
The Selective Serotonin 2A Receptor Antagonist Sarpogrelate Prevents Cardiac Hypertrophy and Systolic Dysfunction via Inhibition of the ERK1/2–GATA4 Signaling Pathway

 , , ,
, , ,
Abstract
:1. Introduction
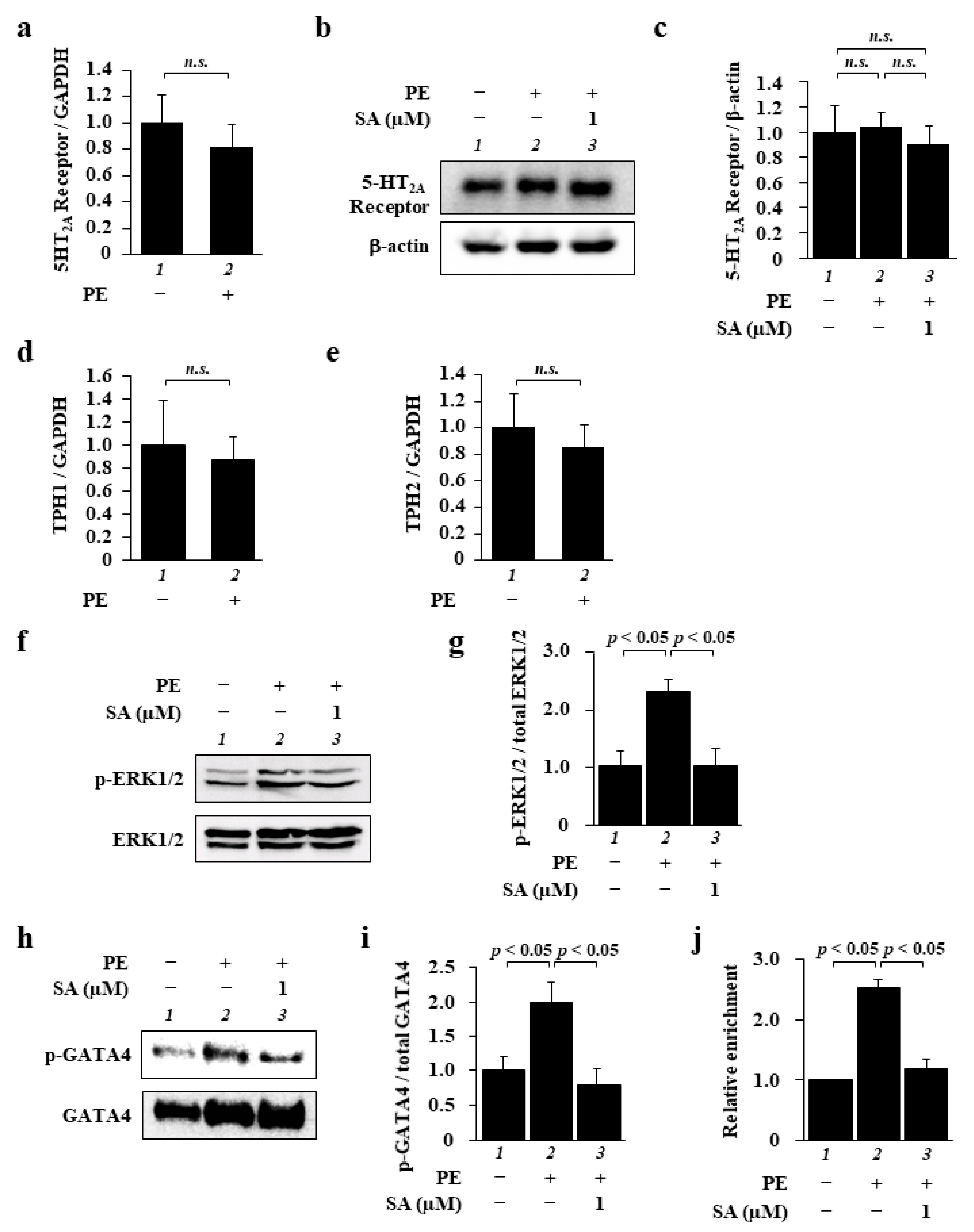
2. Results
2.1. Sarpogrelate Suppressed Cardiomyocyte Hypertrophy Induced by Various Hypertrophic Stimuli
2.2. Sarpogrelate Inhibited the ERK1/2–GATA4 Signaling Pathway in Cardiomyocytes
2.3. Sarpogrelate Suppressed Transverse Aortic Constriction (TAC)-Induced Cardiac Hypertrophy and Systolic Dysfunction
2.4. Sarpogrelate Suppressed TAC-Induced Cardiac Hypertrophy and Fibrosis
2.5. Sarpogrelate Suppressed TAC-Induced Phosphorylation of ERK1/2 and GATA4
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Plasmid Constructs
4.3. Animal Experiments
4.4. Cell Culture
4.5. Sample Preparation
4.6. Immunofluorescence Staining
4.7. Luciferase Reporter Activity
4.8. Western Blotting
4.9. ChIP Assay
4.10. Transverse Aortic Constriction and Drug Treatment
4.11. Echocardiography
4.12. Histological Analysis
4.13. Real-Time PCR
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bragazzi, N.L.; Zhong, W.; Shu, J.; Abu Much, A.; Lotan, D.; Grupper, A.; Younis, A.; Dai, H. Burden of Heart Failure and Underlying Causes in 195 Countries and Territories from 1990 to 2017. Eur. J. Prev. Cardiol. 2021, zwaa147. [Google Scholar] [CrossRef]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and Aetiology of Heart Failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCD Countdown 2030 Collaborators. NCD Countdown 2030: Worldwide Trends in Non-Communicable Disease Mortality and Progress Towards Sustainable Development Goal Target 3.4. Lancet 2018, 392, 1072–1088. [Google Scholar] [CrossRef] [Green Version]
- Callender, T.; Woodward, M.; Roth, G.; Farzadfar, F.; Lemarie, J.C.; Gicquel, S.; Atherton, J.; Rahimzadeh, S.; Ghaziani, M.; Shaikh, M.; et al. Heart Failure Care in Low- and Middle-Income Countries: A Systematic Review and Meta-Analysis. PLoS Med. 2014, 11, e1001699. [Google Scholar] [CrossRef] [Green Version]
- Flather, M.D.; Yusuf, S.; Køber, L.; Pfeffer, M.; Hall, A.; Murray, G.; Torp-Pedersen, C.; Ball, S.; Pogue, J.; Moyé, L.; et al. Long-term ACE-Inhibitor Therapy in Patients with Heart Failure or Left-Ventricular Dysfunction: A Systematic Overview of Data from Individual Patients. Lancet 2000, 355, 1575–1581. [Google Scholar] [CrossRef]
