Co-Clinical Trials: An Innovative Drug Development Platform for Cholangiocarcinoma

 ,
,  ,
, 
 , ,
, ,
Abstract
:
1. Introduction
2. Current Standard-of-Care in CCA Management
3. The Current Landscape of Targeted Therapies in CCA
4. What Are Co-Clinical Trials?
5. Co-Clinical Trials to Accelerate Drug Development in CCA
6. Proof-of-Concept of Co-Clinical Trials Expediting Drug Development in Other Cancers
7. Animal Models Are Used as Pre-Clinical Models in the Current Co-Clinical Trial System
8. Potential Use of Non-Animal Models as Pre-Clinical Models in Co-Clinical Trials for CCA
9. A Proposed Model of Co-Clinical Trials in CCA to Accelerate Drug Development
10. Applications of Co-Clinical Trial in CCA Research and Development
11. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Treeprasertsuk, S.; Poovorawan, K.; Soonthornworasiri, N.; Chaiteerakij, R.; Thanapirom, K.; Mairiang, P.; Sawadpanich, K.; Sonsiri, K.; Mahachai, V.; Phaosawasdi, K. A significant cancer burden and high mortality of intrahepatic cholangiocarcinoma in Thailand: A nationwide database study. BMC Gastroenterol. 2017, 17, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, T. Cholangiocarcinoma--controversies and challenges. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvise, M.; Lamarca, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 98–107. [Google Scholar] [CrossRef] [Green Version]
- Squires, M.H.; Cloyd, J.M.; Dillhoff, M.; Schmidt, C.; Pawlik, T.M. Challenges of surgical management of intrahepatic cholangiocarcinoma. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 671–681. [Google Scholar] [CrossRef]
- Blechacz, B.; Gores, G.J. Cholangiocarcinoma: Advances in pathogenesis, diagnosis, and treatment. Hepatology 2008, 48, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, D.S.; Roberts, L.R. Diagnosis and management of cholangiocarcinoma. Curr. Gastroenterol. Rep. 2008, 10, 43–52. [Google Scholar] [CrossRef]
- Sriputtha, S.; Khuntikeo, N.; Promthet, S.; Kamsa-Ard, S. Survival rate of intrahepatic cholangiocarcinoma patients after surgical treatment in Thailand. Asian Pac. J. Cancer Prev. 2013, 14, 1107–1110. [Google Scholar] [CrossRef] [Green Version]
- Hyder, O.; Hatzaras, I.; Sotiropoulos, G.C.; Paul, A.; Alexandrescu, S.; Marques, H.; Pulitano, C.; Barroso, E.; Clary, B.M.; Aldrighetti, L.; et al. Recurrence after operative management of intrahepatic cholangiocarcinoma. Surgery 2013, 153, 811–818. [Google Scholar] [CrossRef]
- Patel, T.; Singh, P. Cholangiocarcinoma: Emerging approaches to a challenging cancer. Curr. Opin. Gastroenterol. 2007, 23, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Eckmann, K.R.; Patel, D.K.; Landgraf, A.; Slade, J.H.; Lin, E.; Kaur, H.; Loyer, E.; Weatherly, J.M.; Javle, M. Chemotherapy outcomes for the treatment of unresectable intrahepatic and hilar cholangiocarcinoma: A retrospective analysis. Gastrointest. Cancer Res. 2011, 4, 155–160. [Google Scholar] [PubMed]
- Cao, J.; Hu, J.; Liu, S.; Meric-Bernstam, F.; Abdel-Wahab, R.; Xu, J.; Li, Q.; Yan, M.; Feng, Y.; Lin, J.; et al. Intrahepatic Cholangiocarcinoma: Genomic Heterogeneity Between Eastern and Western Patients. JCO Precis. Oncol. 2020, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, V.; Bragazzi, M.C.; Carpino, G.; Torrice, A.; Fraveto, A.; Gentile, R.; Pasqualino, V.; Melandro, F.; Aliberti, C.; Bastianelli, C.; et al. Cholangiocarcinoma: Increasing burden of classifications. Hepatobiliary Surg. Nutr. 2013, 2, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Mian, I.; Blechacz, B. Cancer review: Cholangiocarcinoma. J. Carcinog. 2015, 14, 1. [Google Scholar] [CrossRef]
- Cardinale, V. Classifications and misclassification in cholangiocarcinoma. Liver Int. 2019, 39, 260–262. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, V.; Carpino, G. Multilevel heterogeneity of biliary tract cancers may affect the modelling of prognosis. Liver Int. 2017, 37, 1773–1775. [Google Scholar] [CrossRef] [Green Version]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS ONE 2014, 9, e115383. [Google Scholar] [CrossRef] [Green Version]
