Gastrointestinal Mechanisms Underlying the Cardiovascular Effect of Metformin

 , ,
, ,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
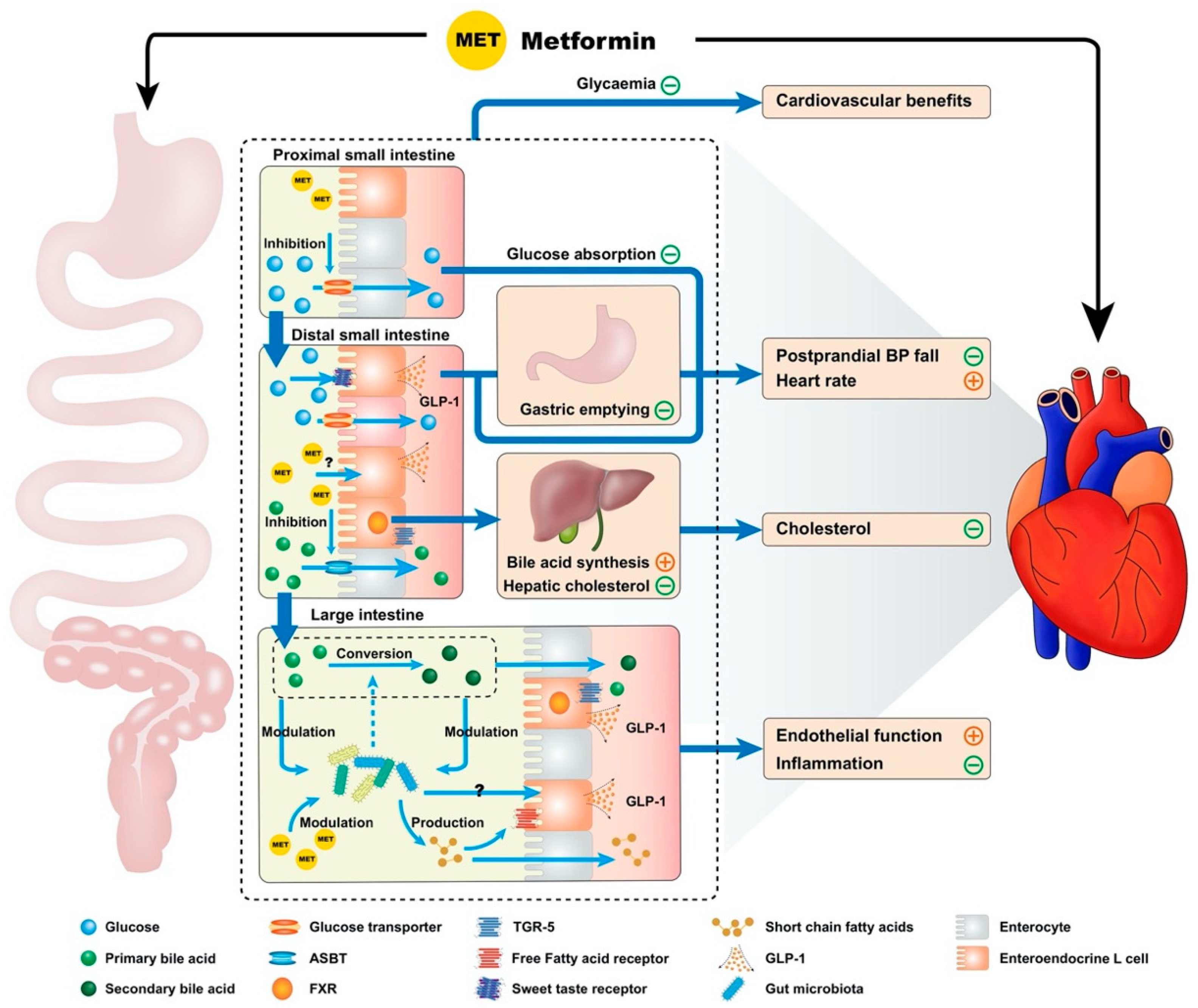
2. Gastrointestinal Actions of Metformin and Cardiovascular Function
2.1. Inhibition of Bile Acid Resorption
2.2. Modulation of the Gut Microbiota
2.3. Reducing the Rate of Glucose Absorption
2.4. Enhanced GLP-1 Secretion and Action
2.5. Slowing of Gastric Emptying
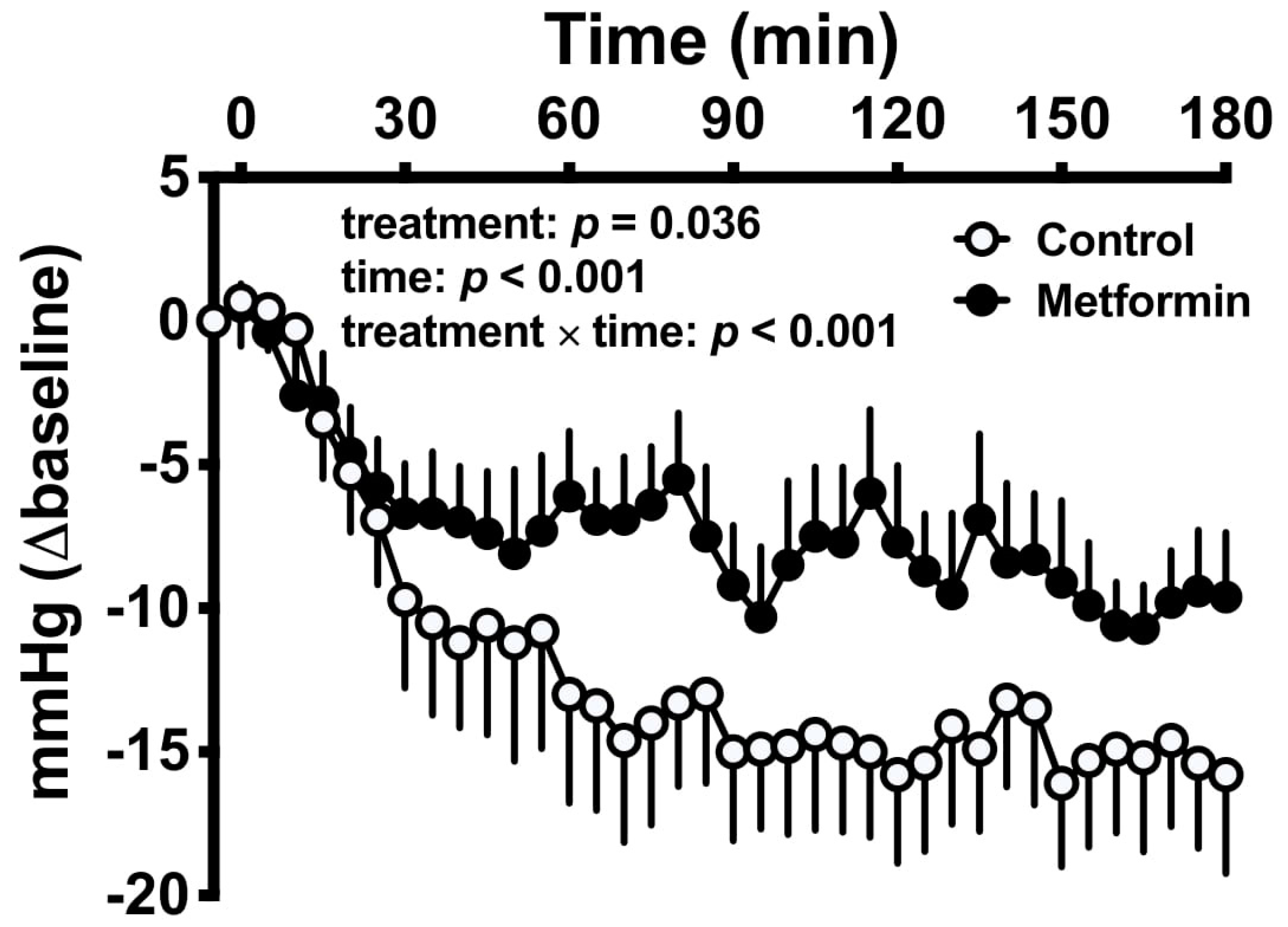
2.6. Attenuation of Postprandial Hypotension
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Ghani, M.; DeFronzo, R.A.; Del Prato, S.; Chilton, R.; Singh, R.; Ryder, R.E.J. Cardiovascular Disease and Type 2 Diabetes: Has the Dawn of a New Era Arrived? Diabetes Care 2017, 40, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UKPDS. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A.W. 10-Year Follow-up of Intensive Glucose Control in Type 2 Diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.H. Metformin Use Is Associated With a Lower Risk of Hospitalization for Heart Failure in Patients With Type 2 Diabetes Mellitus: A Retrospective Cohort Analysis. J. Am. Heart Assoc. 2019, 8, e011640. [Google Scholar] [CrossRef]
- Charytan, D.M.; Solomon, S.D.; Ivanovich, P.; Remuzzi, G.; Cooper, M.E.; McGill, J.B.; Parving, H.H.; Parfrey, P.; Singh, A.K.; Burdmann, E.A.; et al. Metformin use and cardiovascular events in patients with type 2 diabetes and chronic kidney disease. Diabetes Obes. Metab. 2019, 21, 1199–1208. [Google Scholar] [CrossRef]
- Roumie, C.L.; Hung, A.M.; Greevy, R.A.; Grijalva, C.G.; Liu, X.; Murff, H.J.; Elasy, T.A.; Griffin, M.R. Comparative effectiveness of sulfonylurea and metformin monotherapy on cardiovascular events in type 2 diabetes mellitus: A cohort study. Ann. Intern. Med. 2012, 157, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Crowley, M.J.; Williams, J.W., Jr.; Kosinski, A.S.; D’Alessio, D.A.; Buse, J.B. Metformin Use May Moderate the Effect of DPP-4 Inhibitors on Cardiovascular Outcomes. Diabetes Care 2017, 40, 1787–1789. [Google Scholar] [CrossRef] [Green Version]
- Griffin, S.J.; Leaver, J.K.; Irving, G.J. Impact of metformin on cardiovascular disease: A meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia 2017, 60, 1620–1629. [Google Scholar] [CrossRef] [Green Version]
- Fisman, E.Z.; Tenenbaum, A.; Benderly, M.; Goldbourt, U.; Behar, S.; Motro, M. Antihyperglycemic treatment in diabetics with coronary disease: Increased metformin-associated mortality over a 5-year follow-up. Cardiology 1999, 91, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E. Postprandial peaks as a risk factor for cardiovascular disease: Epidemiological perspectives. Int. J. Clin. Pr. Suppl. 2002, 129, 5–11. [Google Scholar]
