
Quantification of 17 Endogenous and Exogenous Steroidal Hormones in Equine and Bovine Blood for Doping Control with UHPLC-MS/MS

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Setup of the Chromatographic and Mass Analyzer Conditions
2.2. Method Validation
2.3. Evaluation of the Stability of Steroids in Glass and Plastic
2.4. Precision and Linearity
2.5. Accuracy
2.6. Recovery
2.7. Matrix Effect
2.8. Specificity
2.9. Application and Testing of the Developed Method to Equine and Bovine Blood
3. Materials and Methods
3.1. Disposable Chemicals and Materials
3.2. Collection of Equine Blood
3.3. Preparation of Steroids-Free Blood and Sample Preparation
3.4. UHPLC-ESI-MS/MS
3.5. Method Validation Settings
3.6. Internal Standards
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Genangeli, M.; Caprioli, G.; Cortese, M.; Laus, F.; Matteucci, M.; Petrelli, R.; Ricciutelli, M.; Sagratini, G.; Sartori, S.; Vittori, S. Development and application of a UHPLC-MS/MS method for the simultaneous determination of 17 steroidal hormones in equine serum. J. Mass Spectrom. 2017, 52, 22–29. [Google Scholar] [CrossRef]
- Andrews, M.A.; Magee, C.D.; Combest, T.M.; Allard, R.J.; Douglas, K.M. Physical Effects of Anabolic-androgenic Steroids in Healthy Exercising Adults. Curr. Sports Med. Rep. 2018, 17, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.M.; Avant, R.A.; Charchenko, C.M.; Westerman, M.E.; Ziegelmann, M.J.; Miest, T.S.; Trost, L.W. Impact of anabolic androgenic steroids on sexual function. Transl. Androl. Urol. 2018, 7, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Melo Junior, A.F.; Dalpiaz, P.L.M.; Sousa, G.J.; Oliveira, P.W.C.; Birocale, A.M.; Andrade, T.U.; Abreu, G.R.; Bissoli, N.S. Nandrolone alter left ventricular contractility and promotes remodelling involving calcium-handling proteins and renin-angiotensin system in male SHR. Life Sci. 2018, 208, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Büttler, R.M.; Martens, F.; Kushnir, M.M.; Ackermans, M.T.; Blankenstein, M.A.; Heijboer, A.C. Simultaneous measurement of testosterone, androstenedione and dehydroepiandrosterone (DHEA) in serum and plasma using Isotope-Dilution 2-Dimension Ultra High Performance Liquid-Chromatography Tandem Mass Spectrometry (ID-LC–MS/MS). Clin. Chim. Acta 2015, 438, 157–159. [Google Scholar] [CrossRef]
- Wong, J.K.Y.; Wan, T.S.M. Doping control analyses in horseracing: A clinician’s guide. Vet. J. 2014, 200, 8–16. [Google Scholar] [CrossRef]
- IFHA. Available online: http://www.ifhaonline.org/default.asp?section=Racing&area=0#a6a (accessed on 25 March 2021).
- Pellegrini, K.G.M.V. Anabolic Steroids. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482418/#article-17499.s9 (accessed on 25 March 2021).
