
An Electroanalytical Solution for the Determination of Pb2+ in Progressive Hair Dyes Using the Cork–Graphite Sensor

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Preparation of Cork-Modified Electrodes
2.3. Preparation of Real Samples
2.4. Electrochemical Measurements
2.5. Inductively Coupled Plasma Optical Emission Spectrometer (ICP–OES) Measurements
3. Results and Discussion
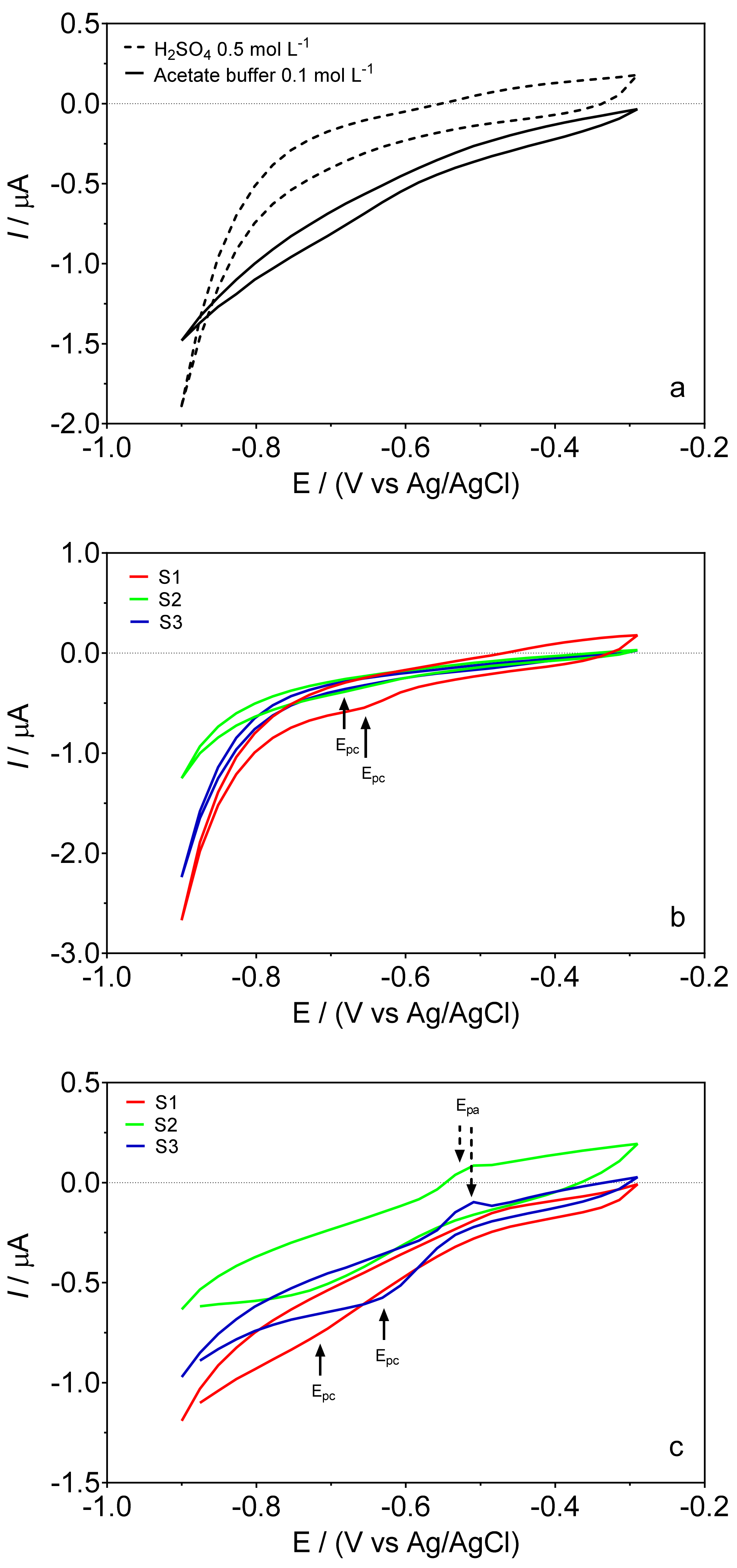