- Metra, M.; Teerlink, J.R. Heart Failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- Elvira, K.S. Microfluidic Technologies for Drug Discovery and Development: Friend or Foe? Trends Pharmacol. Sci. 2021, 42, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Lotfi Shahreza, M.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J.R. A Review of Network-Based Approaches to Drug Repositioning. Brief Bioinform. 2018, 19, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Panchapakesan, U.; Pollock, C. Drug Repurposing in Kidney Disease. Kidney Int. 2018, 94, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Yella, J.K.; Yaddanapudi, S.; Wang, Y.; Jegga, A.G. Changing Trends in Computational Drug Repositioning. Pharmaceuticals 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, G.; Wong, S.T.C. Toward Better Drug Repositioning: Prioritizing and Integrating Existing Methods into Efficient Pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, Y.; Nishimaru, K.; Sawada, T.; Terashi, A.; Handa, S.; Hirai, S.; Hayashi, K.; Tohgi, H.; Fukuuchi, Y.; Uchiyama, S.; et al. Sarpogrelate-Aspirin Comparative Clinical Study for Efficacy and Safety in Secondary Prevention of Cerebral Infarction (S-ACCESS): A Randomized, Double-Blind, Aspirin-Controlled Trial. Stroke 2008, 39, 1827–1833. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, Y.; Nishimaru, K. Sarpogrelate versus Aspirin in Secondary Prevention of Cerebral Infarction: Differential Efficacy in Diabetes?: Subgroup Analysis from S-Access. Stroke 2009, 40, 2862–2865. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Chae, S.; Park, J.; Bae, J.; Go, E.B.; Kim, S.J.; Kim, H.; Hwang, D.; Lee, S.W.; Lee, S.Y. Comprehensive Proteome Profiling of Platelet Identified a Protein Profile Predictive of Responses to an Antiplatelet Agent Sarpogrelate. Mol. Cell Proteom. 2016, 15, 3461–3472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Choi, B.H.; Ku, S.K.; Park, J.H.; Oh, E.; Kwak, M.K.K. Beneficial Effects of Sarpogrelate and Rosuvastatin in High Fat Diet/Streptozotocin-Induced Nephropathy in Mice. PLoS ONE. 2016, 11, e0153965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, C.A.; Ryals, R.C.; Jiang, D.; Coyner, A.S.; Weller, K.K.; Sinha, W.; Robb, B.M.; Yang, P.; Pennesi, M.E. The Role of ERK1/2 Activation in Sarpogrelate-Mediated Neuroprotection. Invest. Ophthalmol. Vis. Sci. 2018, 59, 462–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Mao, N.; Li, M.; Dong, X.; Lin, F.Z.; Xu, Y.; Li, Y.B. Sarpogrelate Inhibits the Expression of ICAM-1 and Monocyte-Endothelial Adhesion Induced by High Glucose in Human Endothelial Cells. Mol. Cell Biochem. 2013, 373, 195–199. [Google Scholar] [CrossRef]
- Sun, Y.M.; Su, Y.; Jin, H.B.; Li, J.; Bi, S. Sarpogrelate Protects against High Glucose-Induced Endothelial Dysfunction and Oxidative Stress. Int. J. Cardiol. 2011, 147, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Tojo, K.; Otsubo, C.; Udagawa, T.; Kumazawa, K.; Ishikawa, M.; Tokudome, G.; Hosoya, T.; Tajima, N.; Claycomb, W.C.; et al. 5-Hydroxytryptamine Synthesis in Hl-1 Cells and Neonatal Rat Cardiocytes. Biochem. Biophys. Res. Commun. 2005, 328, 522–525. [Google Scholar] [CrossRef]
