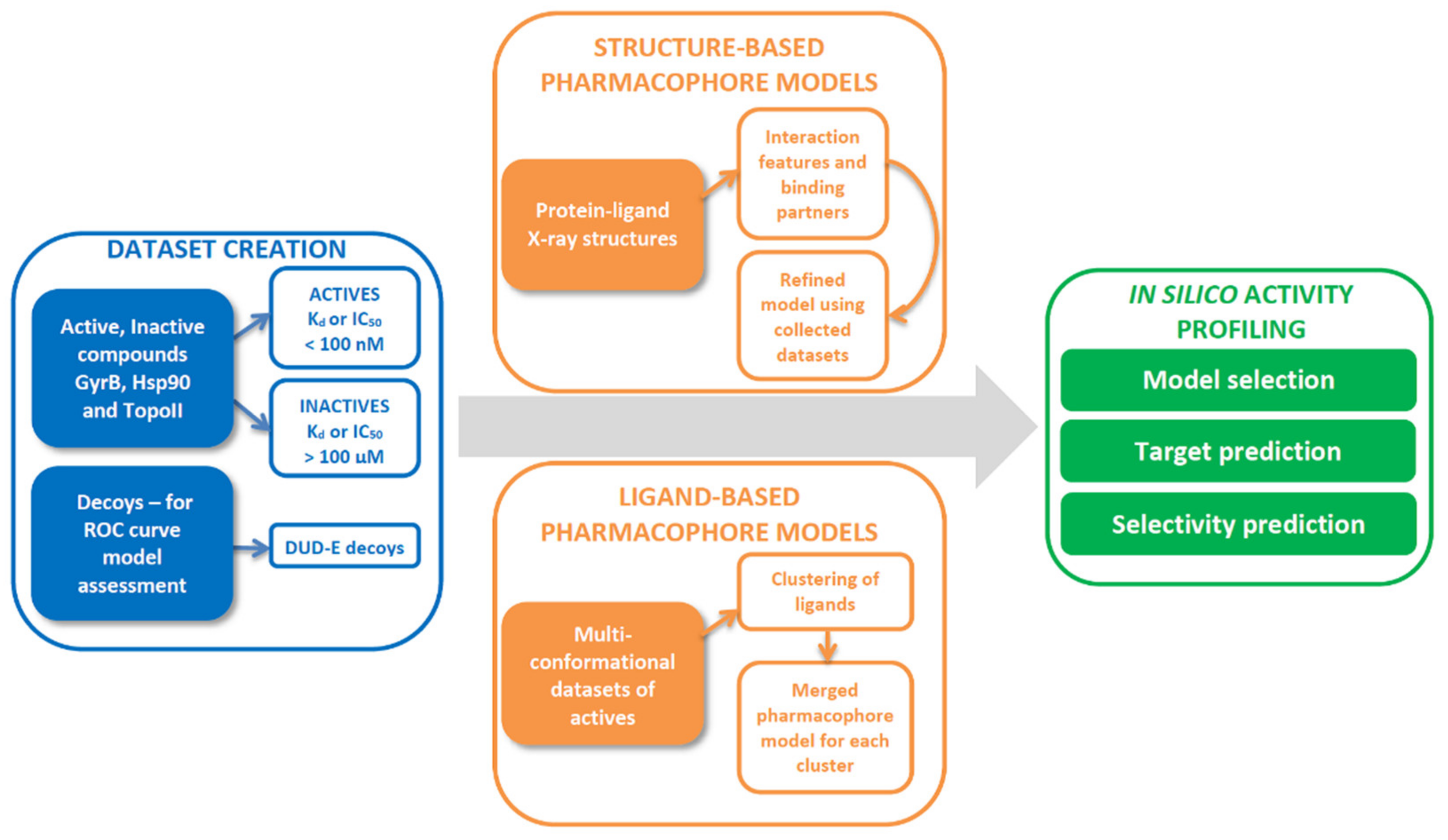
2. Results and Discussion
2.1. Datasets for Model Design
2.2. 3D-Pharmacophore Modelling
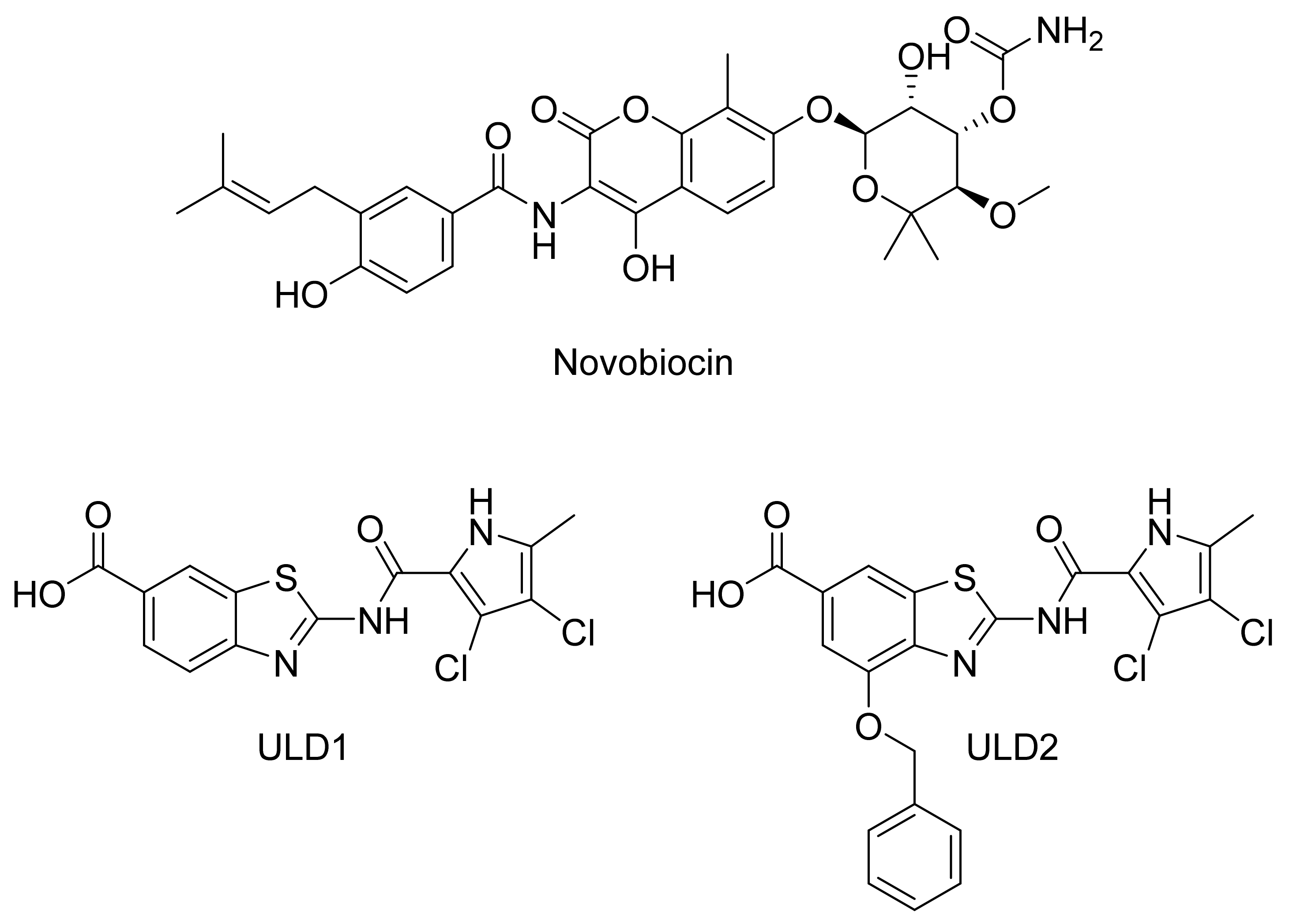
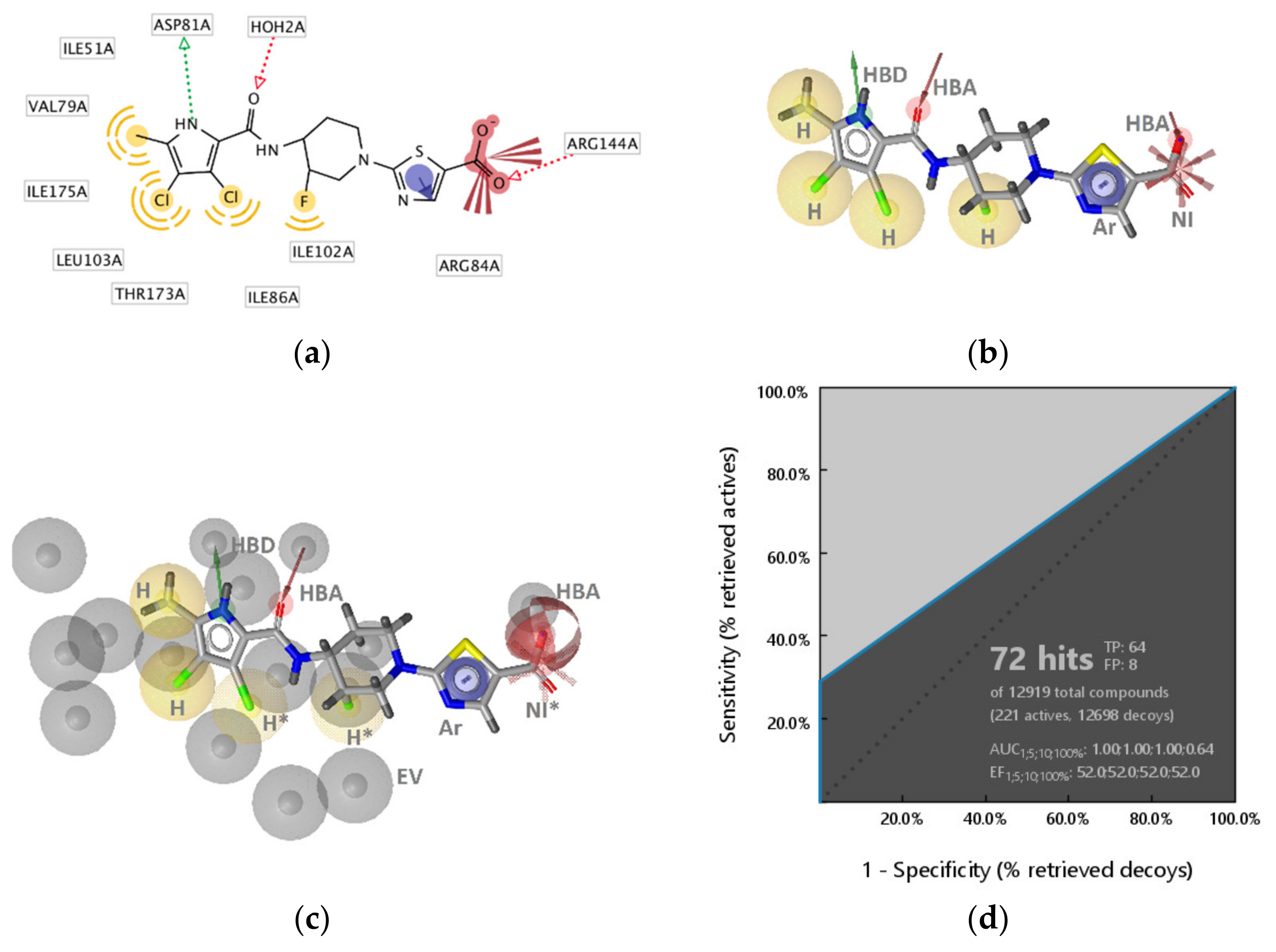
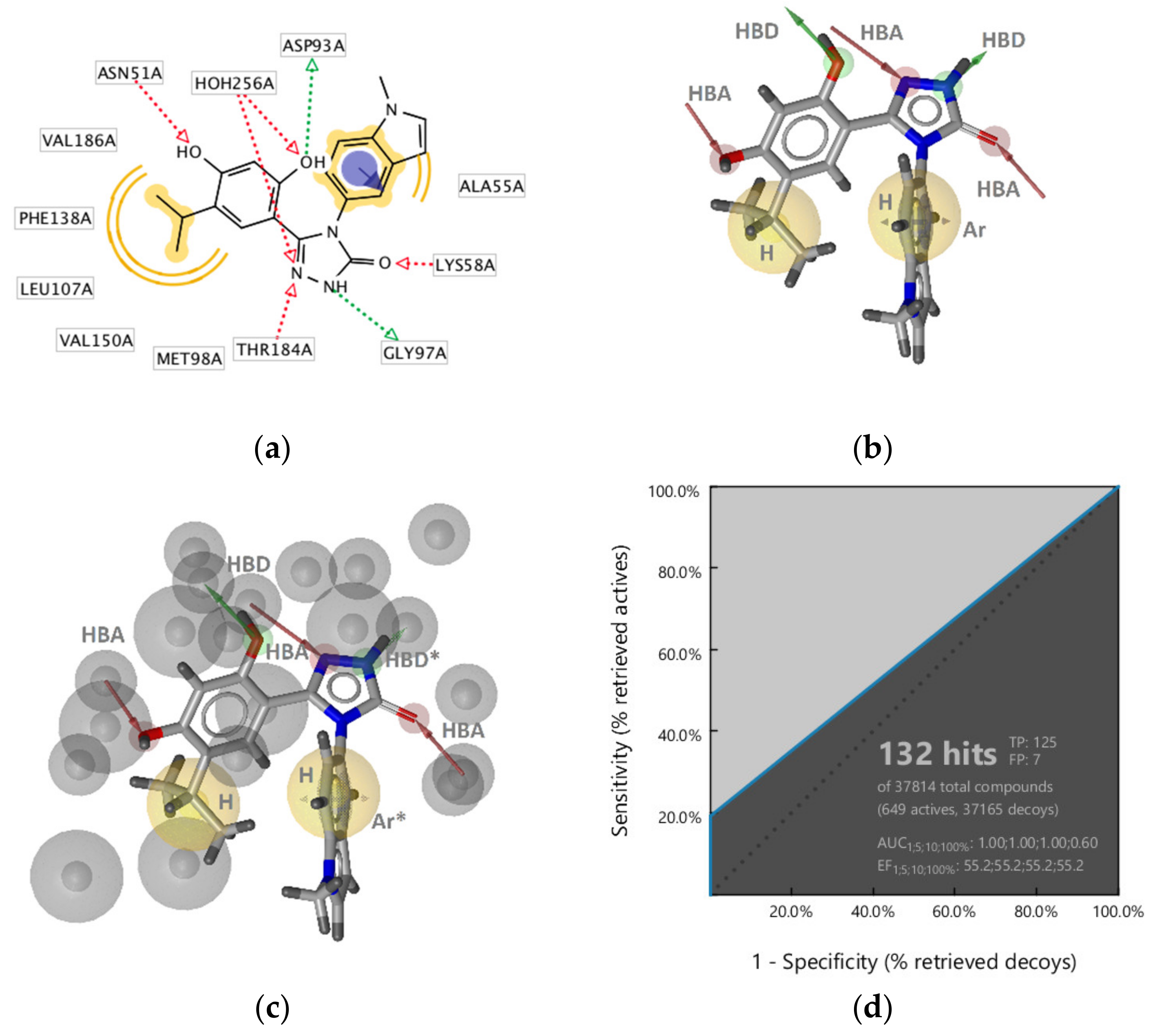
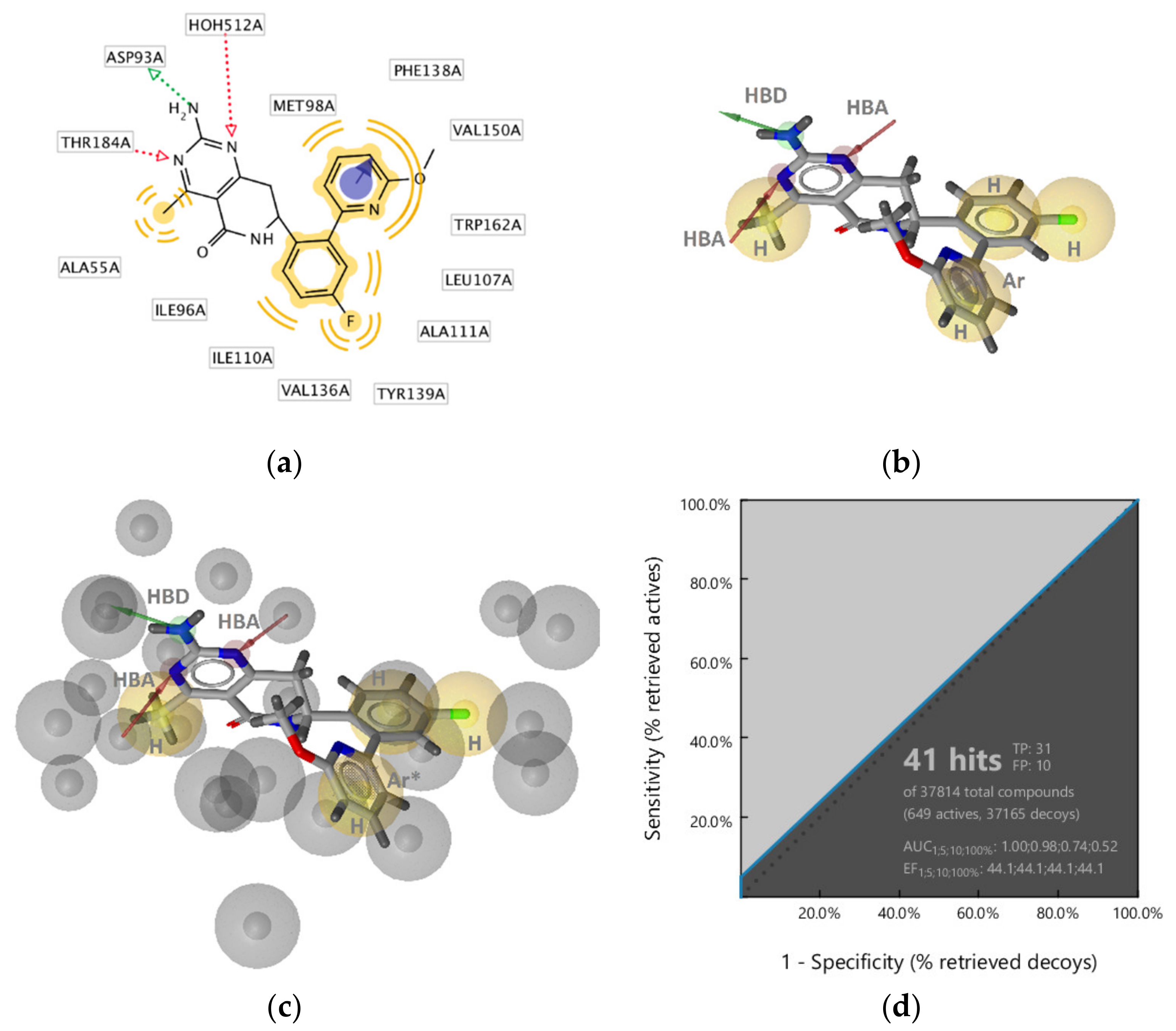
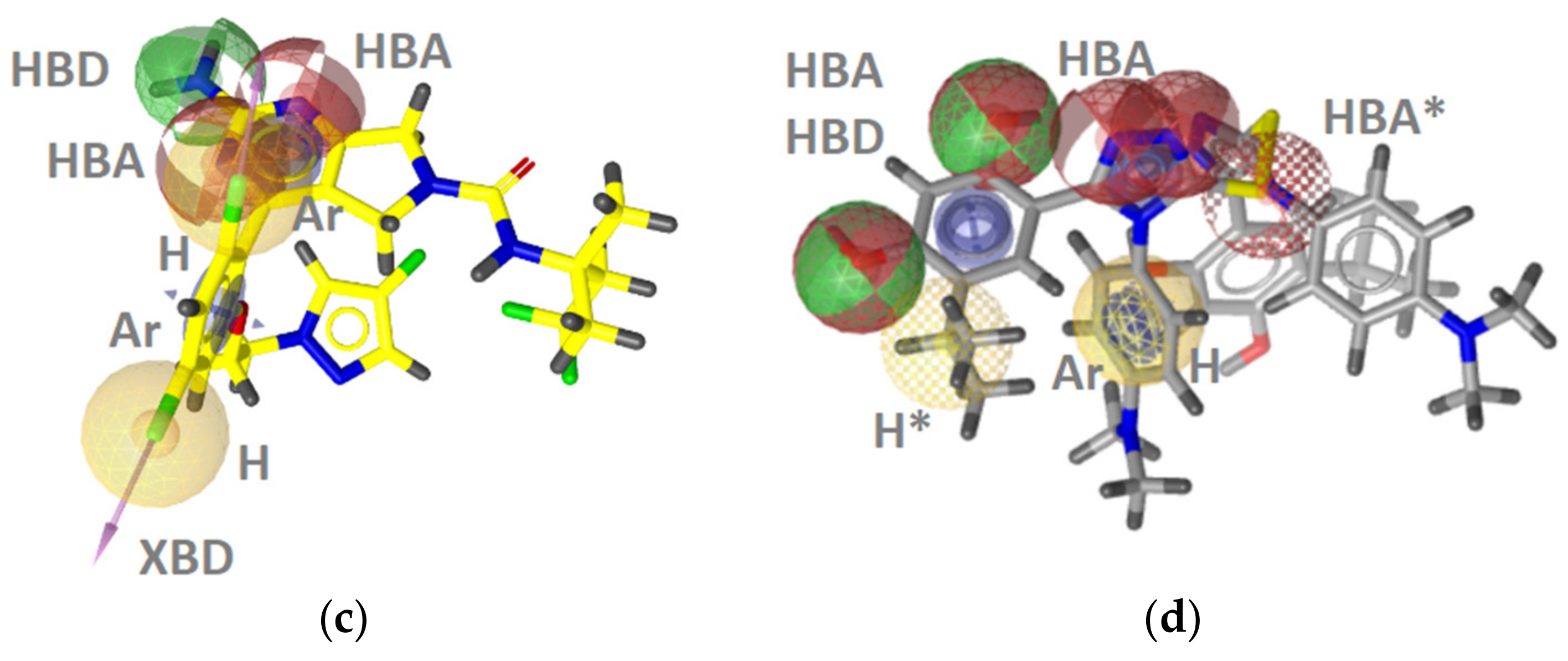
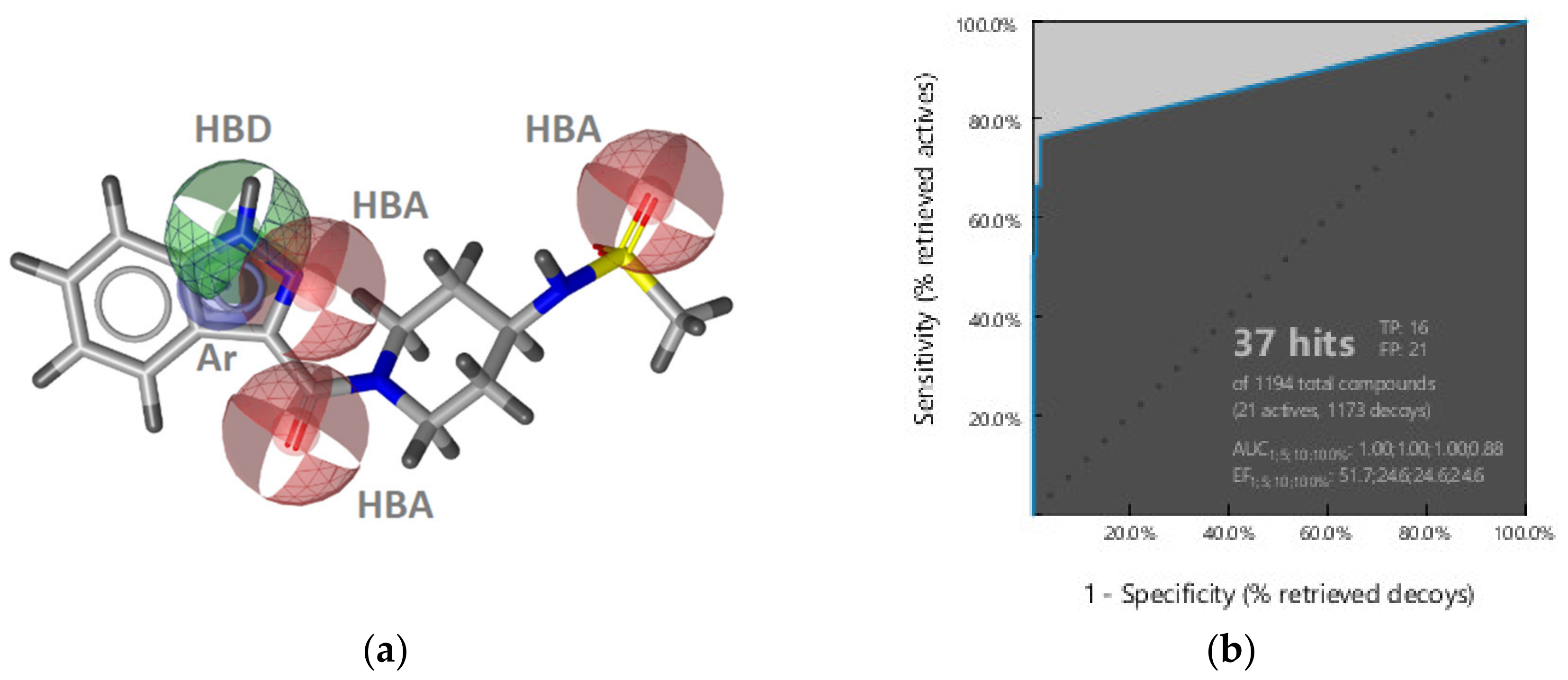
