
2-Phenoxy-3-Trichloromethylquinoxalines Are Antiplasmodial Derivatives with Activity against the Apicoplast of Plasmodium falciparum

 , , , , , , ,
, , , , , , ,
Abstract
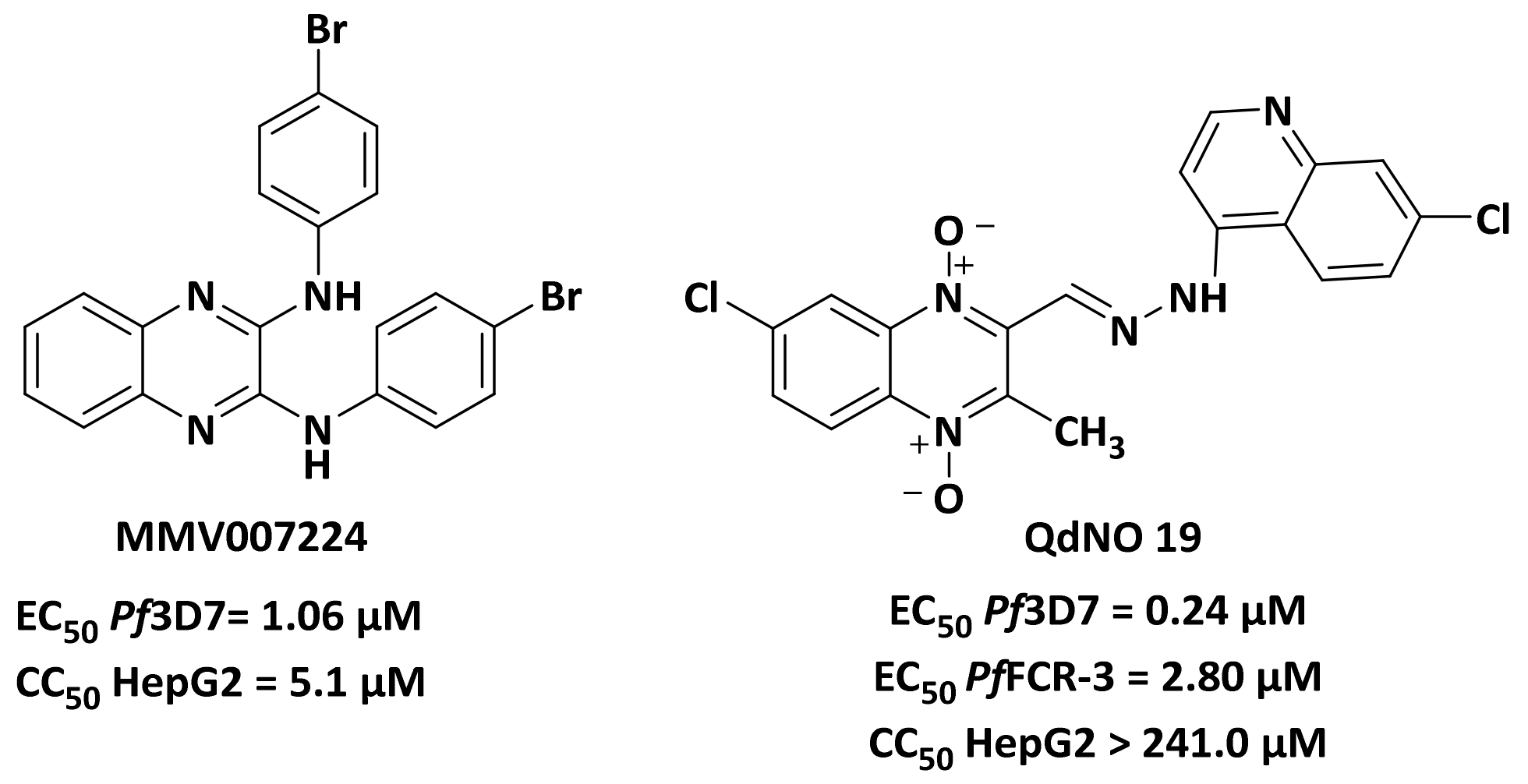
:1. Introduction
2. Results and Discussion
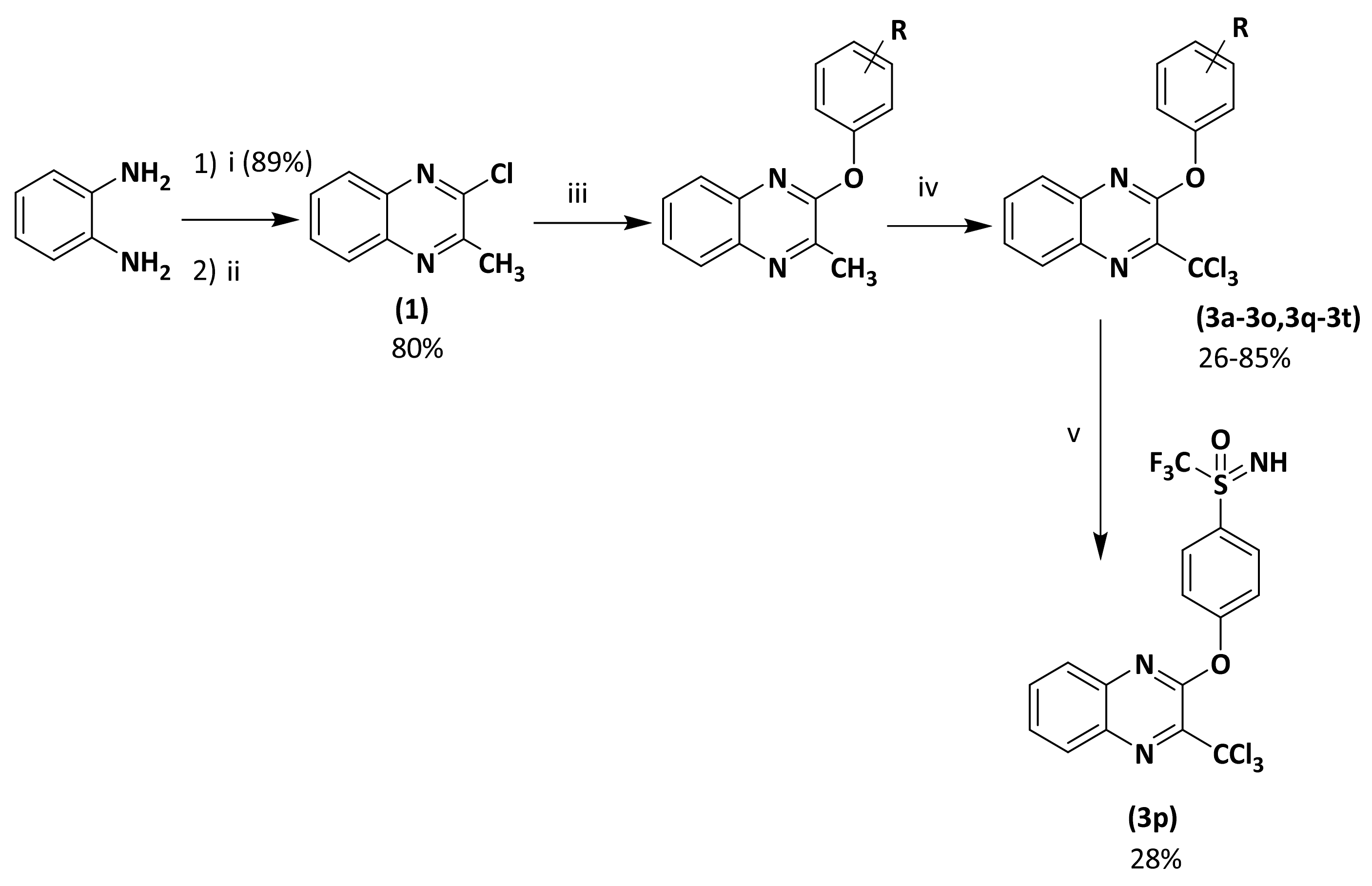
2.1. Synthesis
2.2. Biological Results
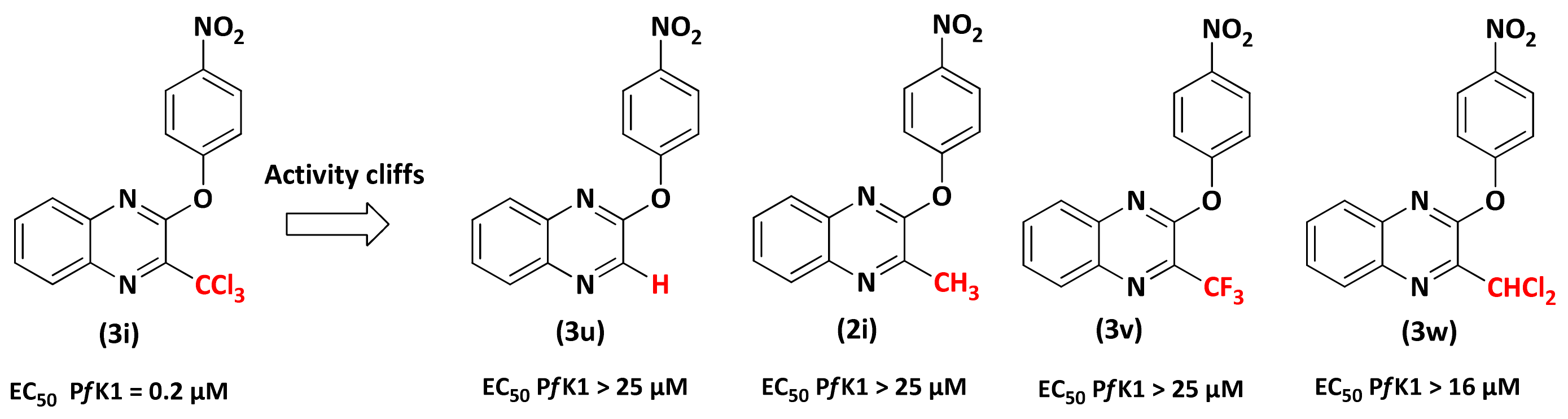
2.2.1. Structure-Activity Relationship (SAR) Study
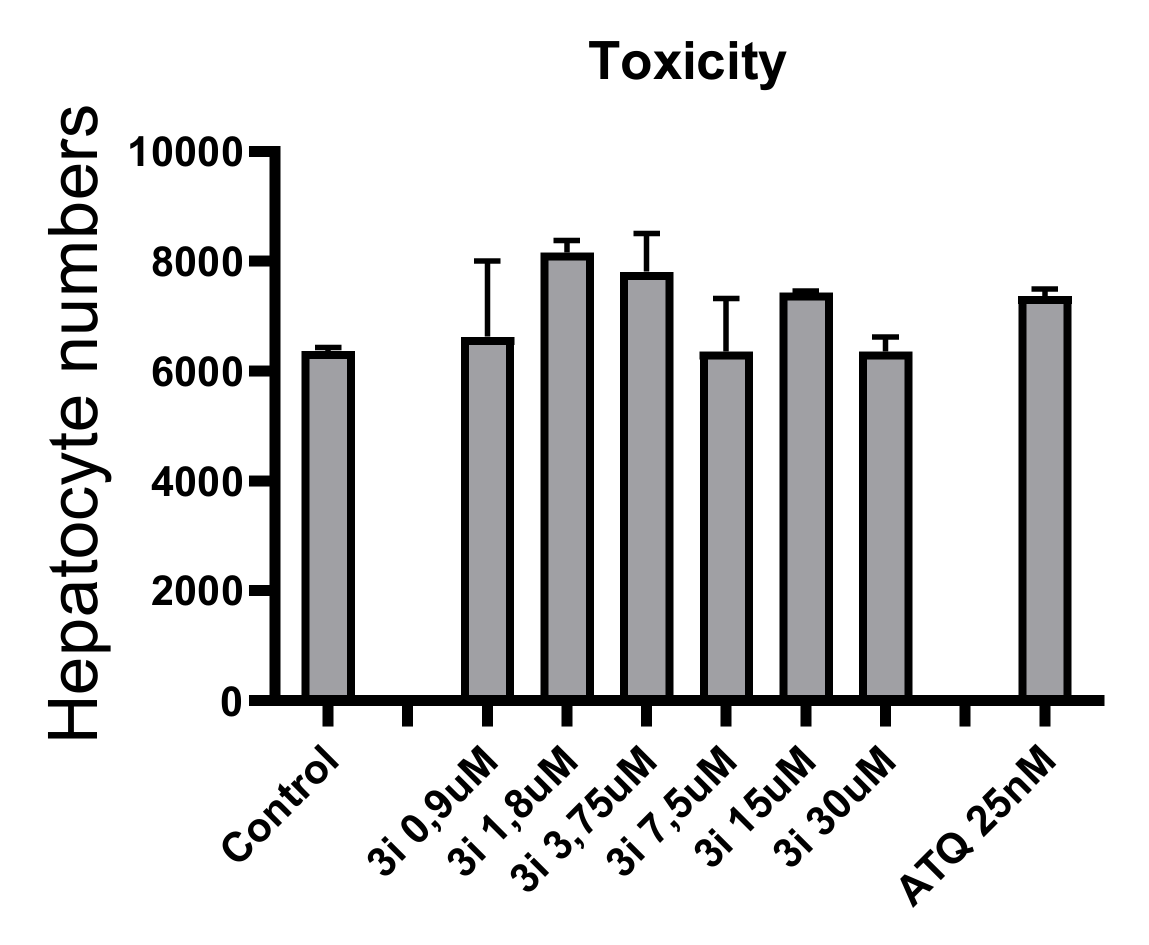
2.2.2. In Vitro Toxicity Data
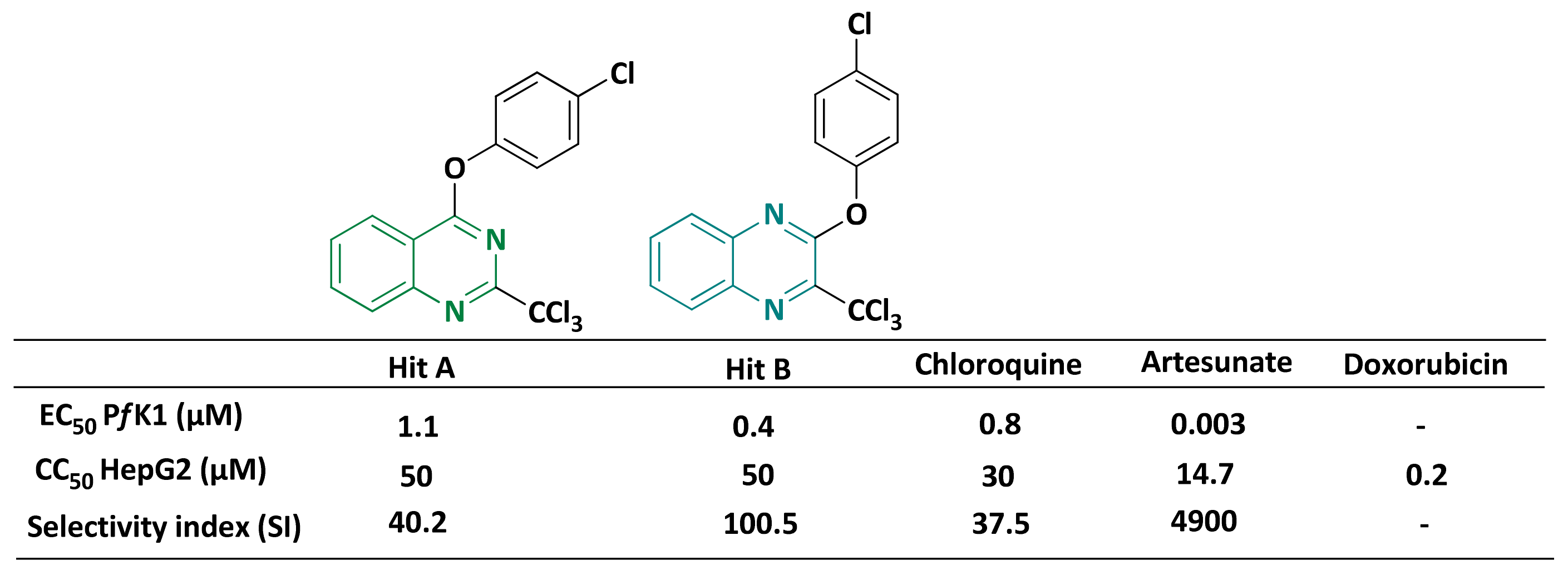
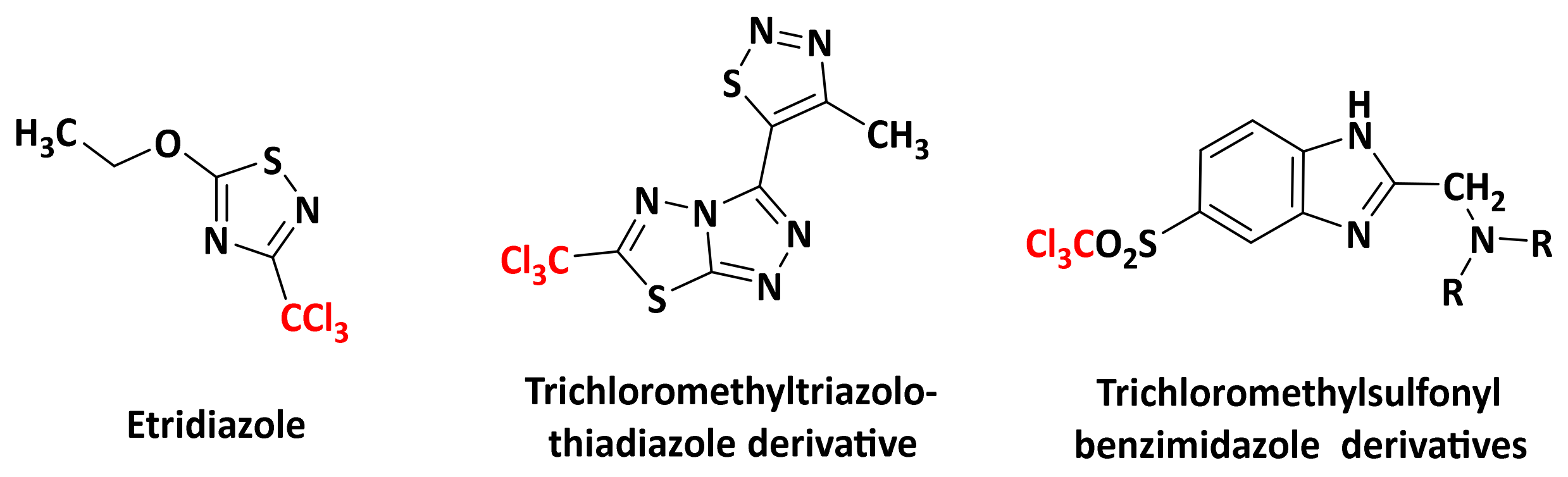
2.2.3. Role of the 3-CCl3 Group
2.2.4. Evaluation on P. falciparum Apicoplast
2.2.5. Evaluation on Artemisinin-Resistant and Artemisinin-Sensitive Parasites
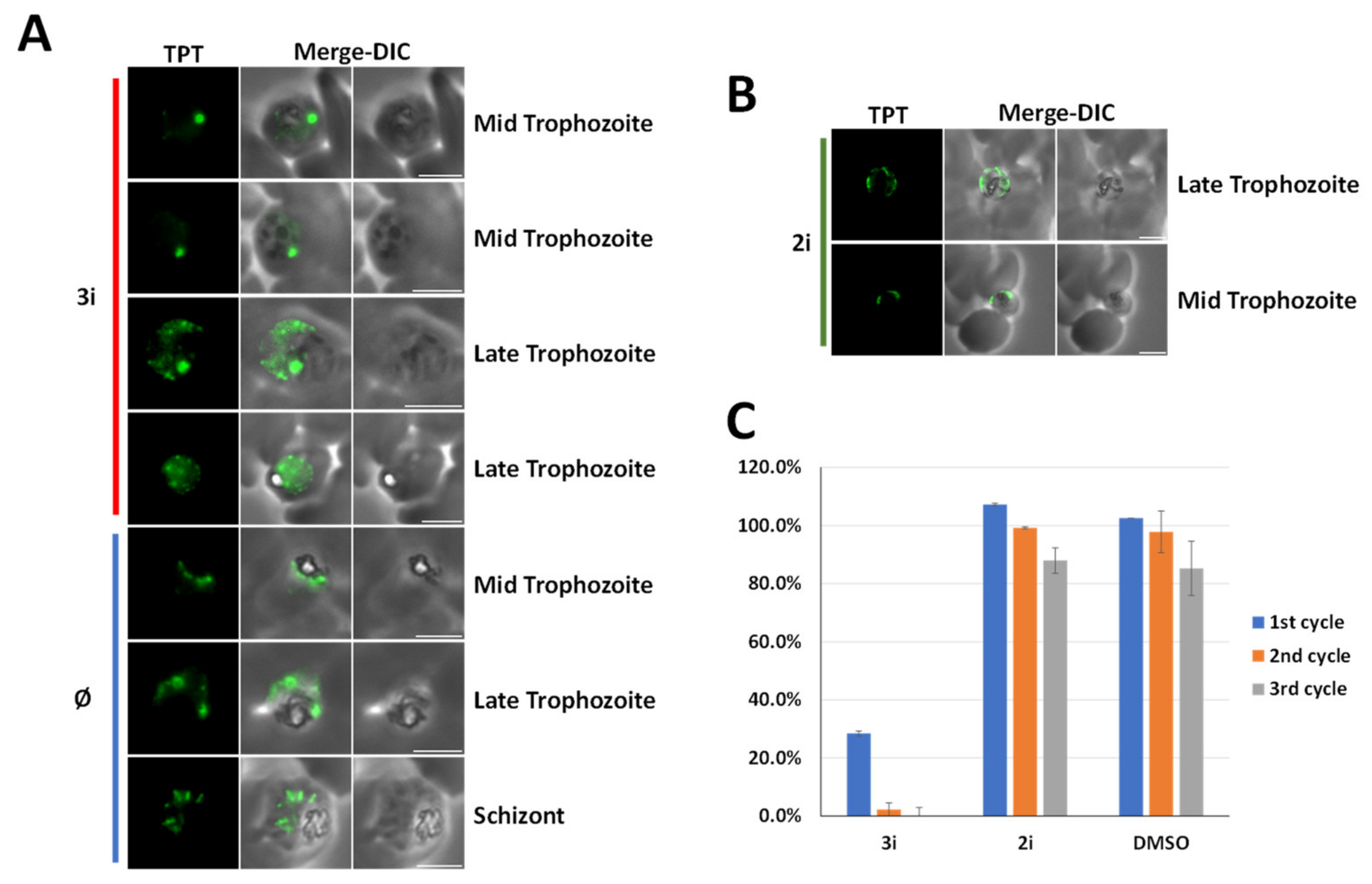
2.2.6. Studying the Effect of (3i) on the Liver Stage Development of P. falciparum
3. Materials and Methods
3.1. Chemistry
3.1.1. Generality
3.1.2. General Procedure for 2-chloro-3-methyl Substituted Quinoxaline (2a–2p)
2-Methyl-3-phenoxyquinoxaline (2a)