- Janiaud, P.; Serghiou, S.; Ioannidis, J.P.A. New clinical trial designs in the era of precision medicine: An overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat. Rev. 2019, 73, 20–30. [Google Scholar] [CrossRef]
- Matchett, K.B.; Lynam-Lennon, N.; Watson, R.W.; Brown, J.A.L. Advances in Precision Medicine: Tailoring Individualized Therapies. Cancers 2017, 9, 146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Shi, J.; Wang, Y.; Zhou, H.; Zhang, Z.; Han, Z.; Li, G.; Yang, B.; Cao, G.; Ke, Y.; et al. Next-generation sequencing-guided molecular-targeted therapy and immunotherapy for biliary tract cancers. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gruenberger, B.; Schueller, J.; Heubrandtner, U.; Wrba, F.; Tamandl, D.; Kaczirek, K.; Roka, R.; Freimann-Pircher, S.; Gruenberger, T. Cetuximab, gemcitabine, and oxaliplatin in patients with unresectable advanced or metastatic biliary tract cancer: A phase 2 study. Lancet Oncol. 2010, 11, 1142–1148. [Google Scholar] [CrossRef]
- Guion-Dusserre, J.F.; Lorgis, V.; Vincent, J.; Bengrine, L.; Ghiringhelli, F. FOLFIRI plus bevacizumab as a second-line therapy for metastatic intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 2096–2101. [Google Scholar] [CrossRef]
- Lubner, S.J.; Mahoney, M.R.; Kolesar, J.L.; Loconte, N.K.; Kim, G.P.; Pitot, H.C.; Philip, P.A.; Picus, J.; Yong, W.P.; Horvath, L.; et al. Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: A phase II Consortium study. J. Clin. Oncol. 2010, 28, 3491–3497. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Meyerhardt, J.A.; Blaszkowsky, L.S.; Kambadakone, A.R.; Muzikansky, A.; Zheng, H.; Clark, J.W.; Abrams, T.A.; Chan, J.A.; Enzinger, P.C.; et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: A phase 2 study. Lancet Oncol. 2010, 11, 48–54. [Google Scholar] [CrossRef]
- Voss, J.S.; Holtegaard, L.M.; Kerr, S.E.; Fritcher, E.G.; Roberts, L.R.; Gores, G.J.; Zhang, J.; Highsmith, W.E.; Halling, K.C.; Kipp, B.R. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum. Pathol. 2013, 44, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.V.; Zhang, S.; Chen, X.; Calvisi, D.F.; Andersen, J.B. Molecular profiling of intrahepatic cholangiocarcinoma: The search for new therapeutic targets. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 349–356. [Google Scholar] [CrossRef]
- Shroff, R.T.; Kennedy, E.B.; Bachini, M.; Bekaii-Saab, T.; Crane, C.; Edeline, J.; El-Khoueiry, A.; Feng, M.; Katz, M.H.G.; Primrose, J.; et al. Adjuvant Therapy for Resected Biliary Tract Cancer: ASCO Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 1015–1027. [Google Scholar] [CrossRef] [Green Version]
- Okusaka, T.; Nakachi, K.; Fukutomi, A.; Mizuno, N.; Ohkawa, S.; Funakoshi, A.; Nagino, M.; Kondo, S.; Nagaoka, S.; Funai, J.; et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: A comparative multicentre study in Japan. Br. J. Cancer 2010, 103, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Marin, J.J.G.; Lozano, E.; Herraez, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Briz, O.; Serrano, M.A.; Efferth, T.; Macias, R.I.R. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1444–1453. [Google Scholar] [CrossRef]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; Gebbia, V.; Pressiani, T.; Testa, A.; Personeni, N.; Arrivas Bajardi, E.; Foa, P.; Buonadonna, A.; Bencardino, K.; Barone, C.; et al. A randomized, multicenter, phase II study of vandetanib monotherapy versus vandetanib in combination with gemcitabine versus gemcitabine plus placebo in subjects with advanced biliary tract cancer: The VanGogh study. Ann. Oncol. 2015, 26, 542–547. [Google Scholar] [CrossRef]
- Krook, M.A.; Lenyo, A.; Wilberding, M.; Barker, H.; Dantuono, M.; Bailey, K.M.; Chen, H.Z.; Reeser, J.W.; Wing, M.R.; Miya, J.; et al. Efficacy of FGFR Inhibitors and Combination Therapies for Acquired Resistance in FGFR2-Fusion Cholangiocarcinoma. Mol. Cancer Ther. 2020, 19, 847–857. [Google Scholar] [CrossRef] [Green Version]
- FDA. FDA Grants Accelerated Approval to Pemigatinib for Cholangiocarcinoma with an FGFR2 Rearrangement or Fusion. 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pemigatinib-cholangiocarcinoma-fgfr2-rearrangement-or-fusion (accessed on 2 July 2020).