2.2.1. DNA Gyrase B Inhibitors
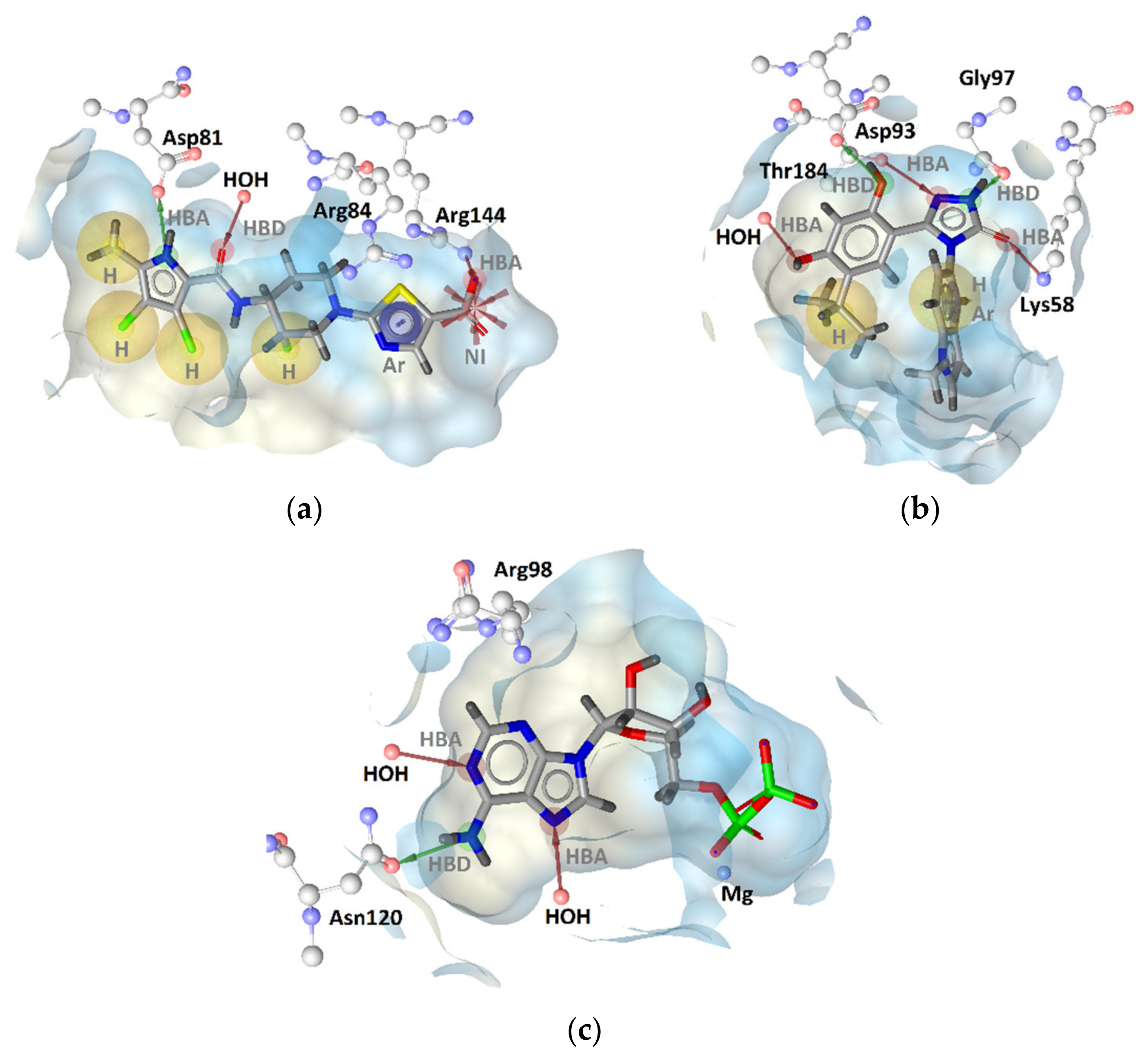
GyrB Pyrrolamide Inhibitor Interaction Features
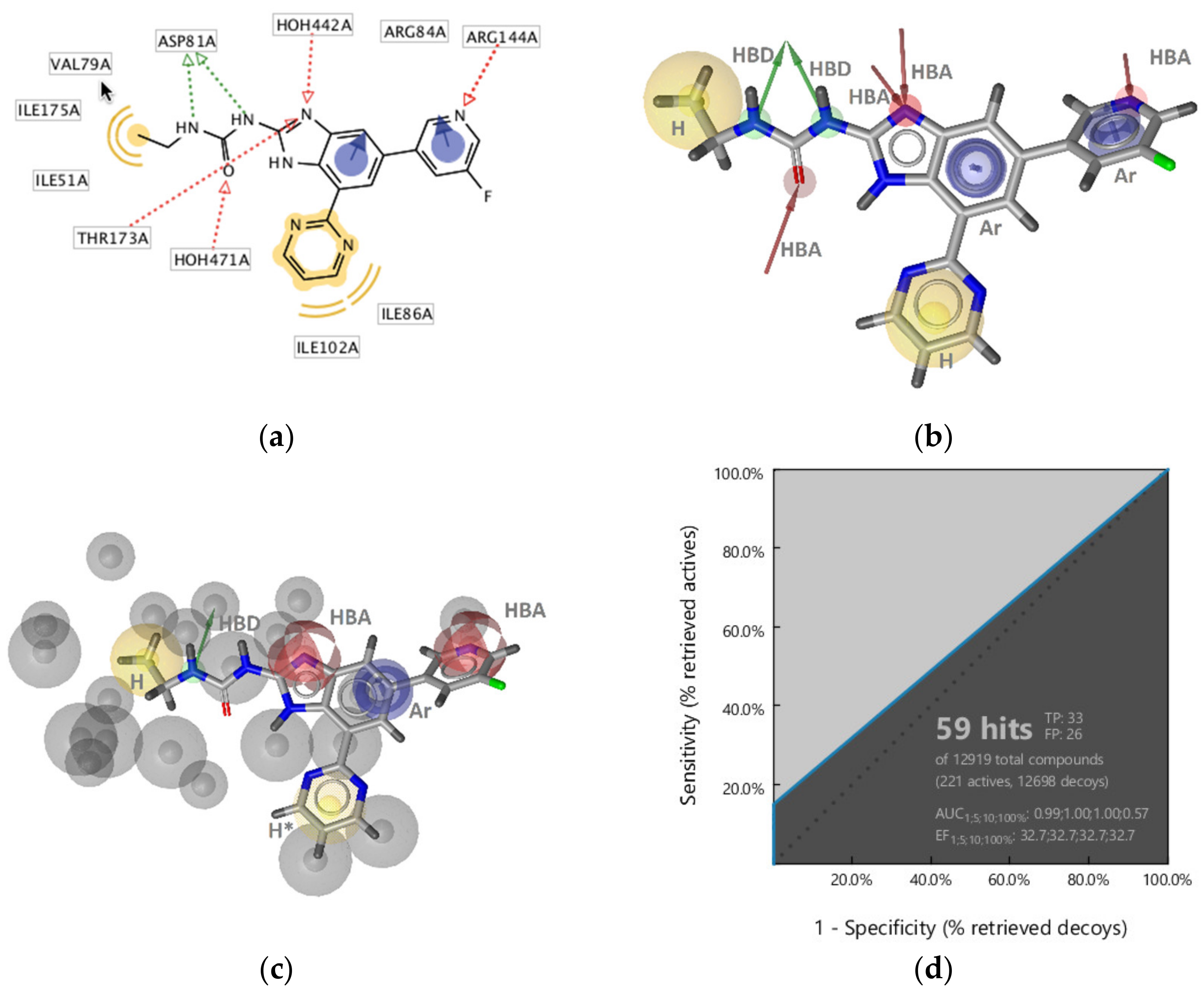
GyrB Ethyl Urea Inhibitor Interaction Features
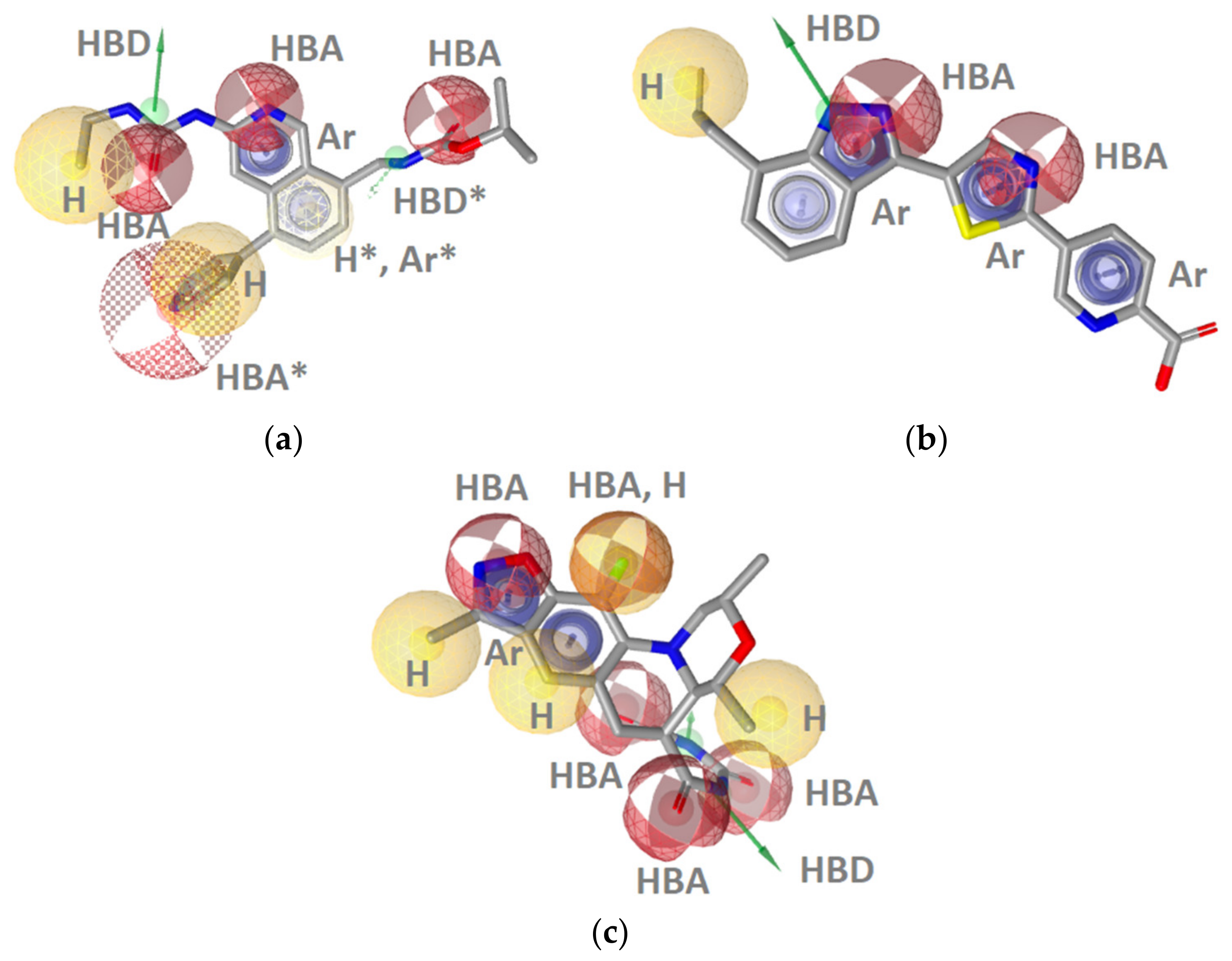
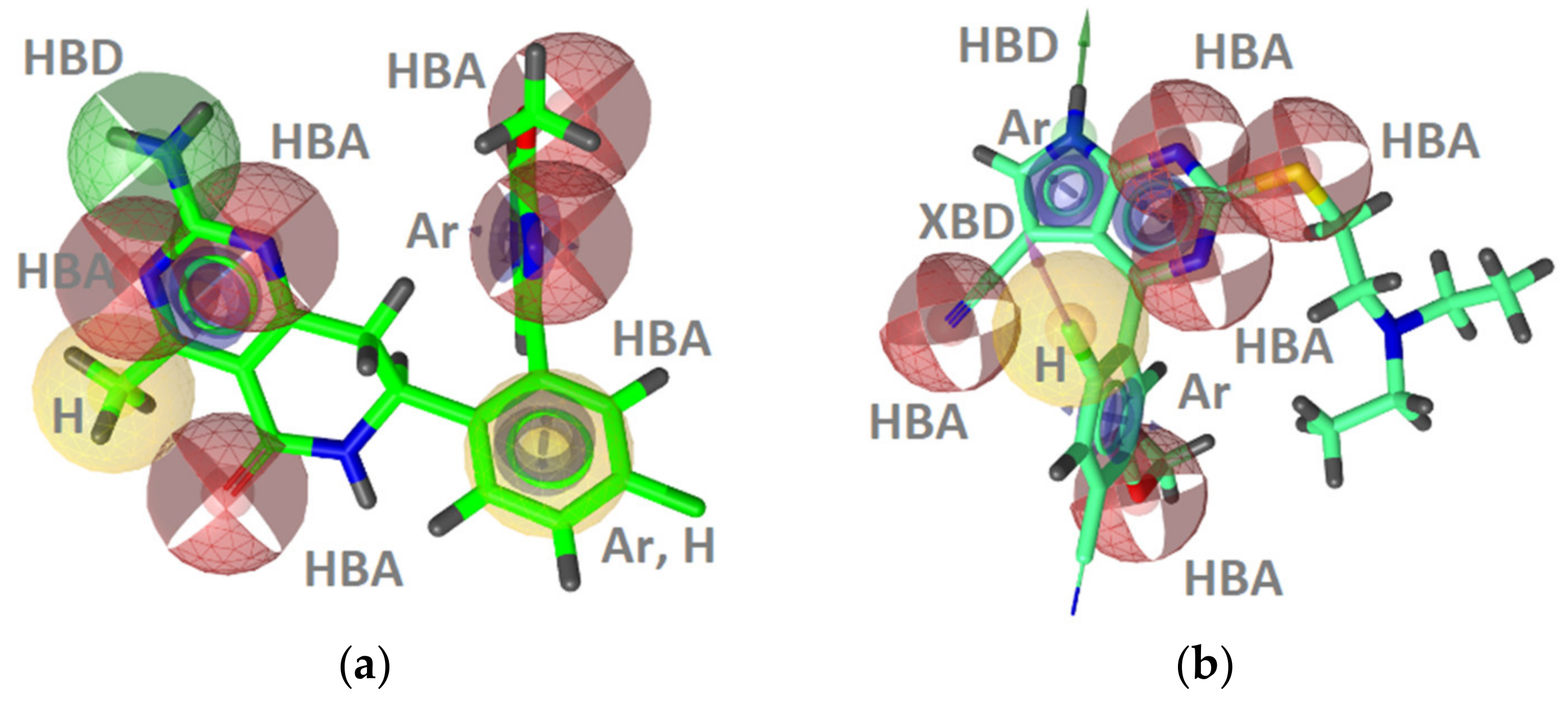
GyrB Inhibitor Ligand-Based Pharmacophore Models