- Heine, R.J.; Balkau, B.; Ceriello, A.; Del Prato, S.; Horton, E.S.; Taskinen, M.R. What does postprandial hyperglycaemia mean? Diabet Med. 2004, 21, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Wascher, T.C.; Schmoelzer, I.; Wiegratz, A.; Stuehlinger, M.; Mueller-Wieland, D.; Kotzka, J.; Enderle, M. Reduction of postchallenge hyperglycaemia prevents acute endothelial dysfunction in subjects with impaired glucose tolerance. Eur. J. Clin. Invest. 2005, 35, 551–557. [Google Scholar] [CrossRef]
- Holman, R.R.; Coleman, R.L.; Chan, J.C.N.; Chiasson, J.L.; Feng, H.; Ge, J.; Gerstein, H.C.; Gray, R.; Huo, Y.; Lang, Z.; et al. Effects of acarbose on cardiovascular and diabetes outcomes in patients with coronary heart disease and impaired glucose tolerance (ACE): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef]
- Stumvoll, M.; Nurjhan, N.; Perriello, G.; Dailey, G.; Gerich, J.E. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 1995, 333, 550–554. [Google Scholar] [CrossRef]
- Sum, C.F.; Webster, J.M.; Johnson, A.B.; Catalano, C.; Cooper, B.G.; Taylor, R. The effect of intravenous metformin on glucose metabolism during hyperglycaemia in type 2 diabetes. Diabet Med. 1992, 9, 61–65. [Google Scholar] [CrossRef]
- Buse, J.B.; DeFronzo, R.A.; Rosenstock, J.; Kim, T.; Burns, C.; Skare, S.; Baron, A.; Fineman, M. The Primary Glucose-Lowering Effect of Metformin Resides in the Gut, Not the Circulation: Results From Short-term Pharmacokinetic and 12-Week Dose-Ranging Studies. Diabetes Care 2016, 39, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Duca, F.A.; Cote, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2015, 21, 506–511. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Buse, J.B.; Kim, T.; Burns, C.; Skare, S.; Baron, A.; Fineman, M. Once-daily delayed-release metformin lowers plasma glucose and enhances fasting and postprandial GLP-1 and PYY: Results from two randomised trials. Diabetologia 2016, 59, 1645–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, M.J.; Jones, K.L.; Sun, Z.; Horowitz, M.; Rayner, C.K.; Wu, T. Metformin attenuates the postprandial fall in blood pressure in type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Horowitz, M.; Rayner, C.K. New insights into the anti-diabetic actions of metformin: From the liver to the gut. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.; Howlett, H.C.; Wiernsperger, N.F.; Bailey, C.J. Differential effects of metformin on bile salt absorption from the jejunum and ileum. Diabetes Obes. Metab. 2003, 5, 120–125. [Google Scholar] [CrossRef]
- Sansome, D.J.; Xie, C.; Veedfald, S.; Horowitz, M.; Rayner, C.K.; Wu, T. Mechanism of glucose-lowering by metformin in type 2 diabetes: Role of bile acids. Diabetes Obes. Metab. 2020, 22, 141–148. [Google Scholar] [CrossRef]
- Nguyen, A.; Bouscarel, B. Bile acids and signal transduction: Role in glucose homeostasis. Cell Signal 2008, 20, 2180–2197. [Google Scholar] [CrossRef]