- Hara, K.; Hirowatari, Y.; Yoshika, M.; Komiyama, Y.; Tsuka, Y.; Takahashi, H. The Ratio of Plasma to Whole-Blood Serotonin may Be a Novel Marker of Atherosclerotic Cardiovascular Disease. J. Lab. Clin. Med. 2004, 144, 31–37. [Google Scholar] [CrossRef]
- Mawe, G.M.; Hoffman, J.M. Serotonin Signalling in the Gut-Functions, Dysfunctions and Therapeutic Targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villeneuve, C.; Caudrillier, A.; Ordener, C.; Pizzinat, N.; Parini, A.; Mialet-Perez, J. Dose-Dependent Activation of Distinct Hypertrophic Pathways by Serotonin in Cardiac Cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Mialet-Perez, J.; D’Angelo, R.; Villeneuve, C.; Ordener, C.; Nègre-Salvayre, A.; Parini, A.; Vindis, C. Serotonin 5-HT2A Receptor-Mediated Hypertrophy is Negatively Regulated by Caveolin-3 in Cardiomyoblasts and Neonatal Cardiomyocytes. J. Mol. Cell Cardiol. 2012, 52, 502–510. [Google Scholar] [CrossRef]
- Park-Windhol, C.; Zhang, P.; Zhu, M.; Su, J.; Chaves, L.; Maldonado, A.E.; King, M.E.; Rickey, L.; Cullen, D.; Mende, U. Gq/11-Mediated Signaling and Hypertrophy in Mice with Cardiac-Specific Transgenic Expression of Regulator of G-Protein Signaling 2. PLoS ONE 2012, 7, e40048. [Google Scholar] [CrossRef] [Green Version]
- Gutkind, J.S.; Offermanns, S. A New Gq-Initiated MAPK Signaling Pathway in the Heart. Dev. Cell 2009, 16, 163–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, O.F.; Molkentin, J.D. Involvement of Extracellular Signal-Regulated Kinases 1/2 in Cardiac Hypertrophy and Cell Death. Circ. Res. 2002, 91, 776–781. [Google Scholar] [CrossRef]
- Streicher, J.M.; Ren, S.; Herschman, H.; Wang, Y. MAPK-Activated Protein Kinase-2 in Cardiac Hypertrophy and Cyclooxygenase-2 Regulation in Heart. Circ. Res. 2010, 106, 1434–1443. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, H.T. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Jagodzik, P.; Tajdel-Zielinska, M.; Ciesla, A.; Marczak, M.; Ludwikow, A. Mitogen-Activated Protein Kinase Cascades in Plant Hormone Signaling. Front. Plant. Sci. 2018, 9, 1387. [Google Scholar] [CrossRef] [PubMed]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic Inhibition of Cardiac ERK1/2 Promotes Stress-Induced Apoptosis and Heart Failure but Has No Effect on Hypertrophy In Vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar] [CrossRef] [Green Version]
