Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach

 ,
,  , ,
, , 
Abstract
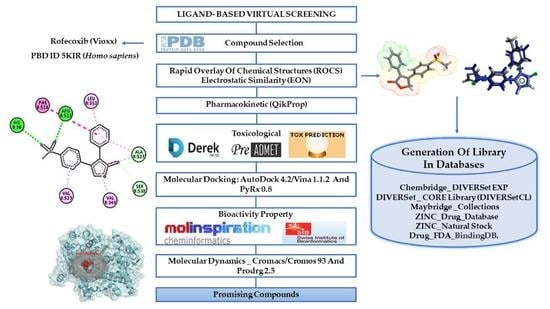
:
1. Introduction
2. Results and Discussion
2.1. Ligand-Based Virtual Screening
2.2. Pharmacokinetic Predictions for the Selected Compounds
2.3. Molecular Docking Simulations Study
2.4. Biological Target Prediction
2.5. Molecular Dynamics (MD) Simulations and Structural Analysis of Systems
Binding Free Energy
2.6. Structure-Activity Relationship of the Promising Molecules
2.7. Prediction of Toxicological Properties
2.8. Predictions of the Cardiotoxicity
3. Materials and Methods
3.1. Template Compound
3.2. Generation of Confomer Library in Databases
3.3. Virtual Screening
3.3.1. Rapid Overlay of Chemical Structures (ROCS)
3.3.2. Electrostatic Similarity (EON)
3.4. In Silico Pharmacokinetic and Toxicological Properties
3.4.1. Pharmacokinetic Predictions
3.4.2. Toxicological Predictions
3.4.3. Prediction of Toxicity Lethal Dose (LD50)
3.4.4. Prediction of the Cardiotoxicity
3.5. Prediction of Biological Target
3.6. Molecular Docking Simulations Study
3.6.1. Selection of Therapeutic Target Structure and Ligand
3.6.2. Docking Study with AutoDock 4.2/Vina 1.1.2 Via Graphical Interface PyRx (Version 0.8.30)
3.7. Molecular Dynamics (MD) Simulation Protocol
3.8. Free Energy Calculation Using MM/GBSA Approach
3.9. Per-Residue Energy Decomposition
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, I. Distinct functions of COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002, 68–69, 165–175. [Google Scholar] [CrossRef]
- Cámara-Lemarroy, C.R.; La Garza, F.J.G.D.; Fernández-Garza, N.E. Molecular inflammatory mediators in peripheral nerve degeneration and regeneration. Neuroimmunomodulation 2010, 17, 314–324. [Google Scholar] [CrossRef]
- Stack, E.; DuBois, R.N. Regulation of cyclo-oxygenase-2. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 787–800. [Google Scholar] [CrossRef]
- Mozziconacci, J.C.; Arnoult, E.; Bernard, P.; Do, Q.T.; Marot, C.; Morin-Allory, L. Optimization and validation of a docking-scoring protocol; application to virtual screening for COX-2 inhibitors. J. Med. Chem. 2005, 48, 1055–1068. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011, 10, 655–683. [Google Scholar]
- Turini, M.E.; DuBois, R.N. Cyclooxygenase-2: A Therapeutic Target. Annu. Rev. Med. 2002, 53, 35–57. [Google Scholar] [CrossRef]
- Sibbald, B. Rofecoxib (Vioxx) voluntarily withdrawn from market. CMAJ 2004, 171, 1027–1028. [Google Scholar] [CrossRef] [Green Version]
- Pasero, C.; McCaffery, M. Selective COX-2 inhibitors. Am. J. Nurs. 2001, 101, 55–56. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Hillson, J.L.; Furst, D.E. Rofecoxib. Expert Opin. Pharmacother. 2000, 1, 1053–1066. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1607, pp. 627–641. [Google Scholar]
- Orlando, B.J.; Malkowski, M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 772–776. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.C.; Poiani, J.G.C.; Ramos, R.S.; Costa, J.S.; Silva, C.H.T.P.; Brasil, D.S.B.; Santos, C.B.R. Ligand- And structure-based virtual screening of 16-((diiso-butylamino)methyl)-6α-hydroxyvouacapane-7β,17β-lactone, a compound with potential anti-prostate cancer activity. J. Serb. Chem. Soc. 2019, 84, 153–174. [Google Scholar] [CrossRef] [Green Version]
- Palheta, I.C.; Ferreira, L.R.; Vale, J.K.L.; Silva, O.P.P.; Herculano, A.M.; Oliveira, K.R.H.M.; Neto, A.M.; Campos, J.M.; Santos, C.B.R.; Borges, R.S. Alkylated Sesamol Derivatives as Potent Antioxidants. Molecules 2020, 25, 3300. [Google Scholar] [CrossRef]
- De Souza, G.C.; Matias Pereira, A.C.; Viana, M.D.; Ferreira, A.M.; Da Silva, I.D.R.; De Oliveira, M.M.R.; Barbosa, W.L.R.; Silva, L.B.; Ferreira, I.M.; Dos Santos, C.B.R.; et al. Acmella oleracea (L) R. K. Jansen Reproductive Toxicity in Zebrafish: An In Vivo and In Silico Assessment. Evid. Based Complement Altern. Med. 2019, 2019. [Google Scholar] [CrossRef]
- PreADMET | Prediction of ADME/Tox—Just another BMDRC Sites. Available online: https://preadmet.bmdrc.kr/ (accessed on 26 May 2020).