- Nardella, C.; Lunardi, A.; Patnaik, A.; Cantley, L.C.; Pandolfi, P.P. The APL paradigm and the “co-clinical trial” project. Cancer Discov. 2011, 1, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Lunardi, A.; Pandolfi, P.P. A co-clinical platform to accelerate cancer treatment optimization. Trends Mol. Med. 2015, 21, 1–5. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Clohessy, J.G.; Pandolfi, P.P. Mouse hospital and co-clinical trial project--from bench to bedside. Nat. Rev. Clin. Oncol. 2015, 12, 491–498. [Google Scholar] [CrossRef]
- Kim, H.R.; Kang, H.N.; Shim, H.S.; Kim, E.Y.; Kim, J.; Kim, D.J.; Lee, J.G.; Lee, C.Y.; Hong, M.H.; Kim, S.M.; et al. Co-clinical trials demonstrate predictive biomarkers for dovitinib, an FGFR inhibitor, in lung squamous cell carcinoma. Ann. Oncol. 2017, 28, 1250–1259. [Google Scholar] [CrossRef]
- Chen, Z.; Akbay, E.; Mikse, O.; Tupper, T.; Cheng, K.; Wang, Y.; Tan, X.; Altabef, A.; Woo, S.A.; Chen, L.; et al. Co-clinical trials demonstrate superiority of crizotinib to chemotherapy in ALK-rearranged non-small cell lung cancer and predict strategies to overcome resistance. Clin. Cancer Res. 2014, 20, 1204–1211. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Cheng, K.; Walton, Z.; Wang, Y.C.; Ebi, H.; Shimamura, T.; Liu, Y.; Tupper, T.; Ouyang, J.; Li, J.; et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 2012, 483, 613–617. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Zhang, G.; Kim, H.S.; Stinson, R.M.; Bechara, R.; Zhang, C.; Chen, Z.; Saba, N.F.; Pakkala, S.; Pillai, R.; et al. Patient-derived xenografts faithfully replicated clinical outcome in a phase II co-clinical trial of arsenic trioxide in relapsed small cell lung cancer. J. Transl. Med. 2016, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Lunardi, A.; Ala, U.; Epping, M.T.; Salmena, L.; Clohessy, J.G.; Webster, K.A.; Wang, G.; Mazzucchelli, R.; Bianconi, M.; Stack, E.C.; et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat. Genet. 2013, 45, 747–755. [Google Scholar] [CrossRef]