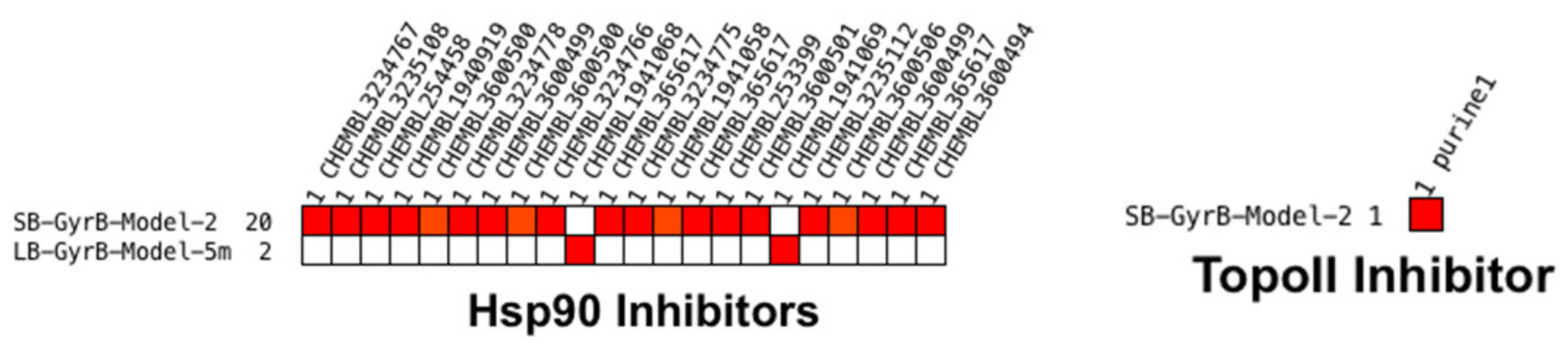
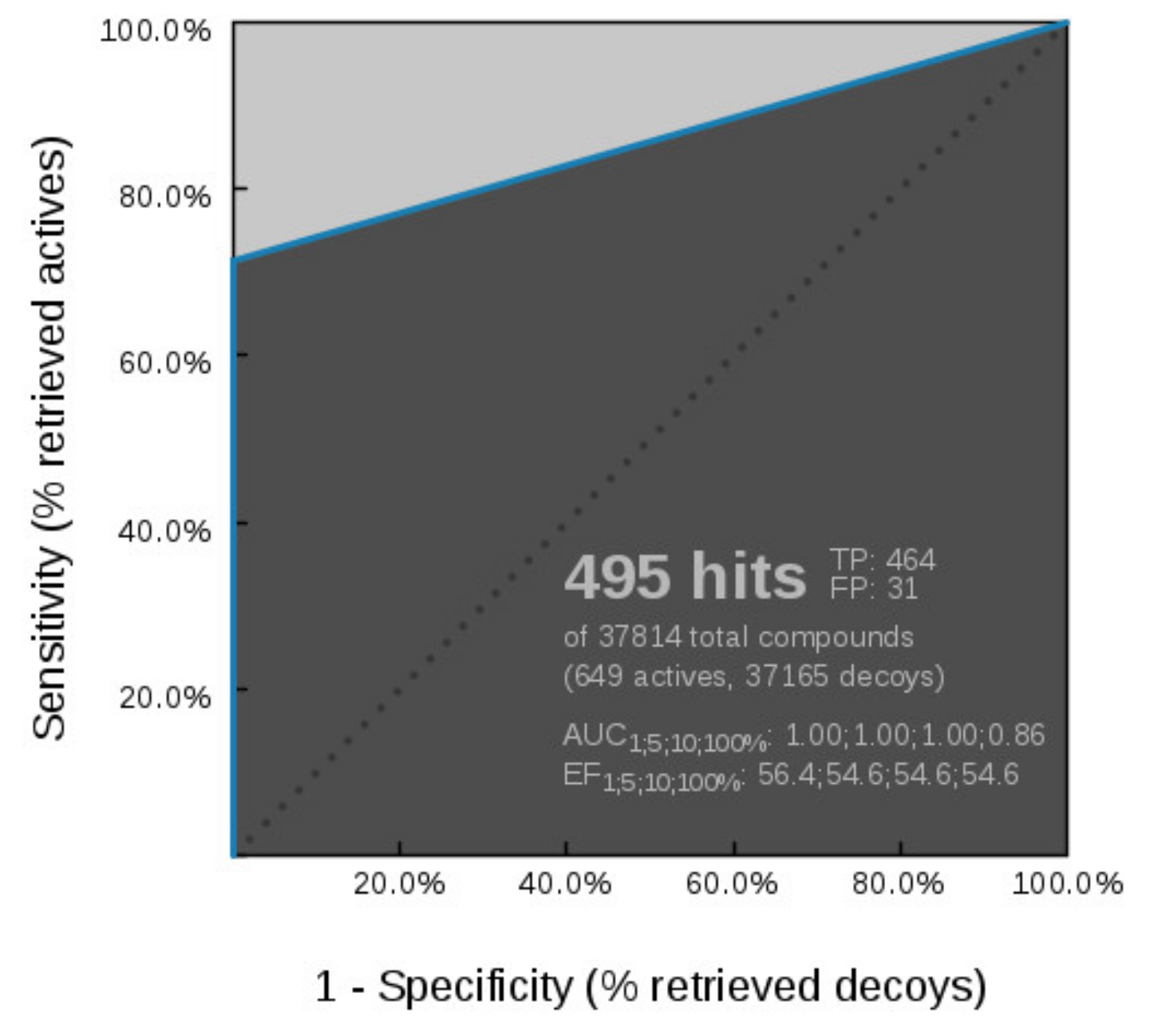
Virtual Screening of Hsp90 and TopoII Inhibitors with GyrB Pharmacophore Models
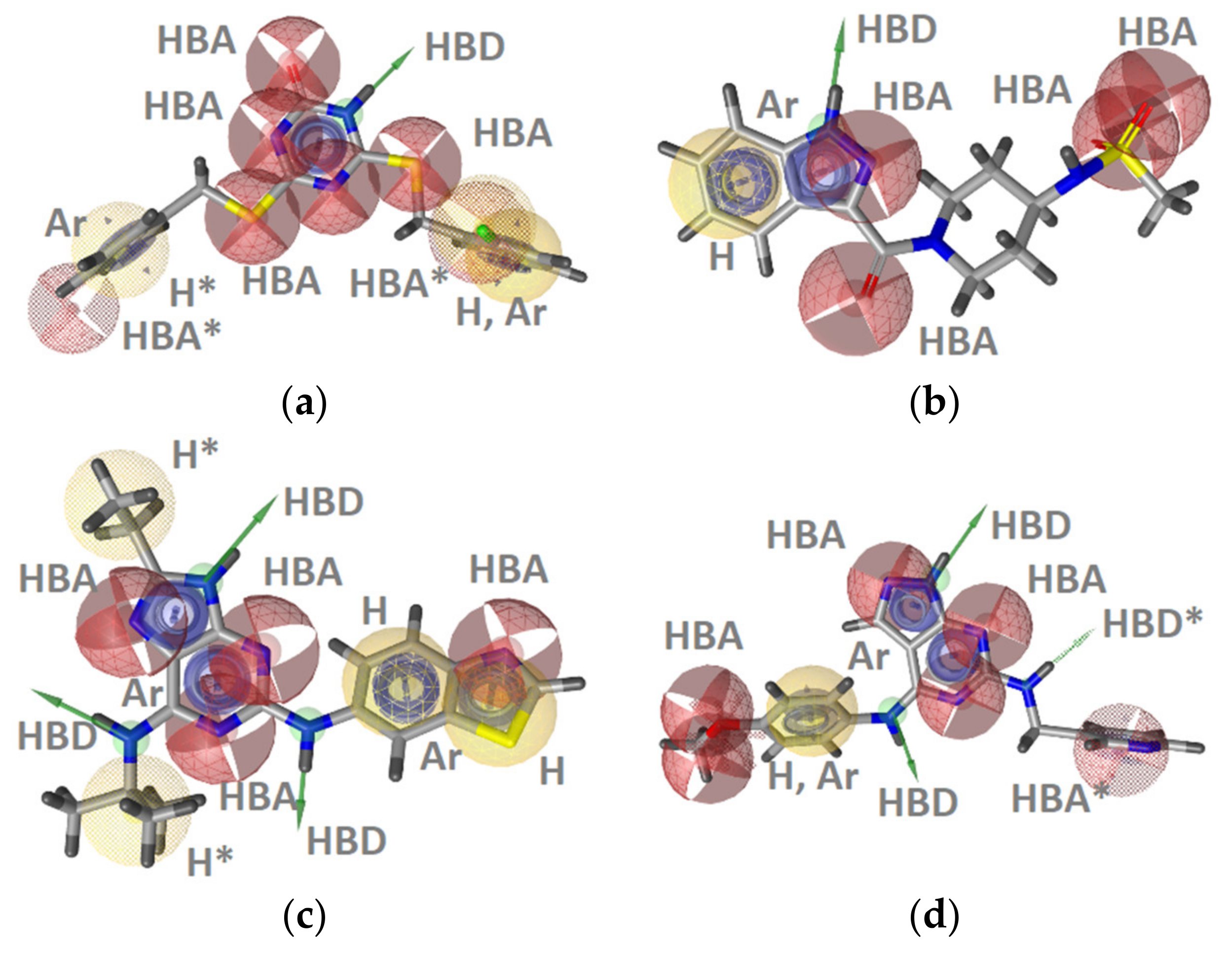
2.2.2. Heat Shock Protein 90 Alpha (Hsp90α) Inhibitors
Hsp90 Resorcinol Interaction Features
Hsp90 Dihydropyridopyrimidinone Interaction Features
Hsp90 Ligand-Based (LB) Pharmacophore Models