- Caron, P.; Turcotte, V.; Guillemette, C. A chromatography/tandem mass spectrometry method for the simultaneous profiling of ten endogenous steroids, including progesterone, adrenal precursors, androgens and estrogens, using low serum volume. Steroids 2015, 104, 16–24. [Google Scholar] [CrossRef]
- Görög, S. Recent Advances in the Analysis of Steroid Hormones and Related Drugs. Anal. Sci. 2004, 20, 767–782. [Google Scholar] [CrossRef] [Green Version]
- Stanczyk, F.Z.; Jurow, J.; Hsing, A.W. Limitations of Direct Immunoassays for Measuring Circulating Estradiol Levels in Postmenopausal Women and Men in Epidemiologic Studies. Cancer Epidemiol. Biomark. Prev. 2010, 19, 903–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taieb, J. Testosterone Measured by 10 Immunoassays and by Isotope-Dilution Gas Chromatography-Mass Spectrometry in Sera from 116 Men, Women, and Children. Clin. Chem. 2003, 49, 1381–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Yuan, D.; Huang, B. Determination of steroid sex hormones in urine matrix by stir bar sorptive extraction based on monolithic material and liquid chromatography with diode array detection. Talanta 2007. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Wu, Y.; Lu, J. Doping control analysis of 13 steroids and structural-like analytes in human urine using Quadrupole-Orbitrap LC–MS/MS with parallel reaction monitoring (PRM) mode. Steroids 2018, 131, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Karatt, T.K.; Nalakath, J.; Perwad, Z.; Albert, P.H.; Abdul Khader, K.K.; Syed Ali Padusha, M.; Laya, S. Mass spectrometric method for distinguishing isomers of dexamethasone via fragment mass ratio: An HRMS approach. J. Mass Spectrom. 2018, 53, 1046–1058. [Google Scholar] [CrossRef]
- Keevil, B.G. Novel liquid chromatography tandem mass spectrometry (LC-MS/MS) methods for measuring steroids. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 663–674. [Google Scholar] [CrossRef]
- You, Y.; Uboh, C.E.; Soma, L.R.; Guan, F.; Li, X.; Liu, Y.; Rudy, J.A.; Chen, J.; Tsang, D. Simultaneous separation and determination of 16 testosterone and nandrolone esters in equine plasma using ultra high performance liquid chromatography–tandem mass spectrometry for doping control. J. Chromatogr. A 2011, 1218, 3982–3993. [Google Scholar] [CrossRef]
- Wong, C.H.F.; Leung, D.K.K.; Tang, F.P.W.; Wong, J.K.Y.; Yu, N.H.; Wan, T.S.M. Rapid screening of anabolic steroids in horse urine with ultra-high-performance liquid chromatography/tandem mass spectrometry after chemical derivatisation. J. Chromatogr. A 2012, 1232, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Tuzimski, T.; Petruczynik, A. Review of Chromatographic Methods Coupled with Modern Detection Techniques Applied in the Therapeutic Drugs Monitoring (TDM). Molecules 2020, 25, 4026. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.L.S.; Kwok, K.Y.; Kwok, W.H.; Tsoi, Y.Y.K.; Wong, J.K.Y.; Wan, T.S.M. Detection of seventy-two anabolic and androgenic steroids and/or their esters in horse hair using ultra-high performance liquid chromatography-high resolution mass spectrometry in multiplexed targeted MS 2 mode and gas chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1566, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.Y.; Choi, T.L.S.; Kwok, K.Y.; Lei, E.N.Y.; Wan, T.S.M. Doping control analysis of 121 prohibited substances in equine hair by liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal. 2018, 158, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Uboh, C.E.; Soma, L.R.; Luo, Y.; Rudy, J.; Tobin, T. Detection, quantification and confirmation of anabolic steroids in equine plasma by liquid chromatography and tandem mass spectrometry. J. Chromatogr. B 2005, 829, 56–68. [Google Scholar] [CrossRef]