- Dutta, A.; Panja, S.; Virk, R.K.; Kim, J.Y.; Zott, R.; Cremers, S.; Golombos, D.M.; Liu, D.; Mosquera, J.M.; Mostaghel, E.A.; et al. Co-clinical Analysis of a Genetically Engineered Mouse Model and Human Prostate Cancer Reveals Significance of NKX3.1 Expression for Response to 5alpha-reductase Inhibition. Eur. Urol. 2017, 72, 499–506. [Google Scholar] [CrossRef]
- Kwong, L.N.; Boland, G.M.; Frederick, D.T.; Helms, T.L.; Akid, A.T.; Miller, J.P.; Jiang, S.; Cooper, Z.A.; Song, X.; Seth, S.; et al. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2015, 125, 1459–1470. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Kang, H.N.; Yun, M.R.; Ju, K.Y.; Choi, J.W.; Jung, D.M.; Pyo, K.H.; Hong, M.H.; Ahn, M.J.; Sun, J.M.; et al. Mouse-human co-clinical trials demonstrate superior anti-tumour effects of buparlisib (BKM120) and cetuximab combination in squamous cell carcinoma of head and neck. Br. J. Cancer 2020, 123, 1720–1729. [Google Scholar] [CrossRef]
- Menachery, S.P.; Laprevote, O.; Nguyen, T.P.; Aravind, U.K.; Gopinathan, P.; Aravindakumar, C.T. Identification of position isomers by energy-resolved mass spectrometry. J. Mass Spectrom 2015, 50, 944–950. [Google Scholar] [CrossRef]
- Jackson, S.J.; Thomas, G.J. Human tissue models in cancer research: Looking beyond the mouse. Dis. Models Mech. 2017, 10, 939. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Li, X.; Liu, P.; Li, M.; Luo, F. Patient-derived xenograft mouse models: A high fidelity tool for individualized medicine (Review). Oncol. Lett. 2018. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Zhao, Z.Y.; An, Q.M.; Dong, B.; Lv, A.; Li, C.P.; Guan, X.Y.; Tian, X.Y.; Wu, J.H.; Hao, C.Y. Comprehensive comparison of patient-derived xenograft models in Hepatocellular Carcinoma and metastatic Liver Cancer. Int. J. Med. Sci 2020, 17, 3073–3081. [Google Scholar] [CrossRef]