2-(2-Chlorophenoxy)-3-methylquinoxaline (2b)
2-(3-Chlorophenoxy)-3-methylquinoxaline (2c)
2-(2,4-Dichlorophenoxy)-3-methylquinoxaline (2d)
2-(2-Fluorophenoxy)-3-methylquinoxaline (2e)
2-(3-Fluorophenoxy)-3-methylquinoxaline (2f)
2-(4-Fluorophenoxy)-3-methylquinoxaline (2g)
2-(4-Methoxyphenoxy)-3-methylquinoxaline (2h)
2-Methyl-3-(4-nitrophenoxy)quinoxaline (2i)
2-Methyl-3-(4-trifluoromethylphenoxy)quinoxaline (2j)
2-Methyl-3-(4-trifluoromethoxyphenoxy)quinoxaline (2k)
2-Methyl-3-[4-(pentafluorosulfanyl)phenoxy]quinoxaline (2l)
2-Methyl-3-[4-(trifluoromethylthio)phenoxy]quinoxaline (2m)
4-[(3-Methylquinoxalin-2-yl)oxy]benzonitrile (2n)
2-Methyl-3-[4-(trifluoromethylsulfonyl)phenoxy]quinoxaline (2o)
2-Methyl-3-(naphthalen-1-yloxy)quinoxaline (2q)
2-Methyl-3-(naphthalen-2-yloxy)quinoxaline (2r)
2-([1,1′-Biphenyl]-4-yloxy)-3-methylquinoxaline (2s)
2-([1,1′-Biphenyl]-3-yloxy)-3-methylquinoxaline (2t)
3.1.3. General Procedure for Preparation of 3-thiophenoxy-2-trichloromethylquinoxaline Derivatives