- PreADMET Version 2.0; Bioinformatics and Molecular Design Research Center: Incheon, Korea, 2020.
- Cruz, J.V.; Neto, M.F.A.; Silva, L.B.; da Ramos, R.S.; da Costa, J.S.; Brasil, D.S.B.; Lobato, C.C.; Da Costa, G.V.; Bittencourt, J.A.H.M.; Da Silva, C.H.T.P.; et al. Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef] [Green Version]
- Bittencourt, J.A.H.M.; Neto, M.F.A.; Lacerda, P.S.; Bittencourt, R.C.V.S.; Silva, R.C.; Lobato, C.C.; Silva, L.B.; Leite, F.H.A.; Zuliani, J.P.; Rosa, J.M.C.; et al. In silico evaluation of ibuprofen and two benzoylpropionic acid derivatives with potential anti-inflammatory activity. Molecules 2019, 24, 1476. [Google Scholar] [CrossRef] [Green Version]
- Enmozhi, S.K.; Raja, K.; Sebastine, I.; Joseph, J. Andrographolide As a Potential Inhibitor of SARS-CoV-2 Main Protease: An In Silico Approach. J. Biomol. Struct. Dyn. 2020, 1–7. [Google Scholar] [CrossRef]
- Mohan, A.C. Determination of Molecular Property, Bioactivity Score and Binding Energy of the Phytochemical Compounds Present in Cassia Auriculata by Molinspiration and DFT Method. Texila Int. J. Basic Med. Sci. 2017, 2, 8–22. [Google Scholar] [CrossRef]
- Desai, P.V.; Patny, A.; Sabnis, Y.; Tekwani, B.; Gut, J.; Rosenthal, P.; Srivastava, A.; Avery, M. Identification of novel parasitic cysteine protease inhibitors using virtual screening. 1. The ChemBridge database. J. Med. Chem. 2004, 47, 6609–6615. [Google Scholar] [CrossRef]
- Ramos, R.S.; Macêdo, W.J.C.; Costa, J.S.; da Silva, C.H.T.d.P.; Rosa, J.M.C.; da Cruz, J.N.; de Oliveira, M.S.; de Aguiar Andrade, E.H.; Silva, R.B.L.E.; Souto, R.N.P.; et al. Potential inhibitors of the enzyme acetylcholinesterase and juvenile hormone with insecticidal activity: Study of the binding mode via docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 1–23. [Google Scholar] [CrossRef]
- Chandra, N.; Bhagavat, R.; Sharma, E.; Sreekanthreddy, P.; Somasundaram, K. Virtual screening, identification and experimental testing of novel inhibitors of PBEF1/Visfatin/NMPRTase for glioma therapy. J. Clin. Bioinforma. 2011, 1, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, 198–201. [Google Scholar] [CrossRef] [Green Version]
- Kearnes, S.; Pande, V. ROCS-derived features for virtual screening. J. Comput. Aided Mol. Des. 2016, 30, 609–617. [Google Scholar] [CrossRef] [Green Version]
- AFITT-CL, version 2.4.1.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2017; Available online: http://www.eyesopen.com/ (accessed on 26 May 2020).
- ROCS OpenEye | ROCS Software | Virtual Screening|Lead Hopping. Available online: https://www.eyesopen.com/rocs (accessed on 26 May 2020).