- Hintikka, L. Development of Mass Spectrometric Methods for Analysis of Anabolic Androgenic Steroids; University of Helsinki: Helsinki, Finland, 2018. [Google Scholar]
- Georgakopoulos, C.G.; Vonaparti, A.; Stamou, M.; Kiousi, P.; Lyris, E.; Angelis, Y.S.; Tsoupras, G.; Wuest, B.; Nielen, M.W.F.; Panderi, I.; et al. Preventive doping control analysis: Liquid and gas chromatography time-of-flight mass spectrometry for detection of designer steroids. Rapid Commun. Mass Spectrom. 2007, 21, 2439–2446. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.; Viljanto, M.; Menzies, E.; Vanhaecke, L. Detection of prohibited substances in equine hair by ultra-high performance liquid chromatography-triple quadrupole mass spectrometry-application to doping control samples. Drug Test. Anal. 2018, 10, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Kaabia, Z.; Dervilly-Pinel, G.; Hanganu, F.; Cesbron, N.; Bichon, E.; Popot, M.A.; Bonnaire, Y.; Le Bizec, B. Ultra high performance liquid chromatography/tandem mass spectrometry based identification of steroid esters in serum and plasma: An efficient strategy to detect natural steroids abuse in breeding and racing animals. J. Chromatogr. A 2013, 1284, 126–140. [Google Scholar] [CrossRef]
- Annesley, T.M. Ion suppression in mass spectrometry. Clin. Chem. 2003, 49, 1041–1044. [Google Scholar] [CrossRef] [Green Version]
- Harrison, A.G. Chemical Ionization Mass Spectrometry, 2nd ed.; CRC Press: Boca Raton, FL, USA, 1992; p. 208. [Google Scholar]
- Shao, B.; Zhao, R.; Meng, J.; Xue, Y.; Wu, G.; Hu, J.; Tu, X. Simultaneous determination of residual hormonal chemicals in meat, kidney, liver tissues and milk by liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2005, 548, 41–50. [Google Scholar] [CrossRef]
- Yamashita, K.; Okuyama, M.; Nakagawa, R.; Honma, S.; Satoh, F.; Morimoto, R.; Ito, S.; Takahashi, M.; Numazawa, M. Development of sensitive derivatization method for aldosterone in liquid chromatography–electrospray ionization tandem mass spectrometry of corticosteroids. J. Chromatogr. A 2008, 1200, 114–121. [Google Scholar] [CrossRef]
- Zuber, J.; Kroll, M.M.; Rathsack, P.; Otto, M. Gas Chromatography/Atmospheric Pressure Chemical Ionization-Fourier Transform Ion Cyclotron Resonance Mass Spectrometry of Pyrolysis Oil from German Brown Coal. Int. J. Anal. Chem. 2016, 2016, 5960916. [Google Scholar] [CrossRef]
- You, Y.; Uboh, C.E.; Soma, L.R.; Guan, F.; Li, X.; Liu, Y.; Chen, J.; Tsang, D. Simultaneous Determination of Testosterone and Testosterone Enanthate in Equine Plasma by UHPLC-MS-MS. Chromatographia 2010, 72, 1097–1106. [Google Scholar] [CrossRef]
- Prokai, L.; Stevens, S.M. Direct Analysis in Real Time (DART) of an Organothiophosphate at Ultrahigh Resolution by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry and Tandem Mass Spectrometry. Int. J. Mol. Sci. 2016, 17, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allis, O.; Dauphard, J.; Hamilton, B.; Ni Shuilleabhain, A.; Lehane, M.; James, K.J.; Furey, A. Liquid Chromatography−Tandem Mass Spectrometry Application, for the Determination of Extracellular Hepatotoxins in Irish Lake and Drinking Waters. Anal. Chem. 2007, 79, 3436–3447. [Google Scholar] [CrossRef]
- Bonfiglio, R.; King, R.C.; Olah, T.V.; Merkle, K. The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun. Mass Spectrom. 1999, 13, 1175–1185. [Google Scholar] [CrossRef]
- Kaabia, Z.; Dervilly-Pinel, G.; Popot, M.A.; Bailly-Chouriberry, L.; Plou, P.; Bonnaire, Y.; Le Bizec, B. Monitoring the endogenous steroid profile disruption in urine and blood upon nandrolone administration: An efficient and innovative strategy to screen for nandrolone abuse in entire male horses. Drug Test. Anal. 2014, 6, 376–388. [Google Scholar] [CrossRef]
- Available online: https://www.comsol.nl/multiphysics/the-joule-heating-effect (accessed on 25 March 2021).