2-Phenoxy-3-trichloromethylquinoxaline (3a)
2-(2-Chlorophenoxy)-3-trichloromethylquinoxaline (3b)
2-(3-Chlorophenoxy)-3-(richloromethylquinoxaline (3c)
2-(2,4-Dichlorophenoxy)-3-trichloromethylquinoxaline (3d)
2-(2-Fluorophenoxy)-3-trichloromethylquinoxaline (3e)
2-(3-Fluorophenoxy)-3-trichloromethylquinoxaline (3f)
2-(4-Fluorophenoxy)-3-trichloromethylquinoxaline (3g)
2-(4-Methoxyphenoxy)-3-trichloromethylquinoxaline (3h)
2-(4-Nitrophenoxy)-3-trichloromethylquinoxaline (3i)
2-Trichloromethyl-3-[4-(trifluoromethyl)phenoxy]quinoxaline (3j)
2-Trichloromethyl-3-[4-(trifluoromethoxy)phenoxy]quinoxaline (3k)
2-Trichloromethyl-3-[4-(pentafluorosulfanyl)phenoxy]quinoxaline (3l)
2-Trichloromethyl-3-[4-(trifluoromethylthio)phenoxy]quinoxaline (3m)
4-[(3-Trichloromethylquinoxalin-2-yl)oxy]benzonitrile (3n)
2-Trichloromethyl-3-[4-(trifluoromethylsulfonyl)phenoxy]quinoxaline (3o)
2-(Naphthalen-2-yloxy)-3-trichloromethylquinoxaline (3q)
2-(Naphthalen-1-yloxy)-3-trichloromethylquinoxaline (3r)