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Grant, J.A.; Pickup, B.T. Gaussian shape methods. Comput. Simul. Biomol. Syst. 1997, 150–176. [Google Scholar] [CrossRef]
- Wiggers, H.J.; Rocha, J.R.; Cheleski, J.; Montanari, C.A. Integration of ligand- and target-based virtual screening for the discovery of cruzain inhibitors. Mol. Inform. 2011, 30, 565–578. [Google Scholar] [CrossRef]
- López-Ramos, M.; Perruccio, F. HPPD: Ligand- and target-based virtual screening on a herbicide target. J. Chem. Inf. Model. 2010, 50, 801–814. [Google Scholar] [CrossRef]
- Markt, P.; Petersen, R.K.; Flindt, E.N.; Kristiansen, K.; Kirchmair, J.; Spitzer, G.; Distinto, S.; Schuster, D.; Wolber, G.; Laggner, C.; et al. Discovery of novel PPAR ligands by a virtual screening approach based on pharmacophore modeling, 3D shape, and electrostatic similarity screening. J. Med. Chem. 2008, 51, 6303–6317. [Google Scholar] [CrossRef]
- da Costa, G.V.; Ferreira, E.F.B.; da Ramos, R.S.; da Silva, L.B.; de Sá, E.M.F.; da Silva, A.K.P.; Lobato, C.M.; Souto, R.N.P.; da Silva, C.H.T.D.P.; Federico, L.B.; et al. Hierarchical Virtual Screening of Potential Insectides Inhibitors of Acetylcholinesterase and Juvenile Hormone from Temephos. Pharmaceuticals 2019, 12, 61. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, E.F.B.; Silva, L.B.; Costa, G.V.; Costa, J.S.; Fujishima, M.A.T.; Leão, R.P.; Ferreira, A.L.S.; Federico, L.B.; Silva, C.H.T.P.; Rosa, J.M.C.; et al. Identification of new inhibitors with potential antitumor activity from polypeptide structures via hierarchical virtual screening. Molecules 2019, 24, 2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Press. QikProp 3.5 User Manual QikProp User Manual; Schrödinger Press: New York, NY, USA, 2012. [Google Scholar]
- Laoui, A.; Polyakov, V.R. Web services as applications’ integration tool: QikProp case study. J. Comput. Chem. 2011, 32, 1944–1951. [Google Scholar] [CrossRef] [PubMed]
- Marchant, C.A.; Briggs, K.A.; Long, A. In silico tools for sharing data and knowledge on toxicity and metabolism: Derek for windows, meteor, and vitic. Toxicol. Mech. Methods 2008, 18, 177–187. [Google Scholar] [CrossRef]
- Reddy, M.; Reddy, C.; Rathore, R.; Erion, M.; Aparoy, P.; Reddy, R.; Reddanna, P. Free Energy Calculations to Estimate Ligand-Binding Affinities in Structure-Based Drug Design. Curr. Pharm. Des. 2014, 20, 3323–3337. [Google Scholar] [CrossRef]
- Meunier, M.; Quirke, N.; Binesti, D. The Calculation of the Electron Affinity of Atoms and Molecules. Mol. Simul. 1999, 23, 109–125. [Google Scholar] [CrossRef]
- Marahatta, A.B. DFT Study on Ground State Electronic Structures of Simple to Complex Molecular Specimens. IJPSAT 2020, 19, 100–112. [Google Scholar]
- Negami, T.; Araki, M.; Okuno, Y.; Terada, T. Calculation of absolute binding free energies between the hERG channel and structurally diverse drugs. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Chagas, C.M.; Moss, S.; Alisaraie, L. Drug metabolites and their effects on the development of adverse reactions: Revisiting Lipinski’s Rule of Five. Int. J. Pharm. 2018, 549, 133–149. [Google Scholar] [CrossRef]
- Ogata, K.; Hatakeyama, M.; Nakamura, S. Effect of atomic charges on octanol–water partition coefficient using alchemical free energy calculation. Molecules 2018, 23, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawski, J.; Popielarska, H.; Myka, A.; Drabi, B.; Bernard, M.K. The log P Parameter as a Molecular Descriptor in the Computer-aided Drug Design—An Overview. Comput. Methods Sci. Technol. 2012, 18, 81–88. [Google Scholar] [CrossRef]
- Sangster, J. Octanol Water Partition Coefficients of Simple Organic Compounds. J. Phys. Chem. Ref. Data 1989, 18, 1111–1229. [Google Scholar] [CrossRef]
- Bennion, B.J.; Be, N.A.; Mcnerney, M.W.; Lao, V.; Carlson, E.M.; Valdez, C.A.; Malfatti, M.A.; Enright, H.A.; Nguyen, T.H.; Lightstone, F.C.; et al. Predicting a Drug’s Membrane Permeability: A Computational Model Validated With in Vitro Permeability Assay Data. J. Phys. Chem. B 2017, 121, 5228–5237. [Google Scholar] [CrossRef] [Green Version]