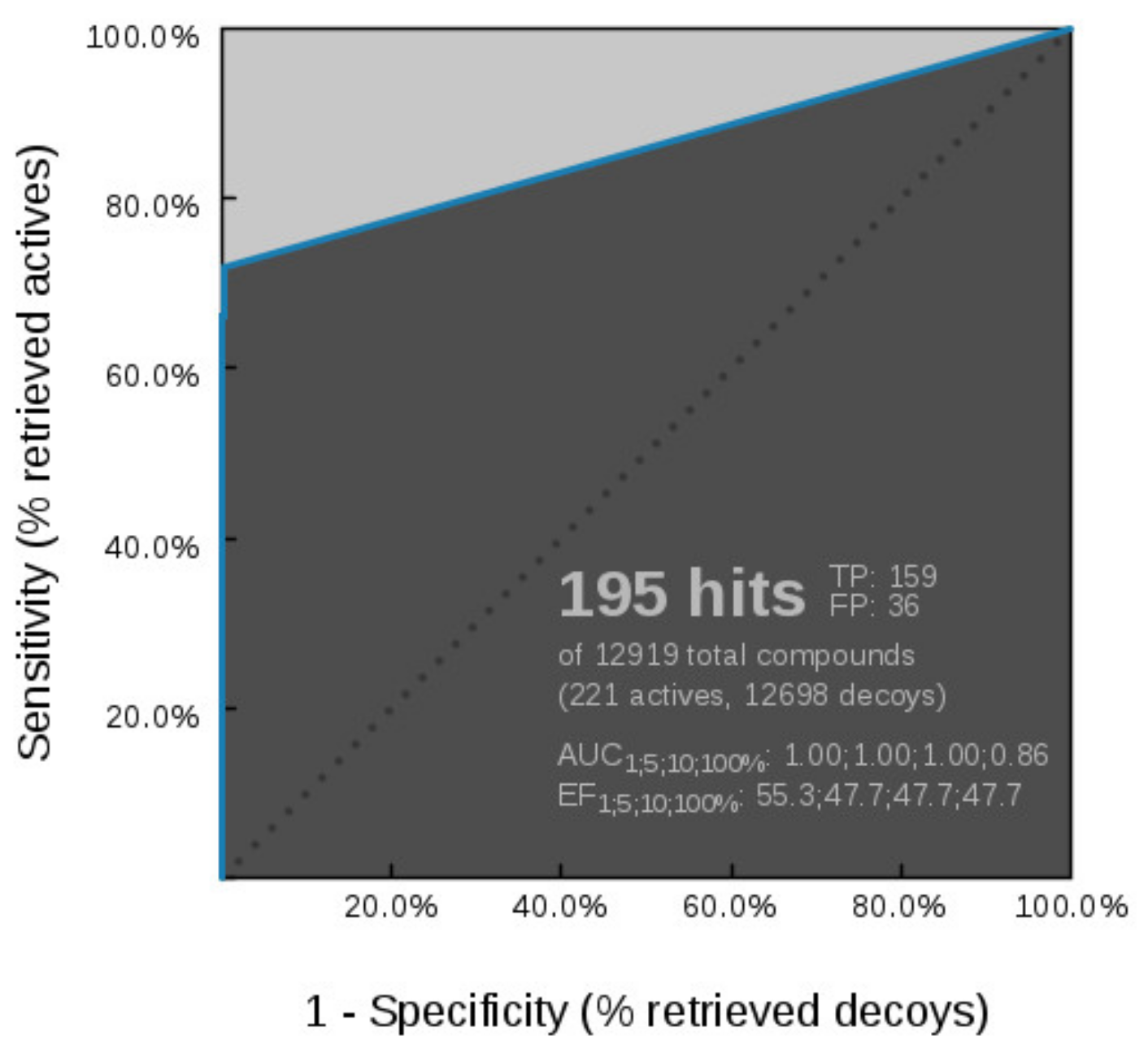
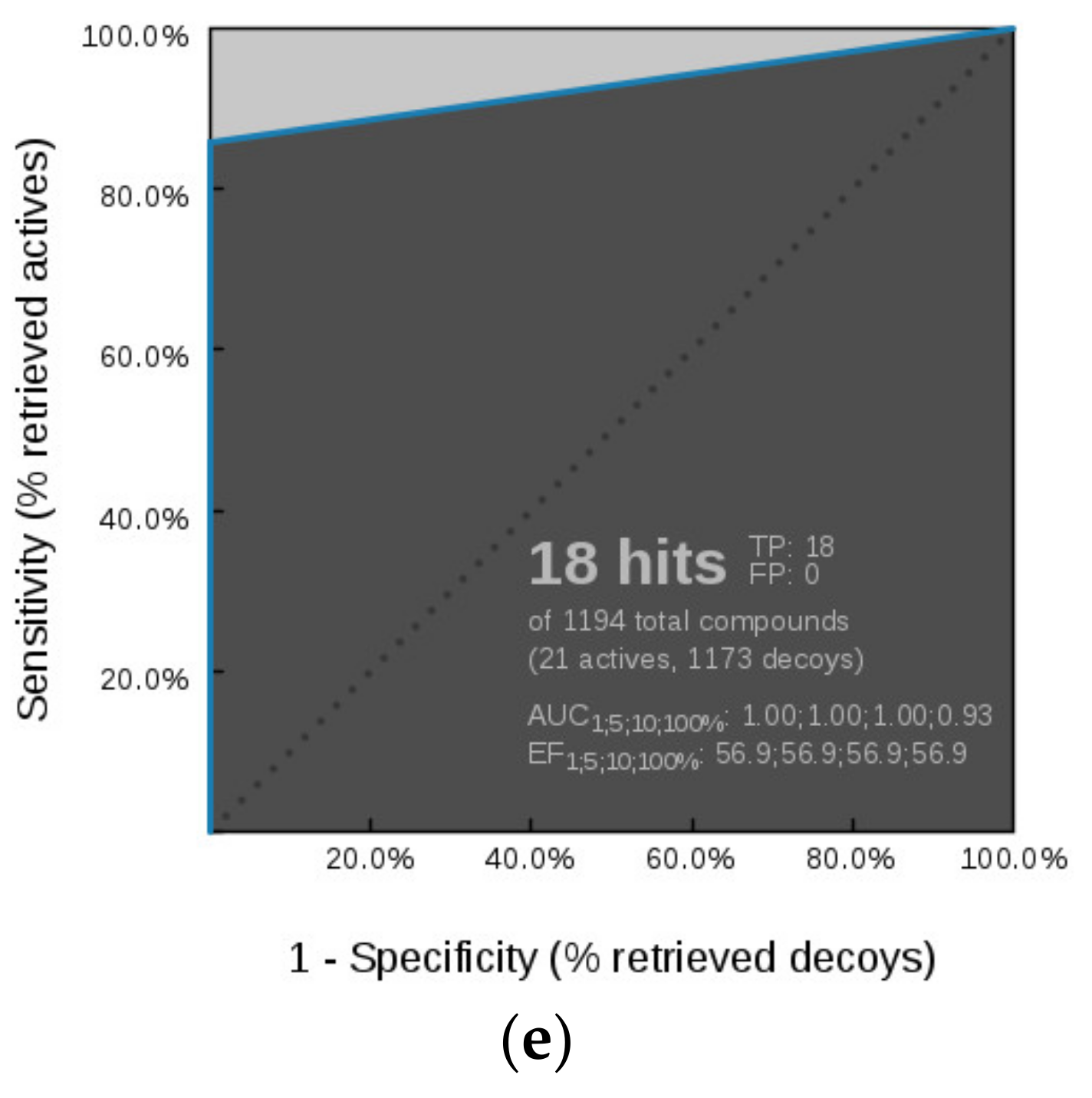
Virtual Screening of GyrB and TopoII Inhibitors with Hsp90 Pharmacophore Models
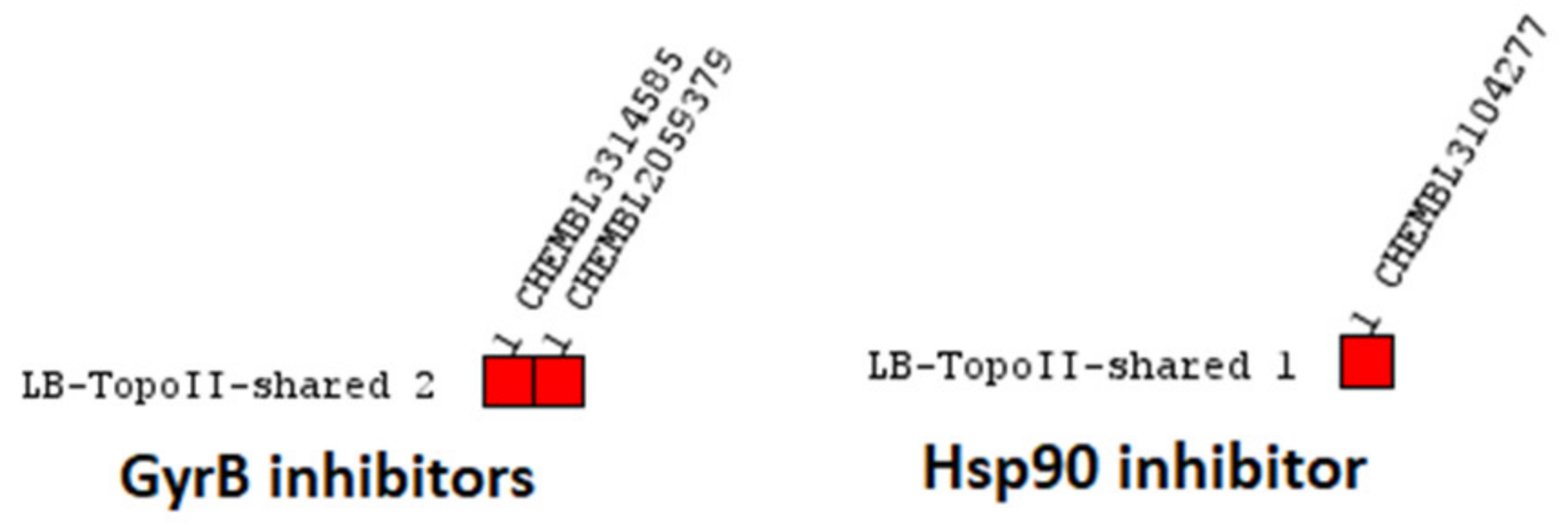
2.2.3. Human Topoisomerase IIα Inhibitors
TopoII Inhibitor Ligand-Based Pharmacophore Models
Virtual Screening of GyrB and Hsp90 Inhibitors with TopoII Pharmacophore Models