2-([1,1′-Biphenyl]-4-yloxy)-3-trichloromethylquinoxaline (3s)
2-([1,1′-Biphenyl]-3-yloxy)-3-trichloromethylquinoxaline (3t)
2-Trichloromethyl-3-[4-(S-trifluoromethylsulfonimidoyl)phenoxy]quinoxaline (3p)
3.1.4. General Procedure for Compounds 3u–3v
Preparation of 2-chloro-3-trifluoromethylquinoxaline
Preparation of compounds (3u, 3v)
2-(4-Nitrophenoxy)quinoxaline (3u)
2-(4-Nitrophenoxy)-3-trifluoromethylquinoxaline (3v)
2-Dichloromethyl-3-(4-nitrophenoxy)quinoxaline (3w)
3.2. Biology
3.2.1. In Vitro Cytotoxicity Evaluation HepG2
3.2.2. In Vitro Antiplasmodial Evaluation
3.2.3. Ames Test
3.2.4. Comet Assay
Cell Culture and Treatment
Comet Assay
Statistics
3.2.5. Evaluation against ART-Resistant Strain
Parasite Culture
Standard In Vitro Chemo-Sensitivity Assay
Recrudescence Assay
Quiescent-Stage Survival Assay
3.2.6. Apicoplast Studies
Culturing Plasmodium-Infected Red Blood Cells
IFA on Treated Parasites
Growth Assay
3.2.7. Study on the Liver Stage
P. falciparum Sporozoite Isolation
Hepatocyte Culture and In Vitro Infection with Plasmodium Sporozoites
Immunostaining and Confocal Microscopy of Infected Hepatocyte Cultures
Quantification of Parasite Size and Numbers in Treated Versus Control Wells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO), World Malaria Report 2020. Available online: https://www.who.int/publications-detail-redirect/world-malaria-report-2020 (accessed on 6 April 2021).
- Talman, A.M.; Clain, J.; Duval, R.; Ménard, R.; Ariey, F. Artemisinin Bioactivity and Resistance in Malaria Parasites. Trends Parasitol. 2019, 35, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Menard, D.; Dondorp, A. Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harb. Perspect. Med. 2017, 7, a025619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, P.J. Has Artemisinin Resistance Emerged in Africa? Lancet Infect. Dis. 2021. [Google Scholar] [CrossRef]
- MMV-Supported Projects|Medicines for Malaria Venture. Available online: https://www.mmv.org/research-development/mmv-supported-projects (accessed on 6 April 2021).