- Bahne, E.; Hansen, M.; Bronden, A.; Sonne, D.P.; Vilsboll, T.; Knop, F.K. Involvement of glucagon-like peptide-1 in the glucose-lowering effect of metformin. Diabetes Obes. Metab. 2016, 18, 955–961. [Google Scholar] [CrossRef]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Rohde, U.; Sonne, D.P.; Christensen, M.; Hansen, M.; Bronden, A.; Torang, S.; Rehfeld, J.F.; Holst, J.J.; Vilsboll, T.; Knop, F.K. Cholecystokinin-Induced Gallbladder Emptying and Metformin Elicit Additive Glucagon-Like Peptide-1 Responses. J. Clin. Endocrinol. Metab. 2016, 101, 2076–2083. [Google Scholar] [CrossRef]
- Xie, C.; Wang, X.; Young, R.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Role of Intestinal Bitter Sensing in Enteroendocrine Hormone Secretion and Metabolic Control. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Bailey, D.; Walsh, D.T.; Warner, T.D. Expression and activation of the farnesoid X receptor in the vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 3668–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabelsi, M.S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liu, X.; Wang, B.; Xu, H.; Xia, Q.; Lu, T.; Wang, F. Farnesoid X receptor deletion improves cardiac function, structure and remodeling following myocardial infarction in mice. Mol. Med. Rep. 2017, 16, 673–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, S.; Raufman, J.-P.; Pallone, T.L. Bile acids regulate cardiovascular function. Clin. Transl. Sci. 2011, 4, 210–218. [Google Scholar] [CrossRef]
- Moller, S.; Henriksen, J.H. Cirrhotic cardiomyopathy. J. Hepatol. 2010, 53, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.S.; Mathur, B.; Eblimit, Z.; Vasquez, H.; Taegtmeyer, H.; Karpen, S.J.; Penny, D.J.; Moore, D.D.; Anakk, S. Bile acid excess induces cardiomyopathy and metabolic dysfunctions in the heart. Hepatology 2017, 65, 189–201. [Google Scholar] [CrossRef]
- Ross, S.; D’Mello, M.; Anand, S.S.; Eikelboom, J.; Stewart, A.F.; Samani, N.J.; Roberts, R.; Paré, G. Effect of Bile Acid Sequestrants on the Risk of Cardiovascular Events: A Mendelian Randomization Analysis. Circ. Cardiovasc. Genet. 2015, 8, 618–627. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Brandmaier, S.; Messias, A.C.; Herder, C.; Draisma, H.H.; Demirkan, A.; Yu, Z.; Ried, J.S.; Haller, T.; Heier, M.; et al. Effects of metformin on metabolite profiles and LDL cholesterol in patients with type 2 diabetes. Diabetes Care 2015, 38, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Solymár, M.; Ivic, I.; Pótó, L.; Hegyi, P.; Garami, A.; Hartmann, P.; Pétervári, E.; Czopf, L.; Hussain, A.; Gyöngyi, Z.; et al. Metformin induces significant reduction of body weight, total cholesterol and LDL levels in the elderly—A meta-analysis. PLoS ONE 2018, 13, e0207947. [Google Scholar] [CrossRef]
- The Lipid Research Clinics Coronary Primary Prevention Trial Results: II. The Relationship of Reduction in Incidence of Coronary Heart Disease to Cholesterol Lowering. JAMA 1984, 251, 365–374. [CrossRef]