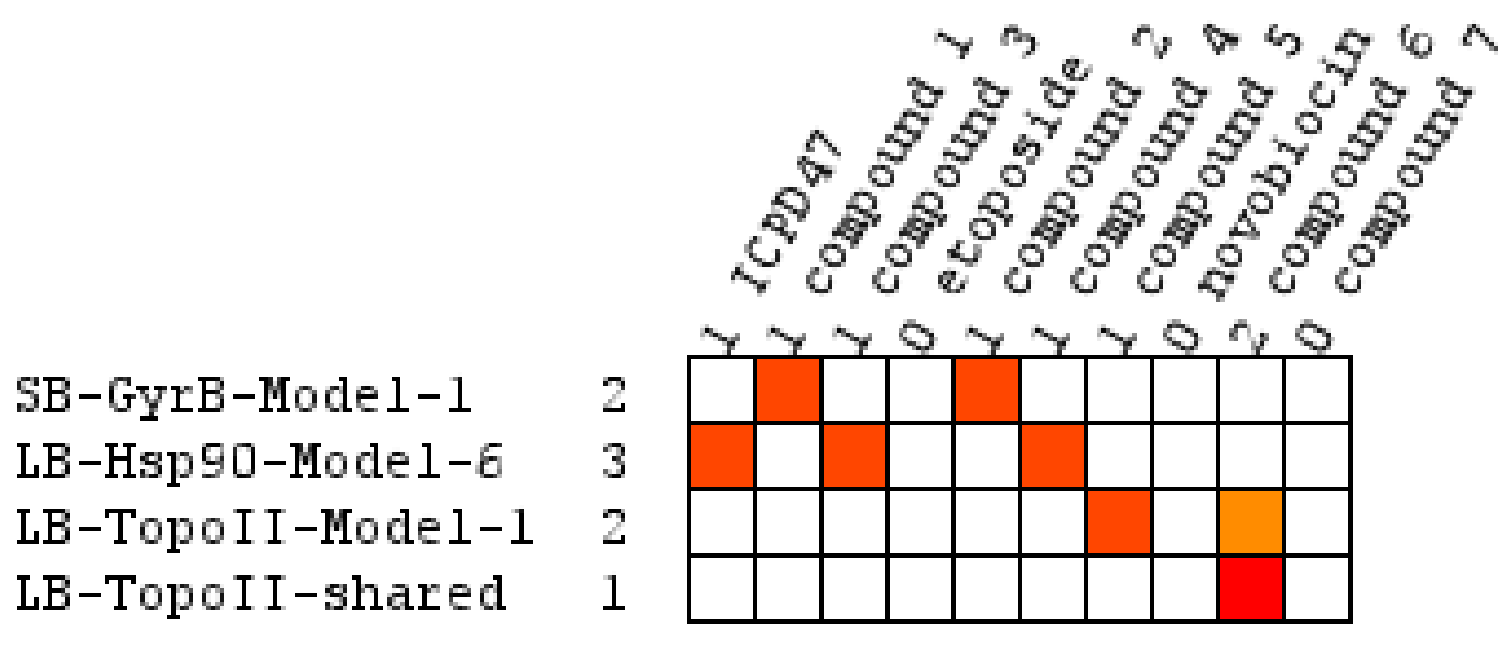
2.3. Biological Evaluation
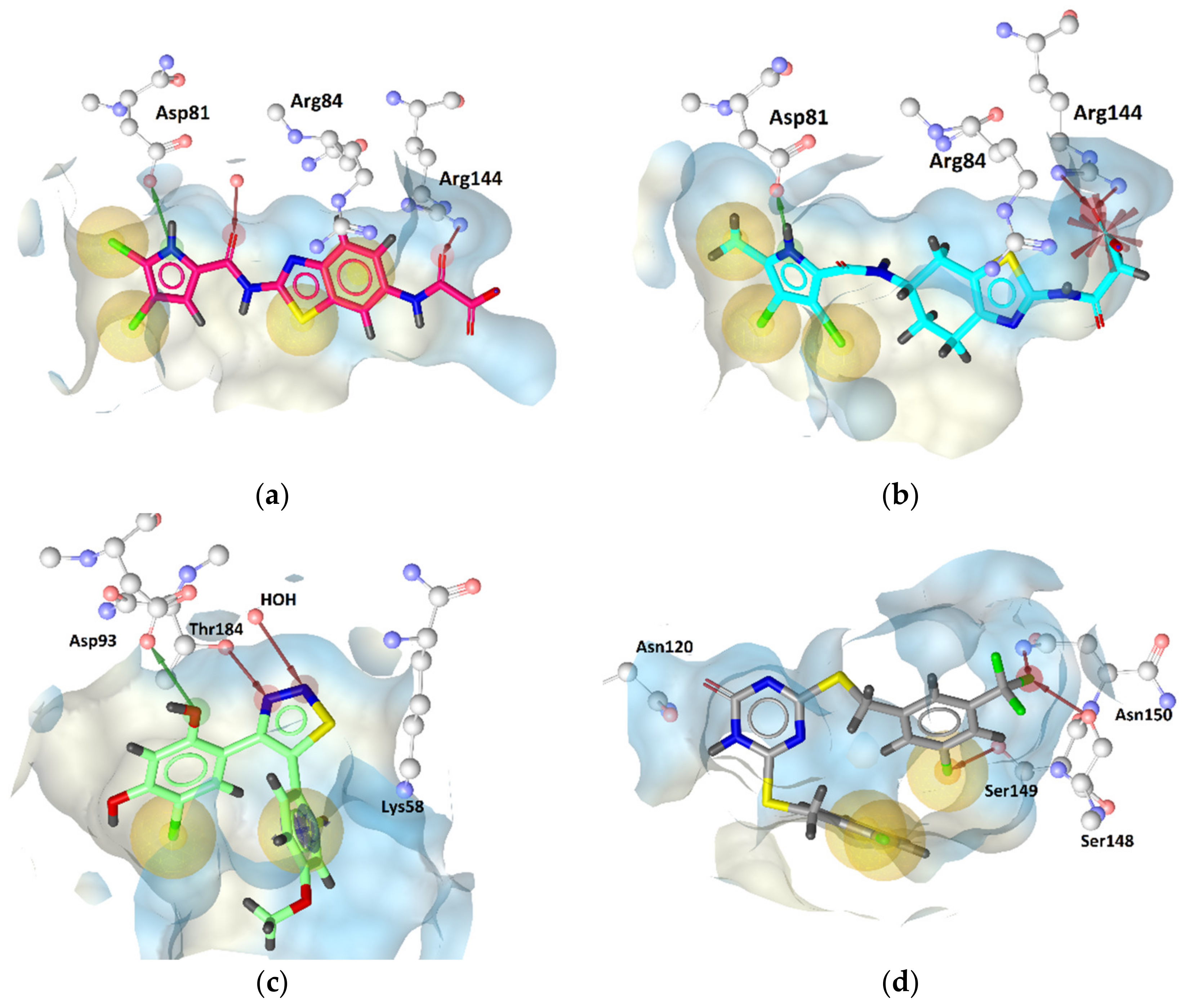
2.4. Molecular Docking
3. Materials and Methods
3.1. Enzymes and Chemical Compounds
3.2. Software
3.3. Identification of Proteins with Similar ATP-Binding Pockets
3.4. Compound Library Preparation
3.5. Structure-Based Pharmacophore Modelling
3.6. Ligand-Based Pharmacophore Modeling
3.7. Pharmacophore Model Optimization
3.8. Molecular Docking
3.9. Determination of Inhibitory Activities on E. coli DNA Gyrase
3.10. Determination of Inhibitory Activities on Human DNA Topoisomerase IIα
3.11. Determination of Binding to Hsp90
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 29 June 2021).