- Mutlak, M.; Kehat, I. Extracellular Signal-Regulated Kinases 1/2 as Regulators of Cardiac Hypertrophy. Front. Pharmacol. 2015, 6, 149. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Xie, Q.; Wang, L.; Lu, Y.; Liu, P.; Yang, P.; Chen, R.; Shao, C.; Qiao, C.; Wang, Z.; et al. The TIR/BB-Loop Mimetic AS-1 Prevents Ang Ii-Induced Hypertensive Cardiac Hypertrophy via NF-κB Dependent Downregulation of miRNA-143. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markou, T.; Lazou, A. Phosphorylation and Activation of Mitogen- and Stress-Activated Protein Kinase-1 in Adult Rat Cardiac Myocytes by G-Protein-Coupled Receptor Agonists Requires Both Extracellular-Signal-Regulated Kinase and p38 Mitogen-Activated Protein Kinase. Biochem. J. 2002, 365, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Mutlak, M.; Kehat, I. Dual Specific Phosphatases (DUSPs) in Cardiac Hypertrophy and Failure. Cell Signal. 2021, 84, 110033. [Google Scholar] [CrossRef]
- Sanna, B.; Bueno, O.F.; Dai, Y.-S.; Wilkins, B.J.; Molkentin, J.D. Direct and Indirect Interactions between Calcineurin-NFAT and MEK1-Extracellular Signal-Regulated Kinase 1/2 Signaling Pathways Regulate Cardiac Gene Expression and Cellular Growth. Mol. Cell Biol. 2005, 25, 865–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rooij, E.; Doevendans, P.A.; De Theije, C.C.; Babiker, F.A.; Molkentin, J.D.; De Windt, L.J. Requirement of Nuclear Factor of Activated T-Cells in Calcineurin-Mediated Cardiomyocyte Hypertrophy. J. Biol. Chem. 2002, 277, 48617–48626. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, T.; Hasegawa, K.; Wada, H.; Kakita, T.; Kaburagi, S.; Yanazume, T.; Sasayama, S. Calcineurin-GATA4 Pathway Is Involved in Beta-Adrenergic Agonist-Responsive Endothelin-1 Transcription in Cardiac Myocytes. J. Biol. Chem. 2001, 276, 34983–34989. [Google Scholar] [CrossRef] [Green Version]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac Remodeling-Concepts and Clinical Implications: A Consensus Paper from an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Archer, C.R.; Robinson, E.L.; Drawnel, F.M.; Roderick, H.L. Endothelin-1 Promotes Hypertrophic Remodelling of Cardiac Myocytes by Activating Sustained Signalling and Transcription Downstream of Endothelin Type A Receptors. Cell Signal. 2017, 36, 240–254. [Google Scholar] [CrossRef]
- Dorn, G.W.; Force, T. Protein Kinase cascades in the Regulation of Cardiac Hypertrophy. J. Clin. Invest. 2005, 115, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Selim, A.M.; Sarswat, N.; Kelesidis, I.; Iqbal, M.; Chandra, R.; Zolty, R. Plasma Serotonin in Heart Failure: Possible Marker and Potential Treatment Target. Heart Lung Circ. 2017, 26, 442–449. [Google Scholar] [CrossRef]