- Pompili, L.; Porru, M.; Caruso, C.; Biroccio, A.; Leonetti, C. Patient-derived xenografts: A relevant preclinical model for drug development. J. Exp. Clin. Cancer Res. 2016, 35, 189. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Pitt, J.M.; Daillere, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef]
- Usai, A.; Di Franco, G.; Colucci, P.; Pollina, L.E.; Vasile, E.; Funel, N.; Palmeri, M.; Dente, L.; Falcone, A.; Morelli, L.; et al. A Model of a Zebrafish Avatar for Co-Clinical Trials. Cancers 2020, 12, 677. [Google Scholar] [CrossRef] [Green Version]
- Robertson, N.; Schook, L.B.; Schachtschneider, K.M. Porcine cancer models: Potential tools to enhance cancer drug trials. Expert Opin. Drug Discov. 2020, 15, 893–902. [Google Scholar] [CrossRef]
- Washburn, A.L.; Shia, W.W.; Lenkeit, K.A.; Lee, S.H.; Bailey, R.C. Multiplexed cancer biomarker detection using chip-integrated silicon photonic sensor arrays. Analyst 2016, 141, 5358–5365. [Google Scholar] [CrossRef] [Green Version]
- Horvath, P.; Aulner, N.; Bickle, M.; Davies, A.M.; Nery, E.D.; Ebner, D.; Montoya, M.C.; Ostling, P.; Pietiainen, V.; Price, L.S.; et al. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 2016, 15, 751–769. [Google Scholar] [CrossRef] [Green Version]
- Eribol, P.; Uguz, A.K.; Ulgen, K.O. Screening applications in drug discovery based on microfluidic technology. Biomicrofluidics 2016, 10, 011502. [Google Scholar] [CrossRef] [Green Version]
- Caplin, J.D.; Granados, N.G.; James, M.R.; Montazami, R.; Hashemi, N. Microfluidic Organ-on-a-Chip Technology for Advancement of Drug Development and Toxicology. Adv. Healthc. Mater. 2015, 4, 1426–1450. [Google Scholar] [CrossRef] [Green Version]