- Tomašič, T.; Katsamakas, S.; Hodnik, Ž.; Ilaš, J.; Brvar, M.; Solmajer, T.; Montalvão, S.; Tammela, P.; Banjanac, M.; Ergović, G.; et al. Discovery of 4,5,6,7-Tetrahydrobenzo[1,2-d]Thiazoles as Novel DNA Gyrase Inhibitors Targeting the ATP-Binding Site. J. Med. Chem. 2015, 58, 5501–5521. [Google Scholar] [CrossRef]
- Zidar, N.; Macut, H.; Tomašič, T.; Brvar, M.; Montalvão, S.; Tammela, P.; Solmajer, T.; Peterlin Mašič, L.; Ilaš, J.; Kikelj, D. N-Phenyl-4,5-Dibromopyrrolamides and N-Phenylindolamides as ATP Competitive DNA Gyrase B Inhibitors: Design, Synthesis, and Evaluation. J. Med. Chem. 2015, 58, 6179–6194. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Tomašič, T.; Barančokova, M.; Katsamakas, S.; Ilaš, J.; Tammela, P.; Peterlin Mašič, L.; Kikelj, D. Discovery of Benzothiazole Scaffold-Based DNA Gyrase B Inhibitors. J. Med. Chem. 2016, 59, 8941–8954. [Google Scholar] [CrossRef]
- Tomašič, T.; Mirt, M.; Barančoková, M.; Ilaš, J.; Zidar, N.; Tammela, P.; Kikelj, D. Design, synthesis and biological evaluation of 4,5-dibromo-N-(thiazol-2-yl)-1H-pyrrole-2-carboxamide derivatives as novel DNA gyrase inhibitors. Bioorganic Med. Chem. 2017, 25, 338–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomašič, T.; Barančoková, M.; Zidar, N.; Ilaš, J.; Tammela, P.; Kikelj, D. Design, synthesis, and biological evaluation of 1-ethyl-3-(thiazol-2-yl)urea derivatives as Escherichia coli DNA gyrase inhibitors. Arch. Pharm. 2017, 351, 1700333. [Google Scholar] [CrossRef] [Green Version]
- Durcik, M.; Lovison, D.; Skok, Ž.; Durante Cruz, C.; Tammela, P.; Tomašič, T.; Benedetto Tiz, D.; Draskovits, G.; Nyerges, Á.; Pál, C.; et al. New N-Phenylpyrrolamide DNA Gyrase B Inhibitors: Optimization of Efficacy and Antibacterial Activity. Eur. J. Med. Chem. 2018, 154, 117–132. [Google Scholar] [CrossRef]
- Lamut, A.; Cruz, C.D.; Skok, Ž.; Barančoková, M.; Zidar, N.; Zega, A.; Mašič, L.P.; Ilaš, J.; Tammela, P.; Kikelj, D.; et al. Design, synthesis and biological evaluation of novel DNA gyrase inhibitors and their siderophore mimic conjugates. Bioorganic Chem. 2019, 95, 103550. [Google Scholar] [CrossRef] [PubMed]
- Bisacchi, G.S.; Manchester, J.I. A New-Class Antibacterial—Almost. Lessons in Drug Discovery and Development: A Critical Analysis of More than 50 Years of Effort toward ATPase Inhibitors of DNA Gyrase and Topoisomerase IV. ACS Infect. Dis. 2014, 1, 4–41. [Google Scholar] [CrossRef]
- Durcik, M.; Tomašič, T.; Zidar, N.; Zega, A.; Kikelj, D.; Peterlin Mašič, L.; Ilaš, J. ATP-Competitive DNA Gyrase and Topoisomerase IV Inhibitors as Antibacterial Agents. Expert Opin. Ther. Pat. 2019, 29, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Challenges of Antibacterial Discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyerges, A.; Tomašič, T.; Durcik, M.; Revesz, T.; Szili, P.; Draskovits, G.; Bogar, F.; Skok, Ž.; Zidar, N.; Ilaš, J.; et al. Rational Design of Balanced Dual-Targeting Antibiotics with Limited Resistance. PLoS Biol. 2020, 18, e3000819. [Google Scholar] [CrossRef]
- Durcik, M.; Nyerges, Á.; Skok, Ž.; Skledar, D.G.; Trontelj, J.; Zidar, N.; Ilaš, J.; Zega, A.; Cruz, C.D.; Tammela, P.; et al. New dual ATP-competitive inhibitors of bacterial DNA gyrase and topoisomerase IV active against ESKAPE pathogens. Eur. J. Med. Chem. 2021, 213, 113200. [Google Scholar] [CrossRef]
- Kedelj, V.; Tomašić, T.; Peterlin Mašič, L.; Zega, A. ATP-Binding Site of Bacterial Enzymes as a Target for Antibacterial Drug Design. J. Med. Chem. 2011, 54, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, A.; Henning-Chubb, C.; Huberman, E. Novobiocin- and phorbol-12-myristate-13-acetate-induced differentiation of human leukemia cells associated with a reduction in topoisomerase II activity. Cancer Res. 1989, 49, 1110–1117. [Google Scholar]
- Marcu, M.G.; Chadli, A.; Bouhouche, I.; Catelli, M.; Neckers, L.M. The Heat Shock Protein 90 Antagonist Novobiocin Interacts with a Previously Unrecognized ATP-binding Domain in the Carboxyl Terminus of the Chaperone. J. Biol. Chem. 2000, 275, 37181–37186. [Google Scholar] [CrossRef] [Green Version]
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and Related Coumarins and Depletion of Heat Shock Protein 90-Dependent Signaling Proteins. J. Natl. Cancer Inst. 2000, 92, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Skok, Ž.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2019, 63, 884–904. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Blagg, B.; Haystead, T.; Trepel, J.B.; Whitesell, L.; Picard, D. Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperon 2018, 23, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; D’Annessa, I.; Moroni, E.; Morra, G.; Paladino, A.; Rinaldi, S.; Compostella, F.; Colombo, G. Allosteric Modulators of HSP90 and HSP70: Dynamics Meets Function through Structure-Based Drug Design. J. Med. Chem. 2018, 62, 60–87. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; You, Q.-D.; Xu, X.-L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2019, 63, 1798–1822. [Google Scholar] [CrossRef] [PubMed]
- Lillsunde, K.-E.; Tomašič, T.; Kikelj, D.; Tammela, P. Marine Alkaloid Oroidin Analogues with Antiviral Potential: A Novel Class of Synthetic Compounds Targeting the Cellular Chaperone Hsp90. Chem. Biol. Drug Des. 2017, 90, 1147–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillsunde, K.-E.; Tomašič, T.; Schult, P.; Lohmann, V.; Kikelj, D.; Tammela, P. Inhibition of Hepatitis C Replication by Targeting the Molecular Chaperone Hsp90: Synthesis and Biological Evaluation of 4,5,6,7-Tetrahydrobenzo[1,2-d]Thiazole Derivatives. Chem. Med. Chem. 2019, 14, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamut, A.; Gjorgjieva, M.; Naesens, L.; Liekens, S.; Lillsunde, K.-E.; Tammela, P.; Kikelj, D.; Tomašič, T. Anti-Influenza Virus Activity of Benzo[d]Thiazoles That Target Heat Shock Protein 90. Bioorg. Chem. 2020, 98, 103733. [Google Scholar] [CrossRef] [PubMed]
- Pugh, K.W.; Zhang, Z.; Wang, J.; Xu, X.; Munthali, V.; Zuo, A.; Blagg, B.S.J. From Bacteria to Cancer: A Benzothiazole-Based DNA Gyrase B Inhibitor Redesigned for Hsp90 C-Terminal Inhibition. ACS Med. Chem. Lett. 2020, 11, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Skok, Ž.; Durcik, M.; Gramec Skledar, D.; Barančoková, M.; Peterlin Mašič, L.; Tomašič, T.; Zega, A.; Kikelj, D.; Zidar, N.; Ilaš, J. Discovery of New ATP-Competitive Inhibitors of Human DNA Topoisomerase IIα through Screening of Bacterial Topoisomerase Inhibitors. Bioorg. Chem. 2020, 102, 104049. [Google Scholar] [CrossRef]
- Tomašić, T.; Peterlin Mašič, L. Prospects for Developing New Antibacterials Targeting Bacterial Type IIA Topoisomerases. Curr. Top. Med. Chem. 2014, 14, 130–151. [Google Scholar] [CrossRef]
- Tari, L.W.; Li, X.; Trzoss, M.; Bensen, D.C.; Chen, Z.; Lam, T.; Zhang, J.; Lee, S.J.; Hough, G.; Phillipson, D.; et al. Tricyclic GyrB/ParE (TriBE) Inhibitors: A New Class of Broad-Spectrum Dual-Targeting Antibacterial Agents. PLoS ONE 2013, 8, e84409. [Google Scholar] [CrossRef] [PubMed]
- Inte:Ligand. LigandScout V.4.4. Available online: https://www.inteligand.com/ligandscout (accessed on 1 July 2021).