- Verhaeghe, P.; Rathelot, P.; Rault, S.; Vanelle, P. Convenient Preparation of Original Vinylic Chlorides with Antiparasitic Potential in Quinoline Series. Lett. Org. Chem. 2006, 3, 891–897. [Google Scholar] [CrossRef]
- Primas, N.; Verhaeghe, P.; Cohen, A.; Kieffer, C.; Dumètre, A.; Hutter, S.; Rault, S.; Rathelot, P.; Azas, N.; Vanelle, P. A New Synthetic Route to Original Sulfonamide Derivatives in 2-Trichloromethylquinazoline Series: A Structure-Activity Relationship Study of Antiplasmodial Activity. Molecules 2012, 17, 8105–8117. [Google Scholar] [CrossRef] [PubMed]
- Gellis, A.; Kieffer, C.; Primas, N.; Lanzada, G.; Giorgi, M.; Verhaeghe, P.; Vanelle, P. A New DMAP-Catalyzed and Microwave-Assisted Approach for Introducing Heteroarylamino Substituents at Position 4 of the Quinazoline Ring. Tetrahedron 2014, 78, 8257–8266. [Google Scholar] [CrossRef] [Green Version]
- Castera-Ducros, C.; Azas, N.; Verhaeghe, P.; Hutter, S.; Garrigue, P.; Dumètre, A.; Mbatchi, L.; Laget, M.; Remusat, V.; Sifredi, F.; et al. Targeting the Human Malaria Parasite Plasmodium Falciparum: In Vitro Identification of a New Antiplasmodial Hit in 4-Phenoxy-2-Trichloromethylquinazoline Series. Eur. J. Med. Chem. 2011, 46, 4184–4191. [Google Scholar] [CrossRef]
- De Souza, G.E.; Bueno, R.V.; de Souza, J.O.; Zanini, C.L.; Cruz, F.C.; Oliva, G.; Guido, R.V.C.; Aguiar, A.C.C. Antiplasmodial Profile of Selected Compounds from Malaria Box: In Vitro Evaluation, Speed of Action and Drug Combination Studies. Malar. J. 2019, 18, 447. [Google Scholar] [CrossRef] [Green Version]
- Quiliano, M.; Pabón, A.; Ramirez-Calderon, G.; Barea, C.; Deharo, E.; Galiano, S.; Aldana, I. New Hydrazine and Hydrazide Quinoxaline 1,4-Di-N-Oxide Derivatives: In Silico ADMET, Antiplasmodial and Antileishmanial Activity. Bioorg. Med. Chem. Lett. 2017, 27, 1820–1825. [Google Scholar] [CrossRef]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; Van Huijsduijnen, R.H.; Kaszubska, W.; Macintyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New Developments in Anti-Malarial Target Candidate and Product Profiles. Malar. J. 2017, 16, 26. [Google Scholar] [CrossRef] [Green Version]
- McFadden, G.I.; Yeh, E. The Apicoplast: Now You See It, Now You Don’t. Int. J. Parasitol. 2017, 47, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Weatherby, K.; Carter, D. Chromera velia: The Missing Link in the Evolution of Parasitism. Adv. Appl. Microbiol. 2013, 85, 119–144. [Google Scholar] [CrossRef] [PubMed]
- Janouskovec, J.; Horák, A.; Oborník, M.; Lukes, J.; Keeling, P.J. A Common Red Algal Origin of the Apicomplexan, Dinoflagellate, and Heterokont Plastids. Proc. Natl. Acad. Sci. USA 2010, 107, 10949–10954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Sadhukhan, G.C. Anti-Malarial Drug Design by Targeting Apicoplasts: New Perspectives. J. Pharmacopunct. 2016, 19, 7–15. [Google Scholar] [CrossRef]
- Botté, C.Y.; Dubar, F.; McFadden, G.I.; Maréchal, E.; Biot, C. Plasmodium falciparum Apicoplast Drugs: Targets or off-Targets? Chem. Rev. 2012, 112, 1269–1283. [Google Scholar] [CrossRef]
- Corral, M.G.; Leroux, J.; Stubbs, K.A.; Mylne, J.S. Herbicidal Properties of Antimalarial Drugs. Sci. Rep. 2017, 7, 45871. [Google Scholar] [CrossRef] [Green Version]
- Saïdani, N.; Botté, C.Y.; Deligny, M.; Bonneau, A.-L.; Reader, J.; Lasselin, R.; Merer, G.; Niepceron, A.; Brossier, F.; Cintrat, J.-C.; et al. Discovery of Compounds Blocking the Proliferation of Toxoplasma Gondii and Plasmodium Falciparum in a Chemical Space Based on Piperidinyl-Benzimidazolone Analogs. Antimicrob. Agents Chemother. 2014, 58, 2586–2597. [Google Scholar] [CrossRef] [Green Version]
- Baschong, W.; Wittlin, S.; Inglis, K.A.; Fairlamb, A.H.; Croft, S.L.; Kumar, T.R.S.; Fidock, D.A.; Brun, R. Triclosan Is Minimally Effective in Rodent Malaria Models. Nat. Med. 2011, 17, 33–34. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, R.R.; Howell, C.D. Toxic Effects of a Fungicide, 5-Ethoxy-3-(Trichloromethyl)-1,2,4-thiadiazole (Terrazole), on the Hepatic Drug Metabolizing Enzyme System in Mice. Bull. Environ. Contam. Toxicol. 1977, 17, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Fan, S.; Fan, Z.; Wang, H.; Zhang, N.; Guo, X.; Yang, D.; Wu, Q.; Yu, B.; Zhou, S. Discovery of Pyruvate Kinase as a Novel Target of New Fungicide Candidate 3-(4-Methyl-1,2,3-Thiadiazolyl)-6-Trichloromethyl-[1,2,4]-Triazolo-[3,4- b][1,3,4]-Thiadizole. J. Agric. Food. Chem. 2018, 66, 12439–12452. [Google Scholar] [CrossRef]