- Wang, Y.N.; Zhao, M.; Yu, Y.B.; Wang, M.; Zhao, C.J. Metabolic profile of Cortex Fraxini in rats using UHPLC combined with Fourier transform ion cyclotron resonance mass spectrometry. RSC Adv. 2016, 6, 39642–39651. [Google Scholar] [CrossRef]
- Magnisali, P.; Dracopoulou, M.; Mataragas, M.; Dacou-Voutetakis, A.; Moutsatsou, P. Routine method for the simultaneous quantification of 17α-hydroxyprogesterone, testosterone, dehydroepiandrosterone, androstenedione, cortisol, and pregnenolone in human serum of neonates using gas chromatography–mass spectrometry. J. Chromatogr. A 2008, 1206, 166–177. [Google Scholar] [CrossRef] [PubMed]


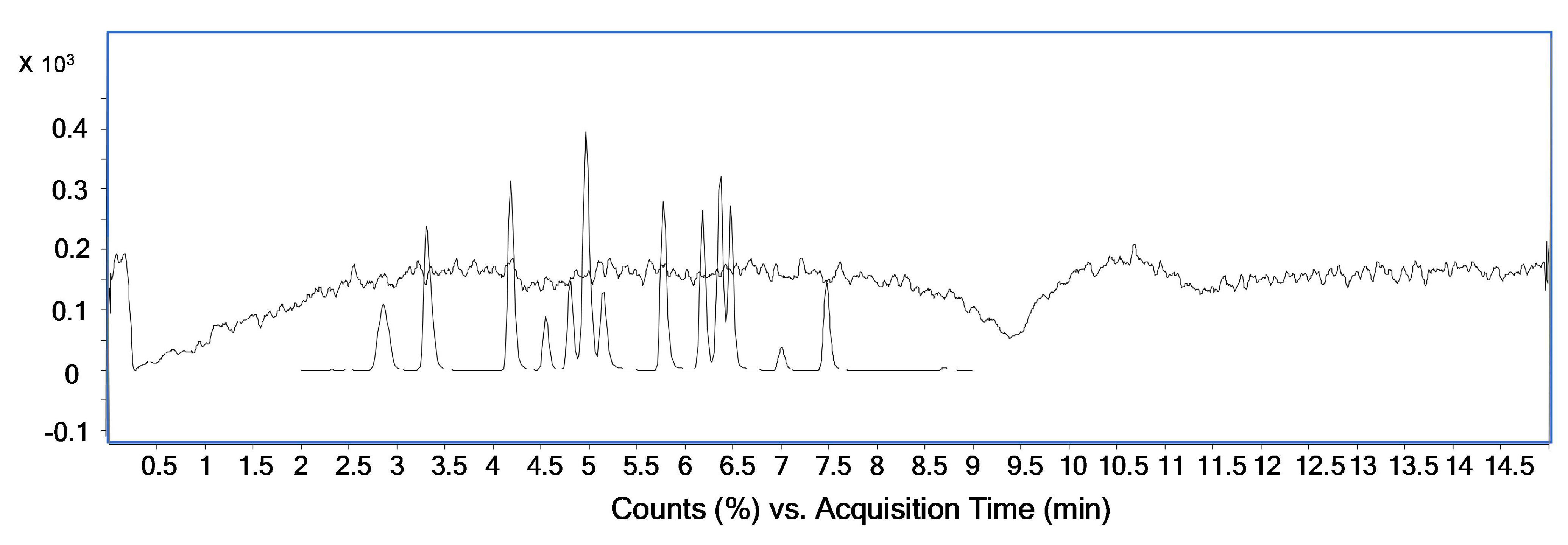{kind=link}
| Compound | Abbreviation | Time Window (Minute) | Precursor Ion a (m/z) | Product Ion (m/z) | Fragmentor (V) | Collision Energy (V) | Dwell Time (Milli- Second) |
|---|---|---|---|---|---|---|---|
| Dexamethasone Sodium Phosphate | Desa-NA-P | 2.0–3.7 | 473.11 | 435.2 355.2 | 97 | 8 | 200 |
| Cortisol | CORT | 2.0–3.7 | 363.01 | 121.1 327.2 | 136 | 24 | 200 |
| Aldosterone | ALDO | 2.0–3.7 | 361.41 | 343.2 315.2 | 116 | 16 | 200 |
| Pregnenolone | PRE | 2.0–3.7 | 361.41 | 343.2 105.0 | 87 | 4 | 200 |
| Methylprednisolone | ME-PRE | 3.7–5.5 | 375.01 | 357.2 323.2 | 92 | 4 | 180 |
| 11-Deoxycortisol | 11-DOC | 3.7–5.5 | 347.51 | 109.10 97.2 | 141 | 32 | 180 |
| Corticosterone | COCO | 3.7–5.5 | 347.01 | 329.2 329.2 | 111 | 12 | 180 |
| Stanozolol | STA | 3.7–5.5 | 329.51 | 81.10 95.10 | 170 | 50 | 180 |
| Boldenone | BOL | 3.7–5.5 | 287.41 | 121.00 135.00 | 107 | 24 | 180 |
| Nandrolone | NAN | 3.7–5.5 | 275.10 | 109.10 82.90 | 100 | 28 | 180 |
| Dexamethasone isonicotinate | DESA-ISO | 5.5–6.8 | 498.61 | 47.20 124.0 | 121 | 8 | 200 |
| 11-Deoxycorticosterone | 11-DCC | 5.5–6.8 | 331.01 | 97.10 109.1 | 117 | 20 | 200 |
| Dihydrotestosterone | DHT | 6.8–9.0 | 273.10 | 255.30 147.0 | 159 | 15 | 200 |
| Testosterone | TESTO | 5.5–6.8 | 289.01 | 97.10 109.1 | 131 | 20 | 100 |