- Wulffelé, M.G.; Kooy, A.; de Zeeuw, D.; Stehouwer, C.D.; Gansevoort, R.T. The effect of metformin on blood pressure, plasma cholesterol and triglycerides in type 2 diabetes mellitus: A systematic review. J. Intern. Med. 2004, 256, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, Z.; Wang, Y.; Dai, Y.; Zhang, X.; Hu, S. Role of Bile Acids in Bariatric Surgery. Front. Physiol. 2019, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.; Gallo, G.A.; Trash, D.B.; Morris, J.S. Diagnositic value of serum bile acid estimations in liver disease. J. Clin. Pathol. 1975, 28, 506–509. [Google Scholar] [CrossRef]
- Tian, J.; Huang, S.; Sun, S.; Ding, L.; Zhang, E.; Liu, Y.; Huang, W. Bile acid signaling and bariatric surgery. Liver Res. 2017, 1, 208–213. [Google Scholar] [CrossRef]
- Patti, M.E.; Houten, S.M.; Bianco, A.C.; Bernier, R.; Larsen, P.R.; Holst, J.J.; Badman, M.K.; Maratos-Flier, E.; Mun, E.C.; Pihlajamaki, J.; et al. Serum bile acids are higher in humans with prior gastric bypass: Potential contribution to improved glucose and lipid metabolism. Obesity 2009, 17, 1671–1677. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.W.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Guo, G.L.; Xie, W. Metformin action through the microbiome and bile acids. Nat. Med. 2018, 24, 1789–1790. [Google Scholar] [CrossRef]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velasquez-Mejia, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin Is Associated With Higher Relative Abundance of Mucin-Degrading Akkermansia muciniphila and Several Short-Chain Fatty Acid-Producing Microbiota in the Gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samah, S.; Ramasamy, K.; Lim, S.M.; Neoh, C.F. Probiotics for the management of type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Res. Clin. Pr. 2016, 118, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Caspary, W.F.; Creutzfeldt, W. Analysis of the inhibitory effect of biguanides on glucose absorption: Inhibition of active sugar transport. Diabetologia 1971, 7, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, C.; Bailey, C.J. Reconsideration of inhibitory effect of metformin on intestinal glucose absorption. J. Pharm. Pharm. 1991, 43, 120–121. [Google Scholar] [CrossRef]
- Cuber, J.C.; Bosshard, A.; Vidal, H.; Vega, F.; Wiernsperger, N.; Rapin, J.R. Metabolic and drug distribution studies do not support direct inhibitory effects of metformin on intestinal glucose absorption. Diabete Metab. 1994, 20, 532–539. [Google Scholar]
- Ikeda, T.; Iwata, K.; Murakami, H. Inhibitory effect of metformin on intestinal glucose absorption in the perfused rat intestine. Biochem. Pharm. 2000, 59, 887–890. [Google Scholar] [CrossRef]
- Sakar, Y.; Meddah, B.; Faouzi, M.A.; Cherrah, Y.; Bado, A.; Ducroc, R. Metformin-induced regulation of the intestinal D-glucose transporters. J. Physiol. Pharm. 2010, 61, 301–307. [Google Scholar]
- Wu, T.; Xie, C.; Wu, H.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Metformin reduces the rate of small intestinal glucose absorption in type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 290–293. [Google Scholar] [CrossRef]
- Wu, T.; Rayner, C.K.; Horowitz, M. Incretins. Handb. Exp. Pharm. 2016, 233, 137–171. [Google Scholar] [CrossRef]