- Ochal, Z.; Ochal, I.; Banach, Ł. Transformations of Trichloromethyl-4-Chlorophenyl Sulfone into New Compounds with Potential Pesticidal Activity. Pol. J. Appl. Chem. 2010, 54, 49–61. [Google Scholar]
- Verhaeghe, P.; Rathelot, P.; Gellis, A.; Vanelle, P. Highly Efficient Microwave Assisted α-Trichlorination Reaction of a -Methylated Nitrogen Containing Heterocycles. Tetrahedron 2006, 62, 8173–8176. [Google Scholar] [CrossRef]
- Strauss, M.J. The Nitroaromatic Group in Drug Design. Pharmacology and Toxicology (for Nonpharmacologists). Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 158–166. [Google Scholar] [CrossRef]
- McCann, J.; Spingarn, N.E.; Kobori, J.; Ames, B.N. Detection of carcinogens as mutagens: Bacterial tester strains with R factor plasmids. Proc. Natl. Acad. Sci. USA 1975, 72, 979–983. [Google Scholar] [CrossRef] [Green Version]
- Primas, N.; Suzanne, P.; Verhaeghe, P.; Cohen, A.; Broggi, J.; Lancelot, J.; Kieffer, C.; Vanelle, P.; Azas, N. Synthesis and in Vitro Evaluation of 4-Trichloromethylpyrrolo [1,2-a]Quinoxalines as New Antiplasmodial Agents. Eur. J. Med. Chem. 2014, 83, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desroches, J.; Kieffer, C.; Primas, N.; Hutter, S.; Gellis, A.; El-Kashef, H.; Rathelot, P.; Verhaeghe, P.; Azas, N.; Vanelle, P. Discovery of New Hit-Molecules Targeting Plasmodium Falciparum through a Global SAR Study of the 4-Substituted-2-Trichloromethylquinazoline Antiplasmodial Scaffold. Eur. J. Med. Chem. 2017, 125, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Amrane, D.; Gellis, A.; Hutter, S.; Prieri, M.; Verhaeghe, P.; Azas, N.; Vanelle, P.; Primas, N. Synthesis and Antiplasmodial Evaluation of 4-Carboxamido- and 4-Alkoxy-2-Trichloromethyl Quinazolines. Molecules 2020, 25, 3929. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Exploring Activity Cliffs in Medicinal Chemistry. J. Med. Chem. 2012, 55, 2932–2942. [Google Scholar] [CrossRef]
- Yeh, E.; DeRisi, J.L. Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium falciparum. PLoS Biol. 2011, 9, e1001138. [Google Scholar] [CrossRef] [Green Version]
- Uddin, T.; McFadden, G.I.; Goodman, C.D. Validation of Putative Apicoplast-Targeting Drugs Using a Chemical Supplementation Assay in Cultured Human Malaria Parasites. Antimicrob. Agents Chemother. 2017, 62, e01161-17. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, B.; Lelièvre, J.; Barragán, M.J.L.; Laurent, V.; Su, X.; Berry, A.; Benoit-Vical, F. Increased Tolerance to Artemisinin in Plasmodium Falciparum Is Mediated by a Quiescence Mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménard, S.; Ben Haddou, T.; Ramadani, A.P.; Ariey, F.; Iriart, X.; Beghain, J.; Bouchier, C.; Witkowski, B.; Berry, A.; Mercereau-Puijalon, O.; et al. Induction of Multidrug Tolerance in Plasmodium Falciparum by Extended Artemisinin Pressure. Emerg. Infect. Dis. 2015, 21, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Reyser, T.; Paloque, L.; Ouji, M.; Nguyen, M.; Ménard, S.; Witkowski, B.; Augereau, J.-M.; Benoit-Vical, F. Identification of Compounds Active against Quiescent Artemisinin-Resistant Plasmodium Falciparum Parasites via the Quiescent-Stage Survival Assay (QSA). J. Antimicrob. Chemother. 2020, 75, 2826–2834. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Qi, S.; Xu, Y.; Liu, H.; Wu, J.; Li, H.; Xia, C.; Duan, G. Visible Light-Induced Photocatalytic C−H Perfluoroalkylation of Quinoxalinones under Aerobic Oxidation Condition. Adv. Synth. Catal. 2019, 361, 5490–5498. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human Malaria Parasites in Continuous Culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Guiguemde, W.A.; Shelat, A.A.; Bouck, D.; Duffy, S.; Crowther, G.J.; Davis, P.H.; Smithson, D.C.; Connelly, M.; Clark, J.; Zhu, F.; et al. Chemical Genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [Google Scholar] [CrossRef]
- De Méo, M.; Laget, M.; Di Giorgio, C.; Guiraud, H.; Botta, A.; Castegnaro, M.; Duménil, G. Optimization of the Salmonella/Mammalian Microsome Assay for Urine Mutagenesis by Experimental Designs. Mutat. Res. 1996, 340, 51–65. [Google Scholar] [CrossRef]
- Benoit-Vical, F.; Lelièvre, J.; Berry, A.; Deymier, C.; Dechy-Cabaret, O.; Cazelles, J.; Loup, C.; Robert, A.; Magnaval, J.-F.; Meunier, B. Trioxaquines Are New Antimalarial Agents Active on All Erythrocytic Forms, Including Gametocytes. Antimicrob. Agents Chemother. 2007, 51, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and Inexpensive Fluorescence-Based Technique for High-Throughput Antimalarial Drug Screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [Green Version]