| Androstenedione | ANDD | 5.5–6.8 | 287.01 | 97.10 109.1 | 131 | 24 | 200 |
| Dehydroepiandrosterone | DHEA | 5.5–6.8 | 271.01 | 253.10 253.2 | 92 | 8 | 200 |
| Androsterone | ANDRO | 6.8–9.0 | 291.41 | 273.20 255.2 | 78 | 4 | 200 |
| Testosterone–d3 | d3-TESTO | 5.5–6.8 | 292.00 | 97.00 | 135 | 25 | 120 |
| Compound | LOD | LOQ | C1 | CM | C2 | CU | U1 |
|---|---|---|---|---|---|---|---|
| ppb | |||||||
| DESA-NA-P | 0.333 | 1.00 | 10 | 50 | 100 | 500 | 1000 |
| COR | 0.183 | 0.55 | 11 | 55 | 110 | 550 | 1100 |
| ALDO | 0.183 | 0.55 | 11 | 55 | 110 | 550 | 1100 |
| PRE | 0.033 | 0.10 | 10 | 50 | 100 | 500 | 1000 |
| ME-PRE | 0.067 | 0.20 | 20 | 100 | 200 | 1000 | 2000 |
| 11-DOC | 0.037 | 0.11 | 11 | 55 | 110 | 550 | 1100 |
| COCO | 0.167 | 0.50 | 10 | 50 | 100 | 500 | 1000 |
| STA | 0.033 | 0.10 | 10 | 50 | 100 | 500 | 1000 |
| BOL | 0.167 | 0.50 | 10 | 50 | 100 | 500 | 1000 |
| NAN | 0.333 | 1.00 | 10 | 50 | 100 | 500 | 1000 |
| DESA-ISO | 0.023 | 0.069 | 6.9 | 34.5 | 69 | 345 | 690 |
| 11-DCC | 0.037 | 0.11 | 11 | 55 | 110 | 550 | 1100 |
| TESTO | 0.037 | 0.05 | 11 | 55 | 110 | 550 | 1100 |
| ANDD | 0.333 | 1.00 | 10 | 50 | 100 | 500 | 1000 |
| DHEA | 1.833 | 5.50 | 11 | 55 | 110 | 550 | 1100 |
| ANDRO | 0.733 | 2.20 | 22 | 110 | 220 | 1100 | 2200 |
| DHT | 1.833 | 5.50 | 11 | 55 | 110 | 550 | 1100 |
| DAY 1 | DAY 2 | |||
|---|---|---|---|---|
| Loss in Plastic (%) | Loss in Glass (%) | Loss in Plastic (%) | Loss in Glass (%) | |
| COR | <1 | 99.63 | <2% | >99.90 |
| ALDO | <1 | 94.29 | <2% | >99.90 |
| 11-DOC | <1 | 99.94 | <2% | >99.90 |
| COCO | <1 | 99.76 | <2% | >99.90 |
| 11-DCC | <1 | 99.88 | <2% | >99.90 |
| PRE | <1 | 99.97 | <2% | >99.90 |
| ANDRO | <1 | 99.52 | <2% | >99.90 |
| TESTO | <1 | 99.85 | <2% | >99.90 |
| ANDD | <1 | 99.83 | <2% | >99.90 |
| DHT | <1 | 99.12 | <2% | >99.90 |
| DHEA | <1 | 99.65 | <2% | >99.90 |
| Compound | Limit of Quantification | Medium Concentration | CU | U1 | Linearity | ||||
|---|---|---|---|---|---|---|---|---|---|