- Portillo-Lara, R.; Annabi, N. Microengineered cancer-on-a-chip platforms to study the metastatic microenvironment. Lab. Chip 2016, 16, 4063–4081. [Google Scholar] [CrossRef] [Green Version]
- Pallante, P.; Pisapia, P.; Bellevicine, C.; Malapelle, U.; Troncone, G. Circulating Tumour Cells in Predictive Molecular Pathology: Focus on Drug-Sensitive Assays and 3D Culture. Acta Cytol. 2019, 63, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Vicent, S.; Lieshout, R.; Saborowski, A.; Verstegen, M.M.A.; Raggi, C.; Recalcati, S.; Invernizzi, P.; van der Laan, L.J.W.; Alvaro, D.; Calvisi, D.F.; et al. Experimental models to unravel the molecular pathogenesis, cell of origin and stem cell properties of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 79–97. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guo, D.; Zhang, Y.; Wang, L.; Sun, T.; Li, Z.; Zhang, X.; Wang, S.; Chen, Y.; Wu, A. Rapid screening for individualized chemotherapy optimization of colorectal cancer: A novel conditional reprogramming technology-based functional diagnostic assay. Transl. Oncol. 2020, 14, 100935. [Google Scholar] [CrossRef]
- Manzella, G.; Schreck, L.D.; Breunis, W.B.; Molenaar, J.; Merks, H.; Barr, F.G.; Sun, W.; Rommele, M.; Zhang, L.; Tchinda, J.; et al. Phenotypic profiling with a living biobank of primary rhabdomyosarcoma unravels disease heterogeneity and AKT sensitivity. Nat. Commun. 2020, 11, 4629. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26. [Google Scholar] [CrossRef]
- Zabron, A.; Edwards, R.J.; Khan, S.A. The challenge of cholangiocarcinoma: Dissecting the molecular mechanisms of an insidious cancer. Dis. Model Mech. 2013, 6, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Shotelersuk, V.; Tongsima, S.; Pithukpakorn, M.; Eu-Ahsunthornwattana, J.; Mahasirimongkol, S. Precision medicine in Thailand. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 245–253. [Google Scholar] [CrossRef]