- Zhang, X.; Jones, K.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Effects of Proximal and Distal Enteral Glucose Infusion on Cardiovascular Response in Health and Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2020, 105, e2877–e2884. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, M. Cardiovascular benefits and safety profile of acarbose therapy in prediabetes and established type 2 diabetes. Cardiovasc. Diabetol. 2007, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalsgaard, N.B.; Gasbjerg, L.S.; Hansen, L.S.; Hansen, N.L.; Stensen, S.; Hartmann, B.; Holst, J.J.; VilsbØLl, T.; Knop, F.K. 1145-P: Acarbose-Induced Glucagon-Like Peptide-1 Secretion Contributes to the Glucose-Lowering Effect of Acarbose. Diabetes 2019, 68, 1145-P. [Google Scholar] [CrossRef]
- Gentilcore, D.; Bryant, B.; Wishart, J.M.; Morris, H.A.; Horowitz, M.; Jones, K.L. Acarbose attenuates the hypotensive response to sucrose and slows gastric emptying in the elderly. Am. J. Med. 2005, 118, 1289. [Google Scholar] [CrossRef] [PubMed]
- Madden, K.M.; Harris, D.E.; Meneilly, G.S. Attenuation of Postprandial Hypotension with Acarbose in Older Adults with Type 2 Diabetes Mellitus. J. Am. Geriatr. Soc. 2015, 63, 1484–1486. [Google Scholar] [CrossRef]
- Gentilcore, D.; Horowitz, M.; Jones, K.L. Acarbose and postprandial hypotension. Hypertension 2007, 50, e159. [Google Scholar] [CrossRef] [Green Version]
- Chiasson, J.L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- Hanefeld, M. Treatment of impaired glucose tolerance with acarbose and its effect on intima-media thickness: A substudy of the STOP-NIDDM trial (study to prevent non-insulin-dependent diabetes mellitus). Endocr. Pr. 2006, 12 (Suppl. 1), 56–59. [Google Scholar] [CrossRef]
- Hanefeld, M.; Cagatay, M.; Petrowitsch, T.; Neuser, D.; Petzinna, D.; Rupp, M. Acarbose reduces the risk for myocardial infarction in type 2 diabetic patients: Meta-analysis of seven long-term studies. Eur. Heart J. 2004, 25, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, T.; Sawicki, P.T. Acarbose for prevention of diabetes, hypertension and cardiovascular events? A critical analysis of the STOP-NIDDM data. Diabetologia 2004, 47, 575–580. [Google Scholar] [CrossRef] [Green Version]
- Holman, R.R. What does the acarbose cardiovascular evaluation (ACE) trial tell us? J. Diabetes 2018, 10, 683–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.L.; Scott, C.A.B.; Lang, Z.; Bethel, M.A.; Tuomilehto, J.; Holman, R.R. Meta-analysis of the impact of alpha-glucosidase inhibitors on incident diabetes and cardiovascular outcomes. Cardiovasc. Diabetol. 2019, 18, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Thazhath, S.S.; Bound, M.J.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Mechanism of increase in plasma intact GLP-1 by metformin in type 2 diabetes: Stimulation of GLP-1 secretion or reduction in plasma DPP-4 activity? Diabetes Res. Clin. Pr. 2014, 106, e3–e6. [Google Scholar] [CrossRef] [PubMed]