| Intra-Day (CV%) | Inter-Day (CV%) | Intra-Day (CV%) | Inter-Day (CV%) | Intra-Day (CV%) | Inter-Day (CV%) | Intra-Day (CV%) | Inter-Day (CV%) | R2 | |
| DESA-NA-P | 10.86 | 14.16 | 6.74 | 7.58 | 2.62 | 4.12 | 1.02 | 1.48 | 0.99326 |
| COR | 16.47 | 18.49 | 9.50 | 10.35 | 6.49 | 8.24 | 3.56 | 4.49 | 0.99939 |
| ALDO | 14.45 | 17.52 | 6.95 | 8.40 | 6.79 | 7.63 | 6.07 | 6.85 | 0.99995 |
| PRE | 15.84 | 17.41 | 10.07 | 10.91 | 2.78 | 3.69 | 1.18 | 2.37 | 0.99922 |
| ME-PRE | 15.93 | 17.45 | 15.04 | 17.85 | 6.32 | 18.40 | 3.26 | 8.12 | 0.99912 |
| 11-DOC | 17.10 | 17.87 | 6.63 | 7.72 | 6.93 | 7.53 | 5.48 | 6.73 | 0.99954 |
| COCO | 10.34 | 14.57 | 7.97 | 10.63 | 1.56 | 4.51 | 0.48 | 2.28 | 0.99991 |
| STA | 13.25 | 14.95 | 5.73 | 9.11 | 3.15 | 5.24 | 1.18 | 4.20 | 0.99941 |
| BOL | 15.83 | 17.89 | 11.85 | 16.66 | 3.71 | 18.05 | 2.03 | 6.97 | 0.99658 |
| NAN | 17.35 | 18.62 | 7.12 | 14.75 | 5.49 | 8.13 | 2.26 | 5.83 | 0.99861 |
| DESA-ISO | 17.43 | 18.37 | 9.77 | 18.99 | 7.88 | 9.65 | 4.49 | 6.41 | 0.99981 |
| 11-DCC | 16.60 | 18.07 | 16.06 | 18.78 | 12.21 | 18.27 | 7.21 | 9.41 | 0.99953 |
| TESTO | 11.76 | 18.17 | 11.40 | 16.89 | 6.35 | 15.06 | 4.39 | 5.72 | 0.99841 |
| ANDD | 10.08 | 16.66 | 14.04 | 18.46 | 8.56 | 9.88 | 6.29 | 8.06 | 0.99987 |
| DHEA | 14.21 | 17.84 | 9.99 | 18.64 | 4.67 | 8.49 | 2.18 | 3.26 | 0.99970 |
| ANDRO | 16.81 | 17.77 | 5.34 | 8.23 | 4.30 | 4.80 | 2.93 | 3.21 | 0.99940 |
| DHT | 13.97 | 17.10 | 9.07 | 9.19 | 6.31 | 6.57 | 4.29 | 5.30 | 0.99997 |
| Exogenous Steroids | Endogenous Steroids | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Animal | DESA-NA-P | PRE | ME-PRE | STA | BOL | NAN | DESA-ISO | 11-DCC | TESTO | ANDD | DHEA | ANDRO | DHT | COR | ALDO | 11-DOC | COCO |
| ppb | |||||||||||||||||
| A 01 | 0.152 | - | 0.470 | 0.395 | - | 0.237 | 0.245 | - | 0.145 | 0.481 | - | - | - | 4.207 | 1.038 | 0.190 | 0.319 |
| A 02 | 0.134 | - | 0.478 | 0.397 | - | - | 0.243 | - | 0.142 | 0.482 | - | - | - | 0.668 | 1.095 | - | 0.330 |
| A 03 | - | 0.444 | 0.558 | 0.392 | - | - | 0.242 | 0.183 | 0.138 | 0.497 | - | - | - | 3.631 | - | 0.179 | 0.288 |