- Bittermann, K.; Goss, K.-U. Predicting apparent passive permeability of Caco-2 and MDCK cell-monolayers: A mechanistic model. PLoS ONE 2017, 12, e0190319. [Google Scholar] [CrossRef]
- Saxena, P.; Hortigon-Vinagre, M.P.; Beyl, S.; Baburin, I.; Andranovits, S.; Iqbal, S.M.; Costa, A.; IJzerman, A.P.; Kügler, P.; Timin, E.; et al. Correlation between human ether-a-go-go-related gene channel inhibition and action potential prolongation. Br. J. Pharmacol. 2017, 174, 3081–3093. [Google Scholar] [CrossRef]
- Meunier, V.; Bourri, M.; Berger, Y.; Fabre, G. The human intestinal epithelial cell line Caco-2; pharmacological and pharmacokinetic applications. Cell Biol. Toxicol. 1995, 11, 187–194. [Google Scholar] [CrossRef]
- Volpe, D.A. Variability in Caco-2 and MDCK Cell-Based Intestinal Permeability Assays. J. Pharm. Sci. 2008, 97, 712–725. [Google Scholar] [CrossRef]
- Norinder, U.; Haeberlein, M. Computational approaches to the prediction of the blood-brain distribution. Adv. Drug Deliv. Rev. 2002, 54, 291–313. [Google Scholar] [CrossRef]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Costa, J.; Da Silva Ramos, R.; Da Silva Lopes Costa, K.; Do Socorro Barros Brasil, D.; De Paula Da Silva, C.H.T.; Ferreira, E.F.B.; Dos Santos Borges, R.; Campos, J.M.; Da Cruz Macêdo, W.J.; Dos Santos, C.B.R. An in silico study of the antioxidant ability for two caffeine analogs using molecular docking and quantum chemical methods. Molecules 2018, 23, 2801. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.B.R.; Santos, K.L.B.; Cruz, J.N.; Leite, F.H.A.; Borges, R.S.; Taft, C.A.; Campos, J.M.; Silva, C.H.T.P. Molecular modeling approaches of selective adenosine receptor type 2A agonists as potential anti-inflammatory drugs. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Gilson, M.K.; Zhou, H.X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. UNIT Using AutoDock for Ligand-Receptor Docking; Elsevier Inc.: Amsterdam, The Netherlands, 2008; ISBN 0471250953. [Google Scholar]
- Borges, R.S.; Palheta, I.C.; Ota, S.S.B.; Morais, R.B.; Barros, V.A.; Ramos, R.S.; Silva, R.C.; Costa, J.S.; Silva, C.H.T.P.; Campos, J.M.; et al. Toward of safer phenylbutazone derivatives by exploration of toxicity mechanism. Molecules 2019, 24, 143. [Google Scholar] [CrossRef] [Green Version]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, 32–38. [Google Scholar] [CrossRef]
- Sun, L.; Ye, R.D. Role of G protein-coupled receptors in inflammation. Acta Pharmacol. Sin. 2012, 33, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.A.; Bennett, M.V.L.; Pelegrin, P.; Fernandez, R. Ion Channels in Inflammatory Processes: What Is Known and What Is Next? Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Tanhehco, E.J. Potassium channel modulators as anti-inflammatory agents. Expert Opin. Ther. Pat. 2001, 11, 1137–1145. [Google Scholar] [CrossRef]
- Bhagwat, S.S. Kinase inhibitors for the treatment of inflammatory and autoimmune disorders. Purinergic Signal. 2009, 5, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Chandra, V.; Rastinejad, F. Structural overview of the nuclear receptor superfamily: Insights into physiology and therapeutics. Annu. Rev. Physiol. 2009, 72, 247–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsatsanis, C.; Androulidaki, A.; Venihaki, M.; Margioris, A.N. Signalling networks regulating cyclooxygenase-2. Int. J. Biochem. Cell Biol. 2006, 38, 1654–1661. [Google Scholar] [CrossRef]
- Armstrong, M.S.; Finn, P.W.; Morris, G.M.; Richards, W.G. Improving the accuracy of ultrafast ligand-based screening: Incorporating lipophilicity into ElectroShape as an extra dimension. J. Comput. Aided Mol. Des. 2011, 25, 785–790. [Google Scholar] [CrossRef]
- ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3245175/ (accessed on 5 August 2020).
- Nowotka, M.M.; Gaulton, A.; Mendez, D.; Bento, A.P.; Hersey, A.; Leach, A. Using ChEMBL web services for building applications and data processing workflows relevant to drug discovery. Expert Opin. Drug Discov. 2017, 12, 757–767. [Google Scholar] [CrossRef] [PubMed]
- DrugBank 3.0: A Comprehensive Resource for ‘Omics’ Research on Drugs. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3013709/ (accessed on 5 August 2020).