- Borg, M.J.; Bound, M.; Grivell, J.; Sun, Z.; Jones, K.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Comparative effects of proximal and distal small intestinal administration of metformin on plasma glucose and glucagon-like peptide-1, and gastric emptying after oral glucose, in type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Bahne, E.; Sun, E.W.L.; Young, R.L.; Hansen, M.; Sonne, D.P.; Hansen, J.S.; Rohde, U.; Liou, A.P.; Jackson, M.L.; de Fontgalland, D.; et al. Metformin-induced glucagon-like peptide-1 secretion contributes to the actions of metformin in type 2 diabetes. Jci. Insight 2018, 3, e93936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noyan-Ashraf, M.H.; Momen, M.A.; Ban, K.; Sadi, A.M.; Zhou, Y.Q.; Riazi, A.M.; Baggio, L.L.; Henkelman, R.M.; Husain, M.; Drucker, D.J. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 2009, 58, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poornima, I.; Brown, S.B.; Bhashyam, S.; Parikh, P.; Bolukoglu, H.; Shannon, R.P. Chronic glucagon-like peptide-1 infusion sustains left ventricular systolic function and prolongs survival in the spontaneously hypertensive, heart failure-prone rat. Circ. Heart Fail. 2008, 1, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Ussher, J.R.; Drucker, D.J. Cardiovascular biology of the incretin system. Endocr. Rev. 2012, 33, 187–215. [Google Scholar] [CrossRef]
- Nyström, T.; Gutniak, M.K.; Zhang, Q.; Zhang, F.; Holst, J.J.; Ahrén, B.; Sjöholm, A. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E1209–E1215. [Google Scholar] [CrossRef]
- Trahair, L.G.; Horowitz, M.; Hausken, T.; Feinle-Bisset, C.; Rayner, C.K.; Jones, K.L. Effects of exogenous glucagon-like peptide-1 on the blood pressure, heart rate, mesenteric blood flow, and glycemic responses to intraduodenal glucose in healthy older subjects. J. Clin. Endocrinol. Metab. 2014, 99, E2628–E2634. [Google Scholar] [CrossRef] [Green Version]
- Ussher, J.R.; Drucker, D.J. Cardiovascular actions of incretin-based therapies. Circ. Res. 2014, 114, 1788–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thazhath, S.S.; Marathe, C.S.; Wu, T.; Chang, J.; Khoo, J.; Kuo, P.; Checklin, H.L.; Bound, M.J.; Rigda, R.S.; Horowitz, M.; et al. Acute effects of the glucagon-like peptide-1 receptor agonist, exenatide, on blood pressure and heart rate responses to intraduodenal glucose infusion in type 2 diabetes. Diabetes Vasc. Dis. Res. 2017, 14, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethel, M.A.; Patel, R.A.; Merrill, P.; Lokhnygina, Y.; Buse, J.B.; Mentz, R.J.; Pagidipati, N.J.; Chan, J.C.; Gustavson, S.M.; Iqbal, N.; et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: A meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 105–113. [Google Scholar] [CrossRef]
- Phillips, L.K.; Rayner, C.K.; Jones, K.L.; Horowitz, M. Measurement of gastric emptying in diabetes. J. Diabetes Complicat. 2014, 28, 894–903. [Google Scholar] [CrossRef]