- Amiar, S.; Katris, N.J.; Berry, L.; Dass, S.; Duley, S.; Arnold, C.-S.; Shears, M.J.; Brunet, C.; Touquet, B.; McFadden, G.I.; et al. Division and Adaptation to Host Environment of Apicomplexan Parasites Depend on Apicoplast Lipid Metabolic Plasticity and Host Organelle Remodeling. Cell Rep. 2020, 30, 3778–3792. [Google Scholar] [CrossRef] [PubMed]
- Dembélé, L.; Franetich, J.-F.; Lorthiois, A.; Gego, A.; Zeeman, A.-M.; Kocken, C.H.M.; Le Grand, R.; Dereuddre-Bosquet, N.; van Gemert, G.-J.; Sauerwein, R.; et al. Persistence and activation of malaria hypnozoites in long-term primary hepatocyte cultures. Nat. Med. 2014, 20, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Bosson-Vanga, H.; Franetich, J.-F.; Soulard, V.; Sossau, D.; Tefit, M.; Kane, B.; Vaillant, J.-C.; Borrmann, S.; Müller, O.; Dereuddre-Bosquet, N.; et al. Differential activity of methylene blue against erythrocytic and hepatic stages of Plasmodium. Malar. J. 2018, 17, 143. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| R | N° | Yield Step 2 (%) | EC50 PfK1 (µM) ± SD | CC50 HepG2 (µM) ± SD | SI d | clogP e |
|---|---|---|---|---|---|---|
| H | 3a | 84 | 0.47 ± 0.10 | 45.3 ± 9.6 | 96.4 | 4.28 |
| 2-Cl | 3b | 67 | 0.55 ± 0.12 | 49.5 ± 10.1 | 90.0 | 4.75 |
| 3-Cl | 3c | 67 | 0.34 ± 0.06 | 37.2 ± 8.1 | 109.4 | 4.77 |
| 2,4-Cl | 3d | 52 | 0.60 ± 0.12 | 37.7 ± 4.7 | 62.8 | 5.28 |
| 2-F | 3e | 43 | 0.64 ± 0.14 | >15.6 c | >24.4 | 4.53 |
| 3-F | 3f | 52 | 0.32 ± 0.07 | 33.5 ± 6.3 | 105.0 | 4.54 |
| 4-F | 3g | 58 | 0.42 ± 0.09 | 35.9 ± 7.5 | 85.5 | 4.56 |
| 4-OMe | 3h | 27 | 0.38 ± 0.08 | >7.8 c | >20.6 | 4.22 |
| 4-NO2 f | 3i | 57 | 0.20 ± 0.04 | 32.0 ± 6.1 | 160.0 | 3.53 |
| 4-CF3 | 3j | 70 | 0.30 ± 0.06 | 28.3 ± 4.4 | 94.3 | 5.27 |
| 4-OCF3 | 3k | 85 | 0.40 ± 0.08 | 27.1 ± 4.7 | 67.8 | 5.15 |
| 4-SF5 | 3l | 84 | 0.20 ± 0.03 | 16.6 ± 3.9 | 83.0 | 5.41 |
| 4-SCF3 | 3m | 58 | 0.20 ± 0.04 | 25.6 ± 4.6 | 128.0 | 5.70 |
| 4-CN | 3n | 81 | 0.25 ± 0.04 | 15.4 ± 2.1 | 77.0 | 4.06 |
| 4-SO2CF3 | 3o | 26 | 0.22 ± 0.07 | 7.2 ± 1.4 | 32.7 | 4.77 |
| 4-SONHCF3 | 3p | 28 | 0.21 ± 0.05 | 6.5 ± 1.4 | 31.0 | 5.11 |
| Chloroquine a | 0.80 | 30.0 | 37.5 | 3.82 | ||
| Artesunate a | 0.003 ± 0.0012 | 14.7 ± 1.4 | 4900 | - | ||
| Doxycycline a | 6.00 | 20.0 | 3.3 | - | ||
| Doxorubicin b | - | 0.2 | - | - | ||

| R’ | N° | Yield Step 2 (%) | EC50 PfK1 (µM) ± SD | CC50 HepG2 (µM) ± SD | SI d | clogP e |
|---|---|---|---|---|---|---|
 | 3q | 43 | 0.60 ± 0.10 | 53.1 ± 7.6 | 88.5 | 5.17 |
 | 3r | 64 | 0.40 ± 0.08 | >31.2 c | >78.1 | 5.19 |
 | 3s | 50 | 0.60 ± 0.10 | >62.5 c | >104.2 | 5.61 |
 | 3t | 70 | 0.50 ± 0.05 | >31.2 c | >62.5 | 5.60 |
| Chloroquine a | 0.80 | 30.0 | 37.5 | 3.82 | ||
| Artesunate a | 0.003 ± 0.0012 | 14.7 ± 1.4 | 4900 | - | ||
| Doxycycline a | 6.00 | 20.0 | 3.3 | - | ||
| Doxorubicin b | - | 0.2 | - | - | ||
| Time (h) | [3i] Concentration | Result | |
|---|---|---|---|
| Ames test a | 48 | 5 mM (w/o S9 mix) 25 mM (w/o S9mix) 5 mM (w/S9 mix) 25 mM (w/S9mix) | negative |
| In vitro comet assay b | 2 | 3.2 µM | negative |
| 2 | 16 µM | ||
| 72 | 3.2 µM | ||
| 72 | 16 µM |
| Mean ± SEM EC50 values | |||
|---|---|---|---|
| F32-ART5 n = 4 * | F32-TEM n = 2 * | p-value $ | |
| 3i | 400 ± 105 nM | 392 ± 34 nM | 0.8 |
| Artemisinin | 11 ± 3 nM | 19 ± 3 nM | 0.267 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amrane, D.; Arnold, C.-S.; Hutter, S.; Sanz-Serrano, J.; Collia, M.; Azqueta, A.; Paloque, L.; Cohen, A.; Amanzougaghene, N.; Tajeri, S.; et al. 2-Phenoxy-3-Trichloromethylquinoxalines Are Antiplasmodial Derivatives with Activity against the Apicoplast of Plasmodium falciparum. Pharmaceuticals 2021, 14, 724. https://doi.org/10.3390/ph14080724
Amrane D, Arnold C-S, Hutter S, Sanz-Serrano J, Collia M, Azqueta A, Paloque L, Cohen A, Amanzougaghene N, Tajeri S, et al. 2-Phenoxy-3-Trichloromethylquinoxalines Are Antiplasmodial Derivatives with Activity against the Apicoplast of Plasmodium falciparum. Pharmaceuticals. 2021; 14(8):724. https://doi.org/10.3390/ph14080724
Chicago/Turabian StyleAmrane, Dyhia, Christophe-Sébastien Arnold, Sébastien Hutter, Julen Sanz-Serrano, Miguel Collia, Amaya Azqueta, Lucie Paloque, Anita Cohen, Nadia Amanzougaghene, Shahin Tajeri, and et al. 2021. "2-Phenoxy-3-Trichloromethylquinoxalines Are Antiplasmodial Derivatives with Activity against the Apicoplast of Plasmodium falciparum" Pharmaceuticals 14, no. 8: 724. https://doi.org/10.3390/ph14080724