- Parry, T.; Ledee, D.; Willis, M.S.; Portman, M.A. Nuclear Receptors and the Adaptive Response of the Heart; Elsevier Inc.: Amsterdam, The Netherlands, 2017; ISBN 9780128031124. [Google Scholar]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gfeller, D.; Michielin, O.; Zoete, V. Systems biology Shaping the interaction landscape of bioactive molecules. Bioinformatics 2013, 29, 3073–3079. [Google Scholar] [CrossRef]
- dos Santos, K.L.B.; Cruz, J.N.; Silva, L.B.; Ramos, R.S.; Neto, M.F.A.; Lobato, C.C.; Ota, S.S.B.; Leite, F.H.A.; Borges, R.S.; da Silva, C.H.T.P.; et al. Identification of novel chemical entities for adenosine receptor type 2a using molecular modeling approaches. Molecules 2020, 25, 1245. [Google Scholar] [CrossRef] [Green Version]
- de Pinto, V.S.; Araújo, J.S.C.; Silva, R.C.; da Costa, G.V.; Cruz, J.N.; Neto, M.F.D.A.; Campos, J.M.; Santos, C.B.R.; Leite, F.H.A.; Junior, M.C.S. In silico study to identify new antituberculosis molecules from natural sources by hierarchical virtual screening and molecular dynamics simulations. Pharmaceuticals 2019, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Cruz, J.N.; Costa, J.F.S.; Khayat, A.S.; Kuca, K. Molecular dynamics simulation and binding free energy studies of novel leads belonging to the benzofuran class inhibitors of Mycobacterium tuberculosis Polyketide Synthase 13. J Biomol. Struct. Dyn. 2018, 37, 1–28. [Google Scholar] [CrossRef]
- Cruz, J.V.; Serafim, R.B.; Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Neto, M.F.A.; Leite, F.H.A.; Taft, C.A.; Silva, C.H.T.P.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.B.R.; da Silva Ramos, R.; Ortiz, B.L.S.; da Silva, G.M.; Giuliatti, S.; Balderas-Lopez, J.L.; Navarrete, A.; Carvalho, J.C.T. Oil from the fruits of Pterodon emarginatus Vog.: A traditional anti-inflammatory. Study combining in vivo and in silico. J. Ethnopharmacol. 2018, 222, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Bhatia, P.; Alam, O.; Javed Naim, M.; Nawaz, F.; Ahmad Sheikh, A.; Jha, M. Recent advancement in the discovery and development of COX-2 inhibitors: Insight into biological activities and SAR studies (2008–2019). Bioorg. Chem. 2019, 89, 103007. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, S.M.; Jain, S.; Dinodia, M.; Kumar, A. Synthesis of Some Thiophene, Imidazole and Pyridine Derivatives Exhibiting Good Anti-Inflammatory and Analgesic Activities. Med. Chem. 2008, 4, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Sharma, P.C.; Yar, M.S.; Kharb, R.; Sharma, P.C.; Yar, M.S. Pharmacological significance of triazole scaffold Pharmacological significance of triazole scaffold. J. Enzyme Inhib. Med. Chem. 2011, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.N.P.; Knaus, E.E.; Road, T.P.; Jolla, L. Evolution of Nonsteroidal Anti-Inflammatory Cyclooxygenase (COX) Inhibition and Beyond Drugs (NSAIDs). J. Pharm. Pharm. Sci. 2008, 11, 81S–110S. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Yu, M.S.; Kazmi, S.R.; Oh, S.Y.; Rhee, K.H.; Bae, M.A.; Lee, B.H.; Shin, D.S.; Oh, K.S.; Ceong, H.; et al. Computational determination of hERG-related cardiotoxicity of drug candidates. BMC Bioinform. 2019, 20, 250. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, P.; Ott, N.; Cooksy, A.L. Benzodiazepines: Electron affinity, receptors and cell signaling—A multifaceted approach. J. Recept. Signal Transduct. 2013, 33, 338–343. [Google Scholar] [CrossRef]
- Lamothe, S.M.; Guo, J.; Li, W.; Yang, T.; Zhang, S. The Human Ether-a-go-go-related Gene (hERG) potassium channel represents an unusual target for protease-mediated damage. J. Biol. Chem. 2016, 291, 20387–20401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K(+) channels: Structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, G.B.; Makkos, A.; Nagy, C.T.; Onódi, Z.; Sayour, N.V.; Gergely, T.G.; Kiss, B.; Görbe, A.; Sághy, É.; Zádori, Z.S.; et al. Hidden Cardiotoxicity of Rofecoxib Can be Revealed in Experimental Models of Ischemia/Reperfusion. Cells 2020, 9, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- EON | Electrostatic Similarity for Lead-Hopping. Available online: https://www.eyesopen.com/eon (accessed on 26 May 2020).
- Muchmore, S.W.; Souers, A.J.; Akritopoulou-Zanze, I. The use of three-dimensional shape and electrostatic similarity searching in the identification of a melanin-concentrating hormone receptor 1 antagonist. Chem. Biol. Drug Des. 2006, 67, 174–176. [Google Scholar] [CrossRef]
- Tosco, P.; Stiefl, N.; Landrum, G. Bringing the MMFF force field to the RDKit: Implementation and validation. J. Cheminform. 2014, 6, 37. [Google Scholar] [CrossRef]
- Sanderson, D.M.; Earnshaw, C.G. Computer Prediction of Possible Toxic Action from Chemical Structure; The DEREK System. Hum. Exp. Toxicol. 1991, 10, 261–273. [Google Scholar] [CrossRef]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, LLC. Release 2017-2: Maestro; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Calculation of Molecular Properties and Bioactivity Score. Available online: https://www.molinspiration.com/cgi-bin/properties (accessed on 26 May 2020).