| A 04 | - | - | - | 0.419 | - | - | 0.247 | - | - | 0.478 | - | - | - | 6.578 | 0.846 | 0.201 | 0.587 |
| A 05 | 0.130 | - | - | 0.393 | - | - | 0.246 | - | - | 0.484 | - | - | - | 2.363 | - | - | - |
| A 06 | 0.100 | - | - | 0.390 | - | - | 0.242 | 0.178 | - | 0.461 | - | - | - | 3.247 | - | 0.180 | - |
| A 07 | - | - | - | - | - | - | 0.241 | 0.187 | - | 0.497 | - | - | - | - | - | - | |
| A 08 | - | - | - | - | - | - | - | 0.194 | 0.142 | 0.436 | - | - | - | 1.670 | - | - | - |
| A 09 | 0.071 | - | - | - | - | - | - | - | - | 0.479 | - | - | - | 0.444 | - | - | 0.334 |
| A 10 | - | - | - | 0.462 | - | - | 0.250 | 0.202 | 0.146 | 0.500 | - | - | - | 0.465 | - | - | 0.351 |
| A 11 | - | - | - | - | - | - | 0.241 | - | 0.137 | 0.532 | - | - | - | 2.082 | - | - | 0.315 |
| A 12 | 0.094 | - | - | - | - | - | 0.243 | - | - | 0.571 | - | - | - | 0.435 | - | - | 0.393 |
| A 13 | 0.170 | - | - | 0.393 | - | - | 0.244 | - | - | 0.561 | - | - | - | 1.956 | - | - | 0.375 |
| A 14 | 0.121 | - | - | 0.387 | - | - | 0.247 | - | - | 0.443 | - | - | - | 3.935 | - | - | 0.491 |
| A 15 | 0.196 | 0,472 | - | - | 0.064 | 0.291 | 0.258 | 0.229 | - | 0.515 | - | - | - | 3.486 | - | 0.197 | 0.383 |
| B 01 | - | - | 0.87 | - | 0.02 | - | - | 0.39 | - | - | - | - | 0.98 | 5.96 | - | 0.53 | 0.49 |
| B 02 | - | - | - | - | - | - | - | 0.39 | 0.23 | 0.60 | - | - | 1.87 | 1.29 | 0.70 | - | 0.40 |
| B 03 | - | - | - | - | - | - | 0.47 | 0.39 | 0.22 | - | - | - | 1.95 | 0.99 | - | - | 0.44 |
| B 04 | - | - | 0.83 | - | - | - | - | - | 0.22 | - | - | - | 1.06 | 5.62 | - | - | 0.41 |
| B 05 | - | - | 0.95 | - | - | - | 0.47 | - | 0.21 | - | - | - | 3.26 | 1.67 | - | - | - |
| B 06 | - | - | 0.86 | - | - | - | 0.47 | - | - | - | - | - | - | 1.35 | - | - | - |
| C 01 | - | - | 1.33 | - | - | - | 0.39 | - | 0.31 | 0.62 | - | - | - | 10.00 | 0.79 | 0.48 | 0.60 |
| C 02 | - | - | 1.05 | - | - | - | 0.40 | - | - | 0.64 | - | - | - | 3.61 | - | 0.45 | 0.54 |