- Khuntikeo, N.; Chamadol, N.; Yongvanit, P.; Loilome, W.; Namwat, N.; Sithithaworn, P.; Andrews, R.H.; Petney, T.N.; Promthet, S.; Thinkhamrop, K.; et al. Cohort profile: Cholangiocarcinoma screening and care program (CASCAP). BMC Cancer 2015, 15, 459. [Google Scholar] [CrossRef] [Green Version]



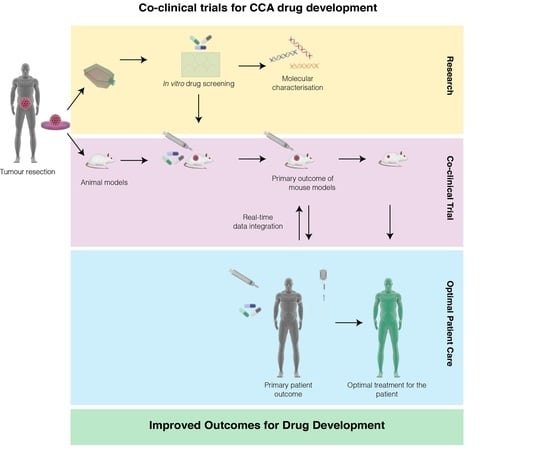
{kind=link}
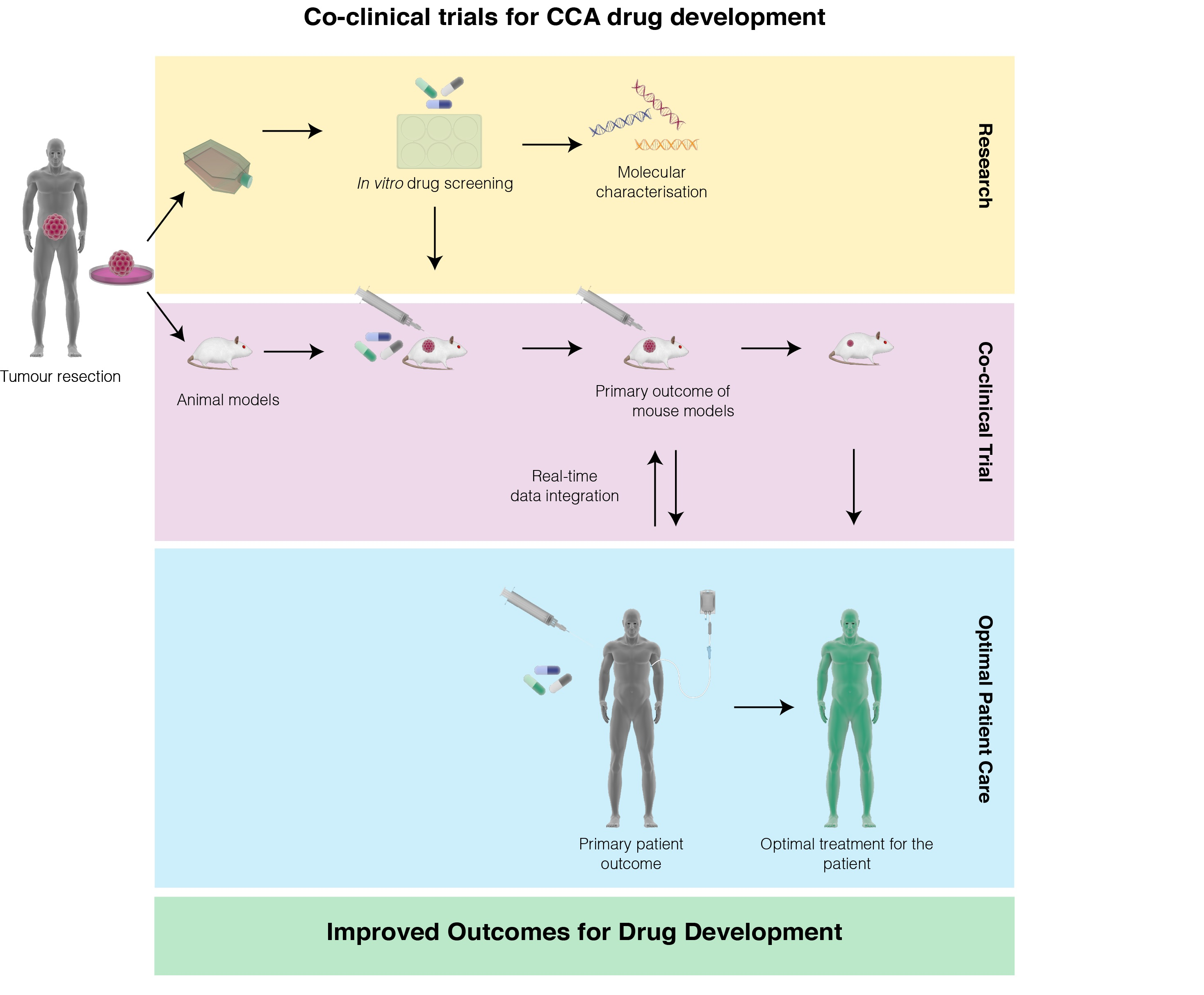
{kind=link}
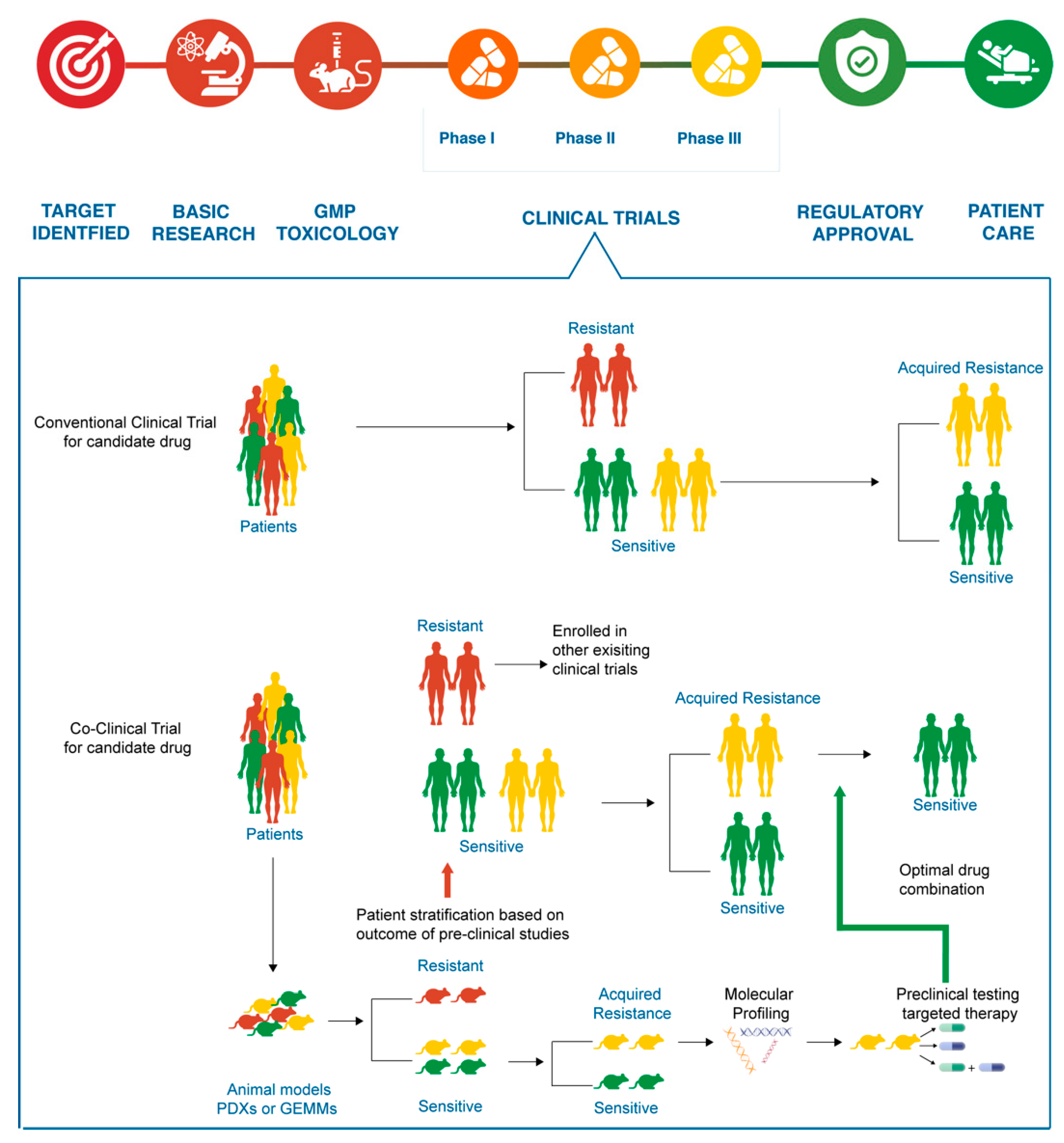
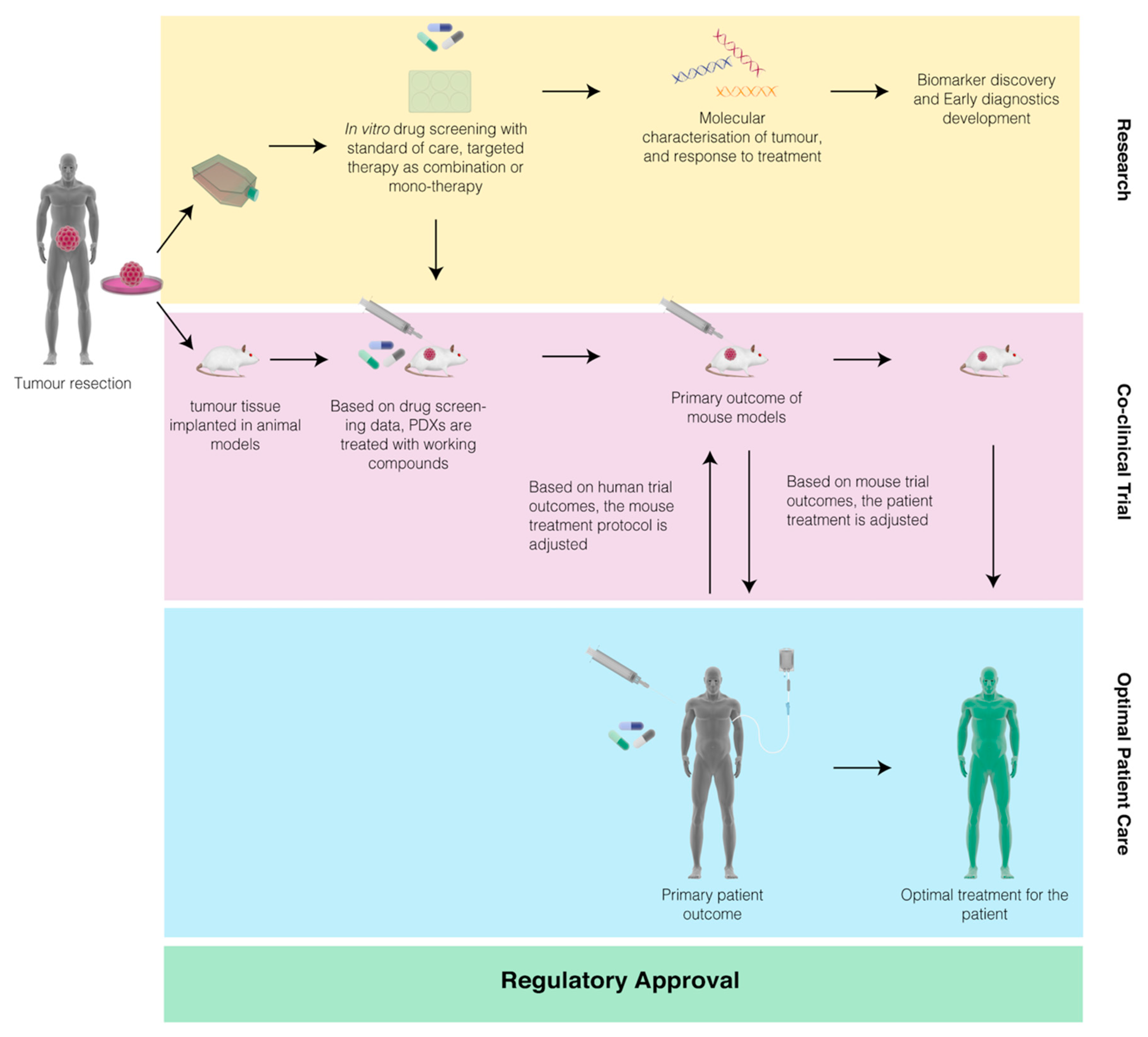{kind=link}
| Cancer Type | Testing Model | Drug | Outcome | Reference |
|---|---|---|---|---|
| Lung squamous cell carcinoma | PDX | dovitinib | FGFR pathways can predict the sensitivity to dovitinib both in cell lines and PDX tumors. | [41] |
| Non-small cell lung cancer | GEMM | crizotinib | ALK inhibitor crizotinib was more effective than standard-of-care drugs in a comparative co-clinical study with patients and GEMM mouse models. The study facilitates the prediction of crizotinib resistance. | [42] |
| KRAS mutant non-small cell lung cancer | GEMM | selumetinib | The addition of selumetinib proved differential response in mice with lung cancer caused by Kras mutation. Mice with Kras + p53 mutations were sensitive but mice with Kras + Lkb1 mutations showed resistance to combination therapy. | [43] |
| Relapsed small cell lung cancer | PDX | arsenic trioxide | Strong correlation between the response of arsenic trioxide and cisplatin in SCLC clinical and PDX model supports the use of PDX models to screen promising anticancer agents prior to clinical testing in patients. | [44] |
| Prostate cancer | GEMM | androgen deprivation therapy | Genetic mouse models along with patients elucidate the mechanism of castration resistance. Results encourage the stratification of patients based for precision medicine. | [45] |