- Linn, C.; Roy, S.; Samant, L.R.; Chowdhary, A. Research Article In silico pharmacokinetics analysis and ADMET of phytochemicals of Datura. J. Chem. Pharm. Res. 2015, 7, 385–388. [Google Scholar]
- Kumar, N.; Mishra, S.S.; Sharma, C.S.; Singh, H.P.; Singh, H. In silico Pharmacokinetic, Bioactivity and Toxicity Evaluation of Some Selected Anti-Ulcer Agents. Int. J. Pharm. Sci. Drug Res. 2017, 9, 68–71. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA. Discovery Studio Modeling Environment; BIOVIA: San Diego, CA, USA, 2015. [Google Scholar]
- Kalia, M.; Yadav, V.K.; Singh, P.K.; Dohare, S.; Sharma, D.; Narvi, S.S.; Agarwal, V. Designing quorum sensing inhibitors of Pseudomonas aeruginosa utilizing FabI: An enzymic drug target from fatty acid synthesis pathway. 3 Biotech 2019, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP Charges To Calculate Conformational Energies, Hydrogen Bond Energies, and Free Energies of Solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Da Costa, K.S.; Galúcio, J.M.; Da Costa, C.H.S.; Santana, A.R.; Dos Santos Carvalho, V.; Do Nascimento, L.D.; Lima E Lima, A.H.; Neves Cruz, J.; Alves, C.N.; Lameira, J. Exploring the Potentiality of Natural Products from Essential Oils as Inhibitors of Odorant-Binding Proteins: A Structure- And Ligand-Based Virtual Screening Approach to Find Novel Mosquito Repellents. ACS Omega 2019, 4, 22475–22486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, F.S.; Rodrigues Do Rego, J.D.A.; Da Costa, M.L.; Lobato Da Silva, L.F.; Da Costa, R.A.; Cruz, J.N.; Brasil, D.D.S.B. Spectroscopic methods and in silico analyses using density functional theory to characterize and identify piperine alkaloid crystals isolated from pepper (Piper Nigrum L.). J. Biomol. Struct. Dyn. 2020, 38, 2792–2799. [Google Scholar] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision 16.A.03; Wallingford CT. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Silva, S.G.; Da Costa, R.A.; De Oliveira, M.S.; Da Cruz, J.N.; Figueiredo, P.L.B.; Do Socorro Barros Brasil, D.; Nascimento, L.D.; De Jesus Chaves Neto, A.M.; De Carvalho, R.N.; De Aguiar Andrade, E.H. Chemical profile of lippia thymoides, evaluation of the acetylcholinesterase inhibitory activity of its essential oil, and molecular docking and molecular dynamics simulations. PLoS ONE 2019, 14, e0213393. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Vale, V.V.; Cruz, J.N.; Viana, G.M.R.; Póvoa, M.M.; do Brasil, D.S.B.; Dolabela, M.F. Naphthoquinones isolated from Eleutherine plicata herb: In vitro antimalarial activity and molecular modeling to investigate their binding modes. Med. Chem. Res. 2020, 29, 487–494. [Google Scholar] [CrossRef]
- de Oliveira, M.S.; da Cruz, J.N.; Gomes Silva, S.; da Costa, W.A.; de Sousa, S.H.B.; Bezerra, F.W.F.; Teixeira, E.; da Silva, N.J.N.; de Aguiar Andrade, E.H.; de Jesus Chaves Neto, A.M.; et al. Phytochemical profile, antioxidant activity, inhibition of acetylcholinesterase and interaction mechanism of the major components of the Piper divaricatum essential oil obtained by supercritical CO2. J. Supercrit. Fluids 2019, 145, 74–84. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Lzaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Chong, L.T.; Pitera, J.W.; Swope, W.C.; Pande, V.S. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. J. Mol. Graph. Model. 