- Lairez, O.; Cognet, T.; Schaak, S.; Calise, D.; Guilbeau-Frugier, C.; Parini, A.; Mialet-Perez, J. Role of Serotonin 5-HT2A Receptors in the Development of Cardiac Hypertrophy in Response to Aortic Constriction in Mice. J. Neural. Transm. 2013, 120, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Guillet-Deniau, I.; Burnol, A.F.; Girard, J. Identification and Localization of a Skeletal Muscle Secrotonin 5-HT2A Receptor Coupled to the Jak/STAT Pathway. J. Biol. Chem. 1997, 272, 14825–14829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Si, L.; Xu, J.; Yi, C.; Xu, X.; Wang, F.; Gu, W.; Zhang, Y.; Wang, X. Asiatic Acid Attenuates Cardiac Hypertrophy by Blocking Transforming Growth Factor-β1-Mediated Hypertrophic Signaling In Vitro and In Vivo. Int. J. Mol. Med. 2014, 34, 499–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehat, I.; Davis, J.; Tiburcy, M.; Accornero, F.; Saba-El-Leil, M.K.; Maillet, M.; York, A.J.; Lorenz, J.N.; Zimmermann, W.H.; Meloche, S.; et al. Extracellular Signal-Regulated Kinases 1 and 2 Regulate the Balance between Eccentric and Concentric Cardiac growth. Circ. Res. 2011, 108, 176–183. [Google Scholar] [CrossRef]
- Bueno, O.F.; De Windt, L.J.; Tymitz, K.M.; Witt, S.A.; Kimball, T.R.; Klevitsky, R.; Hewett, T.E.; Jones, S.P.; Lefer, D.J.; Peng, C.F.; et al. The MEK1-ERK1/2 Signaling Pathway Promotes Compensated Cardiac Hypertrophy in Transgenic Mice. EMBO J. 2000, 19, 6341–6350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.; Lip, G.Y.H. Platelets and Heart Failure. Eur. Heart J. 2006, 27, 2623–2631. [Google Scholar] [CrossRef] [Green Version]
- Tanaka-Totoribe, N.; Hidaka, M.; Gamoh, S.; Yokota, A.; Nakamura, E.; Kuwabara, M.; Tsunezumi, J.; Yamamoto, R. Effects of M-1, a Major Metabolite of Sarpogrelate, on 5-HT-Induced Constriction of Isolated Human Internal Thoracic Artery. Biol. Pharm. Bull. 2020, 43, 1979–1982. [Google Scholar] [CrossRef]
- Sanganalmath, S.K.; Babick, A.P.; Barta, J.; Kumamoto, H.; Takeda, N.; Dhalla, N.S. Antiplatelet Therapy Attenuates Subcellular Remodelling in Congestive Heart Failure. J. Cell Mol. Med. 2008, 12, 1728–1738. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Cho, J.R.; Park, S.M.; Shaha, K.B.; Pierres, F.; Sumiya, T.; Chun, K.J.; Kang, M.K.; Choi, S.; Lee, N. Sarpogrelate Based Triple Antiplatelet Therapy Improved Left Ventricular Systolic Function in Acute Myocardial Infarction: Retrospective Study. Yonsei Med. J. 2017, 58, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Funamoto, M.; Sunagawa, Y.; Katanasaka, Y.; Shimizu, K.; Miyazaki, Y.; Sari, N.; Shimizu, S.; Mori, K.; Wada, H.; Hasegawa, K.; et al. Histone acetylation Domains are Differentially Induced during Development of Heart Failure in Dahl Salt-Sensitive Rats. Int. J. Mol. Sci. 2021, 22, 1771. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Sunagawa, Y.; Kawamura, T.; Takaya, T.; Wada, H.; Nagasawa, A.; Komeda, M.; Fujita, M.; Shimatsu, A.; Kita, T.; et al. The Dietary Compound Curcumin Inhibits p300 Histone Acetyltransferase Activity and Prevents Heart Failure in Rats. J. Clin. Invest. 