- Marathe, C.S.; Horowitz, M.; Trahair, L.G.; Wishart, J.M.; Bound, M.; Lange, K.; Rayner, C.K.; Jones, K.L. Relationships of early and late glycemic responses with gastric emptying during an oral glucose tolerance test. J. Clin. Endocrinol. Metab. 2015, 100, 3565–3571. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Rayner, C.K.; Horowitz, M. Inter-regulation of gastric emptying and incretin hormone secretion: Implications for postprandial glycemic control. Biomark Med. 2016, 10, 1167–1179. [Google Scholar] [CrossRef]
- Jones, K.L.; Huynh, L.Q.; Hatzinikolas, S.; Rigda, R.S.; Phillips, L.K.; Pham, H.T.; Marathe, C.S.; Wu, T.; Malbert, C.H.; Stevens, J.E.; et al. Exenatide once weekly slows gastric emptying of solids and liquids in healthy, overweight people at steady-state concentrations. Diabetes Obes. Metab. 2020, 22, 788–797. [Google Scholar] [CrossRef]
- Park, S.-Y.; Chung, J.O.; Cho, D.H.; Chung, D.J.; Chung, M.Y. Impaired gastric emptying Is associated with a higher hncidence of coronary heart disease in subjects with diabetes. Diabetes 2018, 67, 467-P. [Google Scholar] [CrossRef]
- Trahair, L.G.; Horowitz, M.; Jones, K.L. Postprandial Hypotension Is Associated With More Rapid Gastric Emptying in Healthy Older Individuals. J. Am. Med Dir. Assoc. 2015, 16, 521–523. [Google Scholar] [CrossRef]
- Russo, A.; Stevens, J.E.; Wilson, T.; Wells, F.; Tonkin, A.; Horowitz, M.; Jones, K.L. Guar attenuates fall in postprandial blood pressure and slows gastric emptying of oral glucose in type 2 diabetes. Dig. Dis. Sci. 2003, 48, 1221–1229. [Google Scholar] [CrossRef]
- Trahair, L.G.; Horowitz, M.; Stevens, J.E.; Feinle-Bisset, C.; Standfield, S.; Piscitelli, D.; Rayner, C.K.; Deane, A.M.; Jones, K.L. Effects of exogenous glucagon-like peptide-1 on blood pressure, heart rate, gastric emptying, mesenteric blood flow and glycaemic responses to oral glucose in older individuals with normal glucose tolerance or type 2 diabetes. Diabetologia 2015, 58, 1769–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maida, A.; Lamont, B.J.; Cao, X.; Drucker, D.J. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-alpha in mice. Diabetologia 2011, 54, 339–349. [Google Scholar] [CrossRef]
- Sato, D.; Morino, K.; Nakagawa, F.; Murata, K.; Sekine, O.; Beppu, F.; Gotoh, N.; Ugi, S.; Maegawa, H. Acute Effect of Metformin on Postprandial Hypertriglyceridemia through Delayed Gastric Emptying. Int. J. Mol. Sci. 2017, 18, 1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagell, C.F.; Wettergren, A.; Pedersen, J.F.; Mortensen, D.; Holst, J.J. Glucagon-like peptide-2 inhibits antral emptying in man, but is not as potent as glucagon-like peptide-1. Scand. J. Gastroenterol. 2004, 39, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.B.; Gryback, P.; Holst, J.J.; Hilsted, L.; Hellstrom, P.M.; Jacobsson, H.; Schmidt, P.T. Differential effect of PYY1-36 and PYY3-36 on gastric emptying in man. Regul. Pept. 2009, 158, 57–62. [Google Scholar] [CrossRef]