2009, 27, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Neves Cruz, J.; Santana de Oliveira, M.; Gomes Silva, S.; da Pedro Silva Souza Filho, A.; Santiago Pereira, D.; Lima e Lima, A.H.; de Aguiar Andrade, E.H. Insight into the Interaction Mechanism of Nicotine, NNK, and NNN with Cytochrome P450 2A13 Based on Molecular Dynamics Simulation. J. Chem. Inf. Model. 2020, 60, 766–776. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Neves Cruz, J.; da Costa, K.S.; de Carvalho, T.A.A.; de Alencar, N.A.N. Measuring the structural impact of mutations on cytochrome P450 21A2, the major steroid 21-hydroxylase related to congenital adrenal hyperplasia. J. Biomol. Struct. Dyn. 2020, 38, 1425–1434. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structures | #Stars a | EA (eV) b | RO5 c | %HOA d | QplogPo/w e | QPPCaco f | QPP MDCK g | CNS h | Qplog BB i |
|---|---|---|---|---|---|---|---|---|---|
| Normal range | 0.0 to 5.0 | −0.9 to 1.7 | Max. 4 | 0 to 100 | −2.0 to 6.5 | <25 poor >500 great | <25 poor >500 great | −2 (inactive) +2 (active) | −3.0 to −1.2 |
| Rofecoxib | 1 | 1.99 | 0 | 82.40 | 1.45 | 420.96 | 194.20 | −1 | −0.81 |
| LMQC72 | 0 | 1.37 | 0 | 100.00 | 2.18 | 1470.77 | 900.66 | 0 | −0.17 |
| LMQC36 | 0 | 0.73 | 0 | 100.00 | 3.75 | 1751.71 | 2254.41 | 0 | −0.07 |
| LMQC50 | 0 | 1.44 | 0 | 100.00 | 4.21 | 13,737.5 | 3415.52 | 0 | −0.77 |
| Enzyme | Ligand | Experimental Binding Affinity (kcal/mol) a | Ki (nM) | Docking Predicted Binding Affinity (kcal/mol) | Resolution |
|---|---|---|---|---|---|
| hCOX-2 (PDB 5KIR) | Rofecoxib (RCX) | −9.2 [14] | 310 | −10.4 | 2.69 Å [14] |
| Molecular Docking | Residues | Distance (Å) | Type | ∆G (kcal/mol) |
|---|---|---|---|---|
| Rofecoxib vs. 5KIR | His90 | 2.64213 | Hydrogen Bond | −10.4 |
| Val349 | 4.41806 | Pi-Alkyl | ||
| Leu352 | 5.44011 | Pi-Alkyl | ||
| Arg513 | 2.55259 | Carbon Hydrogen Bond | ||
| Arg513 | 3.07173 | Carbon Hydrogen Bond | ||
| Arg513 | 2.37819 | Hydrogen Bond | ||
| Phe518 | 5.82249 | Pi-Pi Stacked | ||
| Val523 | 3.80966 | Pi-Alkyl | ||
| Ala527 | 4.95368 | Pi-Alkyl | ||
| Ala527 | 3.97589 | Pi-Alkyl | ||
| Ala527 | 2.61541 | Carbon Hydrogen Bond | ||
| Ser530 | 2.84906 | Carbon Hydrogen Bond |
| Molecular Docking | Residues | Distance (Å) | Type | ∆G (kcal/mol) |
|---|---|---|---|---|
| Leu352 | 2.182419 | Hydrogen Bond | ||
| Ser353 | 2.904348 | Hydrogen Bond | ||
| LMQC72 vs. 5KIR | Phe518 | 2.256368 | Hydrogen Bond | |
| Gln192 | 2.826605 | Hydrogen Bond | −11.0 | |
| Val523 | 3.868765 | Pi-Alkyl | ||
| Met522 | 4.589701 | Alkyl | ||
| Ala527 | 4.200118 | Pi-Alkyl | ||
| Ala527 | 3.367419 | Pi-Sigma | ||
| Val349 | 3.784354 | Pi-Sigma | ||
| Val349 | 4.593121 | Alkyl | ||
| Leu351 | 4.422239 | Alkyl | ||
| LMQC36 vs. 5KIR | Leu352 | 3.576441 | Carbon Hydrogen Bond | |
| Val523 | 3.520289 | Pi-Sigma | −10.6 | |
| Val523 | 5.175953 | Pi-Alkyl | ||
| Ser353 | 2.921642 | Carbon Hydrogen Bond | ||
| Arg513 | 4.805970 | Pi-Cation | ||
| Gly192 | 3.057449 | Carbon Hydrogen Bond | ||
| Ile527 | 4.774353 | Alkyl | ||
| Phe518 | 4.346549 | Pi-Alkyl | ||
| Leu351 | 5.018402 | Alkyl | ||
| Leu359 | 4.885905 | Alkyl | ||
| Val116 | 5.139531 | Alkyl | ||
| Try355 | 2.926941 | Hydrogen Bond | ||
| Val349 | 5.180672 | Pi-Alkyl | ||
| LMQC50 vs. 5KIR | Ser353 | 3.396209 | Pi-Sigma | |
| Val523 | 3.868112 | Pi-Sigma | ||