| Prostate cancer | GEMM | 5α-reductase inhibitors | Genetic expression profiling of NKX3.1 mutant mouse models and patients in treatment were compared NKX3.1 expression predicts response to 5α-reductase inhibitors (5-ARI). | [46] |
| Melanoma | GEMM | BRAF inhibitor | Co-clinical analysis of human and mouse melanomas elucidates the patterns of resistance to BRAF inhibitors. This study also identifies biomarkers to predict response. | [47] |
| Head and neck cancer | PDX | buparlisib | Integrated mouse trials helped identify the anti-tumor effects of combination therapy with cetuximab and revise the treatment regime, thus enabling promising outcomes of the clinical trials. | [48] |
| Model | Advantages | Disadvantages | References |
|---|---|---|---|
| PDX |
|
| [40,51,52,54] |
| GEMM |
|
| [54] |
| Model | Advantages | Disadvantages | References |
|---|---|---|---|
| 3D Cell culture (e.g., organoids and spheroids) |
|
| [58,59,64] |
| Micro-engineered systems |
|
| [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balasubramanian, B.; Venkatraman, S.; Myint, K.Z.; Janvilisri, T.; Wongprasert, K.; Kumkate, S.; Bates, D.O.; Tohtong, R. Co-Clinical Trials: An Innovative Drug Development Platform for Cholangiocarcinoma. Pharmaceuticals 2021, 14, 51. https://doi.org/10.3390/ph14010051
Balasubramanian B, Venkatraman S, Myint KZ, Janvilisri T, Wongprasert K, Kumkate S, Bates DO, Tohtong R. Co-Clinical Trials: An Innovative Drug Development Platform for Cholangiocarcinoma. Pharmaceuticals. 2021; 14(1):51. https://doi.org/10.3390/ph14010051
Chicago/Turabian StyleBalasubramanian, Brinda, Simran Venkatraman, Kyaw Zwar Myint, Tavan Janvilisri, Kanokpan Wongprasert, Supeecha Kumkate, David O. Bates, and Rutaiwan Tohtong. 2021. "Co-Clinical Trials: An Innovative Drug Development Platform for Cholangiocarcinoma" Pharmaceuticals 14, no. 1: 51. https://doi.org/10.3390/ph14010051