2008, 118, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Sari, N.; Katanasaka, Y.; Honda, H.; Miyazaki, Y.; Sunagawa, Y.; Funamoto, M.; Shimizu, K.; Shimizu, S.; Wada, H.; Hasegawa, K.; et al. Cacao Bean Polyphenols Inhibit Cardiac Hypertrophy and Systolic Dysfunction in Pressure Overload-induced Heart Failure Model Mice. Planta Med. 2020, 86, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Hasegawa, K.; Kaburagi, S.; Kakita, T.; Wada, H.; Yanazume, T.; Sasayama, S. Phosphorylation of GATA-4 Is Involved in α1-Adrenergic Agonist-Responsive Transcription of the Endothelin-1 Gene in Cardiac Myocytes. J. Biol. Chem. 2000, 275, 13721–13726. [Google Scholar] [CrossRef] [Green Version]
- Dignam, J.D.; Lebovitz, R.M.; Roeder, R.G. Accurate Transcription Initiation by RNA Polymerase II in a Soluble Extract from Isolated Mammalian Nuclei. Nucl. Acid. Res. 1983, 11, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Sunagawa, Y.; Funamoto, M.; Wakabayashi, H.; Genpei, M.; Miyazaki, Y.; Katanasaka, Y.; Sari, N.; Shimizu, S.; Katayama, A.; et al. The Synthetic Curcumin Analogue GO-Y030 Effectively Suppresses the Development of Pressure Overload-induced Heart Failure in Mice. Sci. Rep. 2020, 10, 7172. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Shimizu, K.; Katayama, A.; Funamoto, M.; Shimizu, K.; Nurmila, S.; Shimizu, S.; Miyazaki, Y.; Katanasaka, Y.; Hasegawa, K.; et al. Metformin Suppresses Phenylephrine-Induced Hypertrophic Responses by Inhibiting p300-HAT Activity in Cardiomyocytes. J. Pharmacol. Sci. 2021, 147, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Morimoto, T.; Takaya, T.; Kaichi, S.; Wada, H.; Kawamura, T.; Fujita, M.; Shimatsu, A.; Kita, T.; Hasegawa, K. Cyclin-Dependent Kinase-9 Is a Component of the p300/GATA4 Complex Required for Phenylephrine-Induced Hypertrophy in Cardiomyocytes. J. Biol. Chem. 2010, 285, 9556–9568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Katanasaka, Y.; Sunagawa, Y.; Miyazaki, Y.; Funamoto, M.; Wada, H.; Hasegawa, K.; Morimoto, T. Tyrosine Phosphorylation of RACK1 Triggers Cardiomyocyte Hypertrophy by Regulating the Interaction between p300 and GATA4. Biochim. Biophys. Acta 2016, 1862, 1544–1557. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Funamoto, M.; Shimizu, K.; Shimizu, S.; Sari, N.; Katanasaka, Y.; Miyazaki, Y.; Kakeya, H.; Hasegawa, K.; Morimoto, T. Curcumin, an Inhibitor of p300-HAT Activity, Suppresses the Development of Hypertension-Induced Left Ventricular Hypertrophy with Preserved Ejection Fraction in Dahl Rats. Nutrients 2021, 13, 2608. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Sono, S.; Katanasaka, Y.; Funamoto, M.; Hirano, S.; Miyazaki, Y.; Hojo, Y.; Suzuki, H.; Morimoto, E.; Marui, A.; et al. Optimal Dose-Setting Study of Curcumin for Improvement of Left Ventricular Systolic Function after Myocardial Infarction in Rats. J. Pharmacol. Sci. 2014, 126, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunagawa, Y.; Funamoto, M.; Sono, S.; Shimizu, K.; Shimizu, S.; Genpei, M.; Miyazaki, Y.; Katanasaka, Y.; Morimoto, E.; Ueno, M.; et al. Curcumin and Its Demethoxy Derivatives Possess p300 HAT Inhibitory Activity and Suppress Hypertrophic Responses in Cardiomyocytes. J. Pharmacol. Sci. 2018, 136, 212–217. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham | TAC | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vehicle | Vehicle | SA 1 mg/kg | SA 5 mg/kg | |||||||||
| LVPWd (mm) | 1.50 | ± | 0.07 | 2.05 | ± | 0.09 ** | 1.83 | ± | 0.14 | 1.55 | ± | 0.08 †† |
| LVIDd (mm) | 2.67 | ± | 0.12 | 2.94 | ± | 0.16 | 2.75 | ± | 0.12 | 2.79 | ± | 0.13 |
| FS (%) | 57.4 | ± | 2.2 | 37.7 | ± | 2.4 ** | 41.9 | ± | 3.7 ** | 50.7 | ± | 0.8 †† |
| R-R int (sec) | 0.101 | ± | 0.001 | 0.103 | ± | 0.001 | 0.101 | ± | 0.001 | 0.102 | ± | 0.002 |
| LVMI (mg/mm) | 7.0 | ± | 0.2 | 13.0 | ± | 0.8 ** | 10.8 | ± | 1.0 ** | 9.5 | ± | 1.2 † |
| HW (mg) | 118.3 | ± | 2.2 | 192.1 | ± | 12.7 ** | 166.6 | ± | 2.0 ** | 151.0 | ± | 3.6 † |
| TL (mm) | 22.5 | ± | 0.6 | 22.5 | ± | 0.4 | 22.1 | ± | 0.7 | 23.2 | ± | 0.6 |
| HW/TL (mg/mm) | 5.3 | ± | 0.2 | 8.6 | ± | 0.6 ** | 7.6 | ± | 0.2 ** | 6.5 | ± | 0.2 † |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimizu, K.; Sunagawa, Y.; Funamoto, M.; Honda, H.; Katanasaka, Y.; Murai, N.; Kawase, Y.; Hirako, Y.; Katagiri, T.; Yabe, H.; et al. The Selective Serotonin 2A Receptor Antagonist Sarpogrelate Prevents Cardiac Hypertrophy and Systolic Dysfunction via Inhibition of the ERK1/2–GATA4 Signaling Pathway. Pharmaceuticals 2021, 14, 1268. https://doi.org/10.3390/ph14121268
Shimizu K, Sunagawa Y, Funamoto M, Honda H, Katanasaka Y, Murai N, Kawase Y, Hirako Y, Katagiri T, Yabe H, et al. The Selective Serotonin 2A Receptor Antagonist Sarpogrelate Prevents Cardiac Hypertrophy and Systolic Dysfunction via Inhibition of the ERK1/2–GATA4 Signaling Pathway. Pharmaceuticals. 2021; 14(12):1268. https://doi.org/10.3390/ph14121268
Chicago/Turabian StyleShimizu, Kana, Yoichi Sunagawa, Masafumi Funamoto, Hiroki Honda, Yasufumi Katanasaka, Noriyuki Murai, Yuto Kawase, Yuta Hirako, Takahiro Katagiri, Harumi Yabe, and et al. 2021. "The Selective Serotonin 2A Receptor Antagonist Sarpogrelate Prevents Cardiac Hypertrophy and Systolic Dysfunction via Inhibition of the ERK1/2–GATA4 Signaling Pathway" Pharmaceuticals 14, no. 12: 1268. https://doi.org/10.3390/ph14121268