| Leu352 | 2.176448 | Hydrogen Bond | −10.2 | |
| Arg513 | 3.513371 | Carbon Hydrogen Bond | ||
| Phe518 | 5.858526 | Pi-Pi Stacked | ||
| Gly526 | 4.175181 | Amide-Pi Stacked | ||
| Met522 | 4.690481 | Alkyl | ||
| Ala527 | 3.972980 | Pi-Sigma | ||
| Arg120 | 2.589874 | Hydrogen Bond |
| Number | Compounds | Code ID and Database | Chemical Identification | Code SMILES |
|---|---|---|---|---|
| LMQC72 |  | Chembridge_DIVERSet-CL ZINC 72149848 | C18H15N5 1-{4-[2-(4-methylphenyl)-1H-imidazol-1-yl]phenyl}-1H-1,2,4-triazole | Cc1ccc(cc1)c4nccn4c2ccc(cc2)n3cncn3 |
| LMQC36 |  | Chembridge_DIVERSet-EXP ZINC3615660 | C18H15N5 N-(3-chloro-4-methoxyphenyl)-5-methyl-3-phenyl-1,2-oxazole-4-carboxamide | COc1ccc(cc1Cl)NC(=O)c3c(C)onc3c2ccccc2 |
| LMQC50 |  | Drug@FDA_BindingDB Binding_DB 50224 | C18H15N5 N-(3-chloro-4-methoxyphenyl)-5-methyl-3-phenyl-1,2-oxazole-4-carboxamide 4-(5-(p-tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl) benzenesulfonamide | CS(=O)(=O)c1cccc(c1)n3nc(cc3c2ccc(C)cc2)C(F)(F)F |
| Compound | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor | Enzyme (%) a |
|---|---|---|---|---|---|---|---|
| Rofecoxib | 0.20 | 0.13 | 0.16 | −0.37 | −0.14 | 0.61 | 32% |
| LMQC72 | 0.19 | −0.41 | −0.23 | −0.02 | −0.45 | 0.07 | 16% |
| LMQC36 | −0.29 | −0.40 | 0.04 | −0.04 | −0.10 | −0.43 | 8% |
| LMQC50 | 0.03 | −0.18 | −0.18 | 0.12 | 0.26 | 0.21 | 4% |
| Compound | ΔEvdW a | ΔEele b | ΔGGB c | ΔGNP d | ΔGbind e |
|---|---|---|---|---|---|
| Rofecoxib | −48.12 | −23.66 | 35.74 | −9.27 | −45.31 |
| LMQC72 | −52.92 | −21.45 | 42.71 | −6.92 | −38.58 |
| LMQC36 | −45.93 | −7.79 | 23.28 | −5.66 | −36.10 |
| LMQC50 | −49.80 | −13.86 | 29.93 | −5.67 | −39.40 |
| Compounds | Prediction Alert | Toxicophoric Group | Toxicity Alert | LD50 | Toxicity Class a |
|---|---|---|---|---|---|
| Rofecoxib | Hepatotoxicity in human, mouse and rat |  | Plausible | 4500 mg/kg | V |
| LMQC72 | - | - | No alerts | 674 mg/kg | IV |
| LMQC36 | - | - | No alerts | 6500 mg/kg | VI |
| LMQC50 | - | - | No alerts | 1400 mg/kg | IV |
| Compound | QpLog hERG a | EA (eV) b |
|---|---|---|
| Rofecoxib | Medium risk | 1.997 |
| LMQC72 | Medium risk | 1.374 |
| LMQC36 | Medium risk | 0.739 |
| LMQC50 | Medium risk | 1.446 |
| Enzyme | Ligand | Coordinates of the Grid Center | Grid Size (Points) |
|---|---|---|---|
| COX-2 (PDB code: 5KIR) Homo sapiens | Rofecoxib | X = 24.065 Y = 40.416 Z = 3.057 | 17 x 20 y 27 z |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leão, R.P.; Cruz, J.V.; da Costa, G.V.; Cruz, J.N.; Ferreira, E.F.B.; Silva, R.C.; de Lima, L.R.; Borges, R.S.; dos Santos, G.B.; Santos, C.B.R. Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach. Pharmaceuticals 2020, 13, 209. https://doi.org/10.3390/ph13090209
Leão RP, Cruz JV, da Costa GV, Cruz JN, Ferreira EFB, Silva RC, de Lima LR, Borges RS, dos Santos GB, Santos CBR. Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach. Pharmaceuticals. 2020; 13(9):209. https://doi.org/10.3390/ph13090209
Chicago/Turabian StyleLeão, Rozires P., Josiane V. Cruz, Glauber V. da Costa, Jorddy N. Cruz, Elenilze F. B. Ferreira, Raí C. Silva, Lúcio R. de Lima, Rosivaldo S. Borges, Gabriela B. dos Santos, and Cleydson B. R. Santos. 2020. "Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach" Pharmaceuticals 13, no. 9: 209. https://doi.org/10.3390/ph13090209