| C 03 | - | - | 1.07 | 0.27 | - | - | 0.40 | - | - | - | - | - | - | 1.17 | - | - | - |
| C 04 | - | - | 1.08 | - | - | - | - | - | 0.31 | - | - | - | - | 2.36 | - | - | 0.54 |
| C 05 | - | - | 1.08 | - | - | - | 0.40 | - | - | - | - | - | - | 0.88 | - | - | 0.45 |
| C 06 | - | - | 1.02 | - | - | - | 0.40 | - | - | 0.62 | - | - | - | 4.96 | - | 0.46 | 0.58 |
| D 01 | - | - | 0.876 | - | 0.024 | 0.531 | - | 0.397 | - | - | - | - | 0.986 | 5.967 | - | 0.531 | 0.497 |
| D 02 | - | - | - | - | - | - | - | 0.396 | 0.236 | 0.604 | - | - | 1.872 | 1.296 | 0.702 | - | 0.401 |
| D 03 | - | - | - | - | - | - | 0.3 | 0.394 | 0.221 | - | - | - | 0.959 | 0.992 | - | - | 0.448 |
| D 04 | - | - | 0.834 | - | - | - | - | - | 0.226 | - | - | - | 1.060 | 5.62 | - | - | 0.418 |
| D 05 | - | - | 0.953 | - | - | - | - | - | 0.219 | - | - | - | - | 1.671 | - | - | - |
| D 06 | - | - | 0.866 | - | - | - | 0.4 | - | 0.213 | - | - | - | - | 1.356 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caprioli, G.; Genangeli, M.; Mustafa, A.M.; Petrelli, R.; Ricciutelli, M.; Sagratini, G.; Sartori, S.; Laus, F.; Vittori, S.; Cortese, M. Quantification of 17 Endogenous and Exogenous Steroidal Hormones in Equine and Bovine Blood for Doping Control with UHPLC-MS/MS. Pharmaceuticals 2021, 14, 393. https://doi.org/10.3390/ph14050393
Caprioli G, Genangeli M, Mustafa AM, Petrelli R, Ricciutelli M, Sagratini G, Sartori S, Laus F, Vittori S, Cortese M. Quantification of 17 Endogenous and Exogenous Steroidal Hormones in Equine and Bovine Blood for Doping Control with UHPLC-MS/MS. Pharmaceuticals. 2021; 14(5):393. https://doi.org/10.3390/ph14050393
Chicago/Turabian StyleCaprioli, Giovanni, Michele Genangeli, Ahmed M. Mustafa, Riccardo Petrelli, Massimo Ricciutelli, Gianni Sagratini, Stefano Sartori, Fulvio Laus, Sauro Vittori, and Manuela Cortese. 2021. "Quantification of 17 Endogenous and Exogenous Steroidal Hormones in Equine and Bovine Blood for Doping Control with UHPLC-MS/MS" Pharmaceuticals 14, no. 5: 393. https://doi.org/10.3390/ph14050393