- Yamazaki, K.; Yasuda, N.; Inoue, T.; Nagakura, T.; Kira, K.; Saeki, T.; Tanaka, I. The combination of metformin and a dipeptidyl peptidase IV inhibitor prevents 5-fluorouracil-induced reduction of small intestine weight. Eur. J. Pharm. 2004, 488, 213–218. [Google Scholar] [CrossRef]
- Park, H.-M.; Park, S.-Y.; Chung, J.O.; Cho, D.H.; Park, C.-H.; Kim, H.-S.; Chung, D.J.; Choi, S.-K.; Rew, J.-S.; Chung, M.Y. Association between gastric emptying time and incidence of cardiovascular diseases in subjects with diabetes. J. Neurogastroenterol. Motil. 2019, 25, 387–393. [Google Scholar] [CrossRef]
- Kong, M.F.; Horowitz, M.; Jones, K.L.; Wishart, J.M.; Harding, P.E. Natural history of diabetic gastroparesis. Diabetes Care 1999, 22, 503–507. [Google Scholar] [CrossRef]
- Trahair, L.G.; Horowitz, M.; Jones, K.L. Postprandial hypotension: A systematic review. J. Am. Med. Dir. Assoc. 2014, 15, 394–409. [Google Scholar] [CrossRef]
- Aronow Wilbert, S.; Ahn, C. Association of postprandial hypotension with incidence of falls, syncope, coronary events, stroke, and total mortality at 29-month follow-up in 499 older nursing home residents. J. Am. Geriatr. Soc. 1997, 45, 1051–1053. [Google Scholar] [CrossRef]
- Vanis, L.; Gentilcore, D.; Rayner, C.K.; Wishart, J.M.; Horowitz, M.; Feinle-Bisset, C.; Jones, K.L. Effects of small intestinal glucose load on blood pressure, splanchnic blood flow, glycemia, and GLP-1 release in healthy older subjects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1524–R1531. [Google Scholar] [CrossRef] [PubMed]
- Heseltine, D.; Dakkak, M.; Macdonald, I.A.; Bloom, S.R.; Potter, J.F. Effects of carbohydrate type on postprandial blood pressure, neuroendocrine and gastrointestinal hormone changes in the elderly. Clin. Auton. Res. 1991, 1, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Barragan, J.M.; Rodriguez, R.E.; Blazquez, E. Changes in arterial blood pressure and heart rate induced by glucagon-like peptide-1-(7-36) amide in rats. Am. J. Physiol. 1994, 266, E459–E466. [Google Scholar] [CrossRef] [PubMed]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peuler, J.D. Opposing adrenergic actions of intravenous metformin on arterial pressure in female spontaneously hypertensive rats. Cardiovasc. Res. 1999, 43, 237–247. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borg, M.J.; Rayner, C.K.; Jones, K.L.; Horowitz, M.; Xie, C.; Wu, T. Gastrointestinal Mechanisms Underlying the Cardiovascular Effect of Metformin. Pharmaceuticals 2020, 13, 410. https://doi.org/10.3390/ph13110410
Borg MJ, Rayner CK, Jones KL, Horowitz M, Xie C, Wu T. Gastrointestinal Mechanisms Underlying the Cardiovascular Effect of Metformin. Pharmaceuticals. 2020; 13(11):410. https://doi.org/10.3390/ph13110410
Chicago/Turabian StyleBorg, Malcolm J., Christopher K. Rayner, Karen L. Jones, Michael Horowitz, Cong Xie, and Tongzhi Wu. 2020. "Gastrointestinal Mechanisms Underlying the Cardiovascular Effect of Metformin" Pharmaceuticals 13, no. 11: 410. https://doi.org/10.3390/ph13110410