- Bryant, S.D.; Langer, T. Data Mining Using Ligand Profiling and Target Fishing. In Data Mining in Drug Discovery; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; pp. 257–270. [Google Scholar] [CrossRef]
- Langer, T.; Bryant, S.D. In Silico Screening: Hit Finding from Database Mining. In The Practice of Medicinal Chemistry, 3rd ed.; Wermuth, C.G., Ed.; Academic Press: New York, NY, USA, 2008; pp. 210–227. [Google Scholar]
- Langer, T. Pharmacophores in Drug Research. Mol. Inform. 2010, 29, 470–475. [Google Scholar] [CrossRef]
- Karaboga, A.S.; Planesas, J.M.; Petronin, F.; Teixidó, J.; Souchet, M.; Pérez-Nueno, V.I. Highly SpecIfic and Sensitive Pharmacophore Model for Identifying CXCR4 Antagonists. Comparison with Docking and Shape-Matching Virtual Screening Performance. J. Chem. Inf. Model. 2013, 53, 1043–1056. [Google Scholar] [CrossRef]
- Brvar, M.; Perdih, A.; Oblak, M.; Mašič, L.P.; Solmajer, T. In silico discovery of 2-amino-4-(2,4-dihydroxyphenyl)thiazoles as novel inhibitors of DNA gyrase B. Bioorganic Med. Chem. Lett. 2010, 20, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Doddareddy, M.R.; Thorat, D.A.; Seo, S.H.; Hong, T.-J.; Cho, Y.S.; Hahn, J.-S.; Pae, A.N. Structure based design of heat shock protein 90 inhibitors acting as anticancer agents. Bioorganic Med. Chem. 2011, 19, 1714–1720. [Google Scholar] [CrossRef]
- Tomašič, T.; Durcik, M.; Keegan, B.M.; Skledar, D.G.; Zajec, Ž.; Blagg, B.S.J.; Bryant, S.D. Discovery of Novel Hsp90 C-Terminal Inhibitors Using 3D-Pharmacophores Derived from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2020, 21, 6898. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Janežič, D. ProBiS Algorithm for Detection of Structurally Similar Protein Binding Sites by Local Structural Alignment. Bioinformatics 2010, 26, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Janežič, D. ProBiS: A Web Server for Detection of Structurally Similar Protein Binding Sites. Nucleic Acids Res. 2010, 38, W436–W440. [Google Scholar] [CrossRef]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jupp, S.; Malone, J.; Bolleman, J.; Brandizi, M.; Davies, M.; Garcia, L.; Gaulton, A.; Gehant, S.; Laibe, C.; Redaschi, N.; et al. The EBI RDF Platform: Linked Open Data for the Life Sciences. Bioinformatics 2014, 30, 1338–1339. [Google Scholar] [CrossRef] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.S.F.F.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.; Uhalte, E.C.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2016, 45, D945–D954. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Sherer, B.A.; Hull, K.; Green, O.; Basarab, G.; Hauck, S.; Hill, P.; Loch, J.T.; Mullen, G.; Bist, S.; Bryant, J.; et al. Pyrrolamide DNA gyrase inhibitors: Optimization of antibacterial activity and efficacy. Bioorganic Med. Chem. Lett. 2011, 21, 7416–7420. [Google Scholar] [CrossRef] [PubMed]
- Seidel, T.; Bryant, S.D.; Ibis, G.; Poli, G.; Langer, T. 3D Pharmacophore Modeling Techniques in Computer-Aided Molecular Design Using LigandScout. In Tutorials in Chemoinformatics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 279–309. [Google Scholar] [CrossRef]
- Grillot, A.-L.; Le Tiran, A.; Shannon, D.; Krueger, E.; Liao, Y.; O’Dowd, H.; Tang, Q.; Ronkin, S.; Wang, T.; Waal, N.; et al. Second-Generation Antibacterial Benzimidazole Ureas: Discovery of a Preclinical Candidate with Reduced Metabolic Liability. J. Med. Chem. 2014, 57, 8792–8816. [Google Scholar] [CrossRef] [PubMed]
- McBride, C.M.; Levine, B.; Xia, Y.; Bellamacina, C.; Machajewski, T.; Gao, Z.; Renhowe, P.; Antonios-McCrea, W.; Barsanti, P.; Brinner, K.; et al. Design, Structure–Activity Relationship, and in Vivo Characterization of the Development Candidate NVP-HSP990. J. Med. Chem. 2014, 57, 9124–9129. [Google Scholar] [CrossRef]
- Lamut, A.; Skok, Z.; Barančoková, M.; Gutierrez, L.J.; Cruz, C.D.; Tammela, P.; Draskovits, G.; Szili, P.; Nyerges, A.; Pál, C.; et al. Second-generation 4,5,6,7-tetrahydrobenzo[d]thiazoles as novel DNA gyrase inhibitors. Future Med. Chem. 2020, 12, 277–297. [Google Scholar] [CrossRef]
- Cikotiene, I.; Kazlauskas, E.; Matuliene, J.; Michailoviene, V.; Torresan, J.; Jachno, J.; Matulis, D. 5-Aryl-4-(5-substituted-2,4-dihydroxyphenyl)-1,2,3-thiadiazoles as inhibitors of Hsp90 chaperone. Bioorganic Med. Chem. Lett. 2009, 19, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, E.; Petrikaitė, V.; Michailovienė, V.; Revuckienė, J.; Matulienė, J.; Grinius, L.; Matulis, D. Thermodynamics of Aryl-Dihydroxyphenyl-Thiadiazole Binding to Human Hsp90. PLoS ONE 2012, 7, e36899. [Google Scholar] [CrossRef]
- Kazlauskas, E.; Brukstus, A.; Petrikas, H.; Petrikaite, V.; Cikotiene, I.; Matulis, D. Improving the Hsp90 Inhibitors Containing 4-(2,4-Dihydroxyphenyl)-1,2,3-thiadiazole Scaffold: Synthesis, Affinity and Effect on Cancer Cells. Anticancer Agents Med. Chem. 2017, 17, 1593–1603. [Google Scholar] [CrossRef]
- Bergant, K.; Janežič, M.; Valjavec, K.; Sosič, I.; Pajk, S.; Štampar, M.; Žegura, B.; Gobec, S.; Filipič, M.; Perdih, A. Structure-Guided Optimization of 4,6-Substituted-1,3,5-triazin-2(1H)-ones as Catalytic Inhibitors of Human DNA Topoisomerase IIα. Eur. J. Med. Chem. 2019, 175, 330–348. [Google Scholar] [CrossRef] [PubMed]
- Dudutienė, V.; Matulienė, J.; Smirnov, A.; Timm, D.D.; Zubrienė, A.; Baranauskienė, L.; Morkūnaitė, V.; Smirnovienė, J.; Michailovienė, V.; Juozapaitienė, V.; et al. Discovery and Characterization of Novel Selective Inhibitors of Carbonic Anhydrase IX. J. Med. Chem. 2014, 57, 9435–9446. [Google Scholar] [CrossRef]
- Zubrienė, A.; Gutkowska, M.; Matulienė, J.; Chaleckis, R.; Michailovienė, V.; Voroncova, A.; Venclovas, C.; Zylicz, A.; Zylicz, M.; Matulis, D. Thermodynamics of radicicol binding to human Hsp90 alpha and beta isoforms. Biophys. Chem. 2010, 152, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2004, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, N.-O.; de Bruyn Kops, C.; Flachsenberg, F.; Sommer, K.; Rarey, M.; Kirchmair, J. Benchmarking Commercial Conformer Ensemble Generators. J. Chem. Inf. Model. 2017, 57, 2719–2728. [Google Scholar] [CrossRef]
- Poli, G.; Seidel, T.; Langer, T. Conformational Sampling of Small Molecules With iCon: Performance Assessment in Comparison With OMEGA. Front. Chem. 2018, 6, 229. [Google Scholar] [CrossRef] [PubMed]
- Sieb, C.; Meinl, T.; Berthold, M.R. Parallel and Distributed Data Pipelining with KNIME. Mediterr. J. Comput. Net. 2007, 3, 43–51. [Google Scholar]
- Inte:Ligand GmbH. Inte:Ligand Expert KNIME Extensions. Available online: http://www.inteligand.com/knime-nodes (accessed on 1 July 2021).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Petrauskas, V.; Zubrienė, A.; Todd, M.J.; Matulis, D. Inhibitor Binding to Carbonic Anhydrases by Fluorescent Thermal Shift Assay. In Carbonic Anhydrase as Drug Target: Thermodynamics and Structure of Inhibitor Binding; Matulis, D., Ed.; Springer International Publishing: Basel, Switzerland, 2019; pp. 63–78. [Google Scholar]
- Linkuvienė, V.; Zubrienė, A.; Manakova, E.; Petrauskas, V.; Baranauskienė, L.; Zakšauskas, A.; Smirnov, A.; Gražulis, S.; Ladbury, J.E.; Matulis, D. Thermodynamic, Kinetic, and Structural Parameterization of Human Carbonic Anhydrase Interactions toward Enhanced Inhibitor Design. Q. Rev. Biophys. 2018, 51, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrauskas, V.; Baranauskienė, L.; Zubrienė, A.; Matulis, D. Isothermal Titration Calorimetry and Fluorescent Thermal and Pressure Shift Assays in Protein–Ligand Interactions. In Biocalorimetry; Bastos, M., Ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 261–280. [Google Scholar]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Human Hsp90α NTD Kd [μM] | Human TopoII IC50 [μM] | E. coli DNA gyrase IC50 [μM] |
|---|---|---|---|---|
| ICPD47 | 0.0125 [49,50] | / | / | |
| novobiocin | / | / | 0.17 | |
| etoposide | / | 71 | / | |
| 1 |  | >100 | >100 | 0.087 [4] |
| 2 |  | >100 | >100 | 0.020 [48] |
| 3 |  | 0.026 [50] | >100 | >100 |
| 4 |  | 0.0066 [51] | >100 | >100 |
| 5 |  | >100 | 8.1 [52] | >100 |
| 6 |  | >100 | 8.4 [52] | >100 |
| 7 [53] |  | >100 | >100 | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomašič, T.; Zubrienė, A.; Skok, Ž.; Martini, R.; Pajk, S.; Sosič, I.; Ilaš, J.; Matulis, D.; Bryant, S.D. Selective DNA Gyrase Inhibitors: Multi-Target in Silico Profiling with 3D-Pharmacophores. Pharmaceuticals 2021, 14, 789. https://doi.org/10.3390/ph14080789
Tomašič T, Zubrienė A, Skok Ž, Martini R, Pajk S, Sosič I, Ilaš J, Matulis D, Bryant SD. Selective DNA Gyrase Inhibitors: Multi-Target in Silico Profiling with 3D-Pharmacophores. Pharmaceuticals. 2021; 14(8):789. https://doi.org/10.3390/ph14080789
Chicago/Turabian StyleTomašič, Tihomir, Asta Zubrienė, Žiga Skok, Riccardo Martini, Stane Pajk, Izidor Sosič, Janez Ilaš, Daumantas Matulis, and Sharon D. Bryant. 2021. "Selective DNA Gyrase Inhibitors: Multi-Target in Silico Profiling with 3D-Pharmacophores" Pharmaceuticals 14, no. 8: 789. https://doi.org/10.3390/ph14080789