3.1. Effect of the Supporting Electrolyte
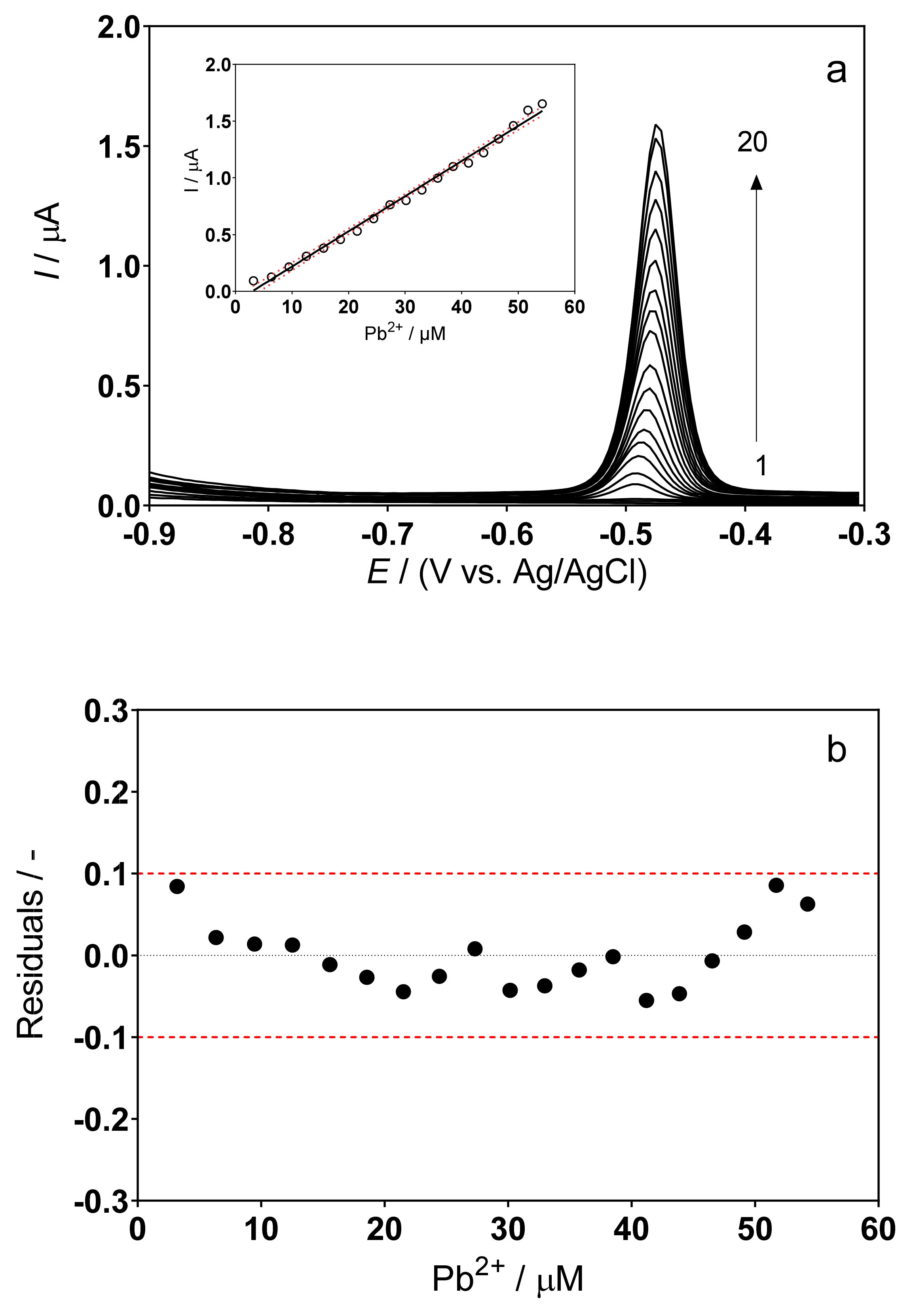
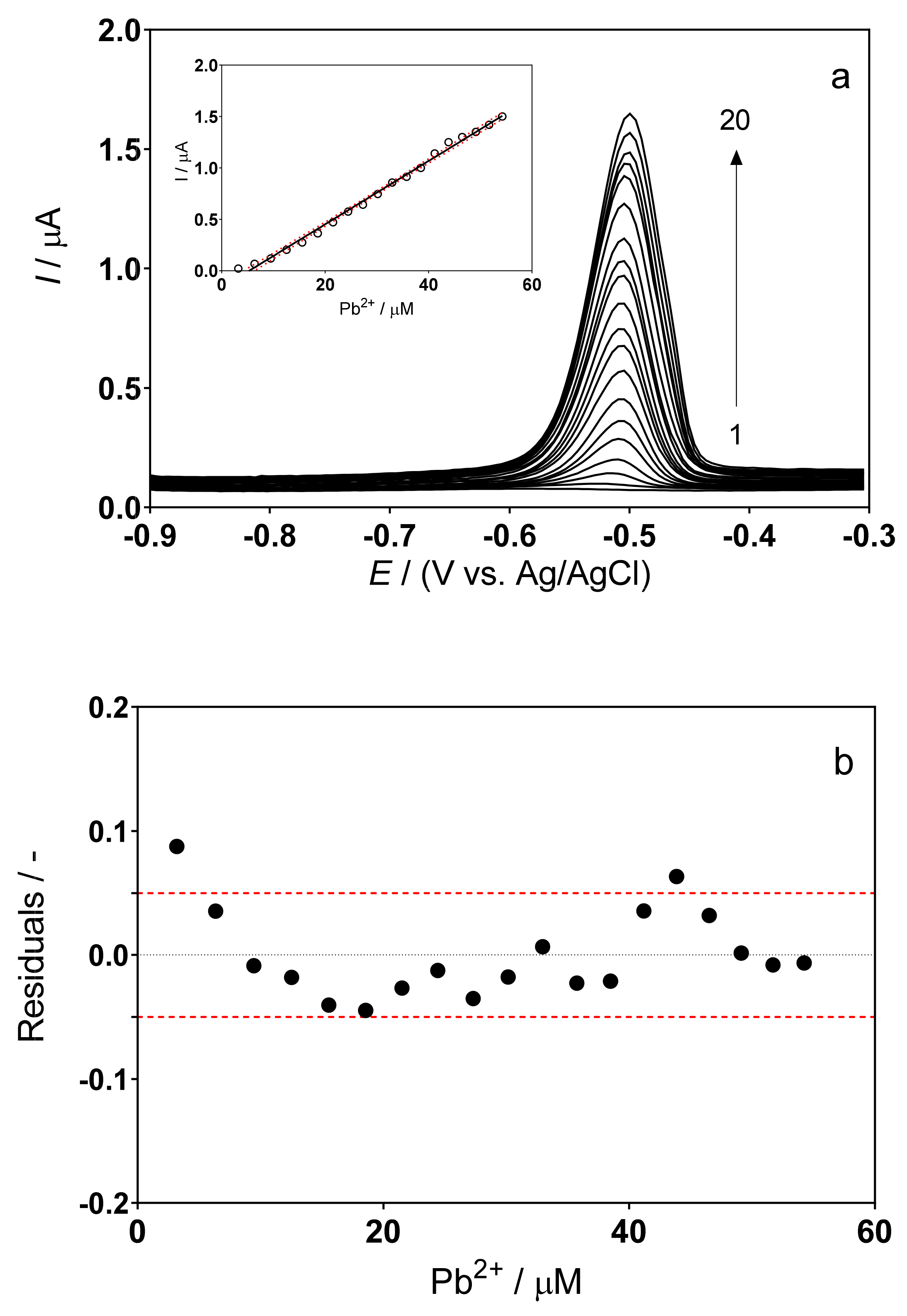
3.2. DPSV Determinations: Effect of the Supporting Electrolyte
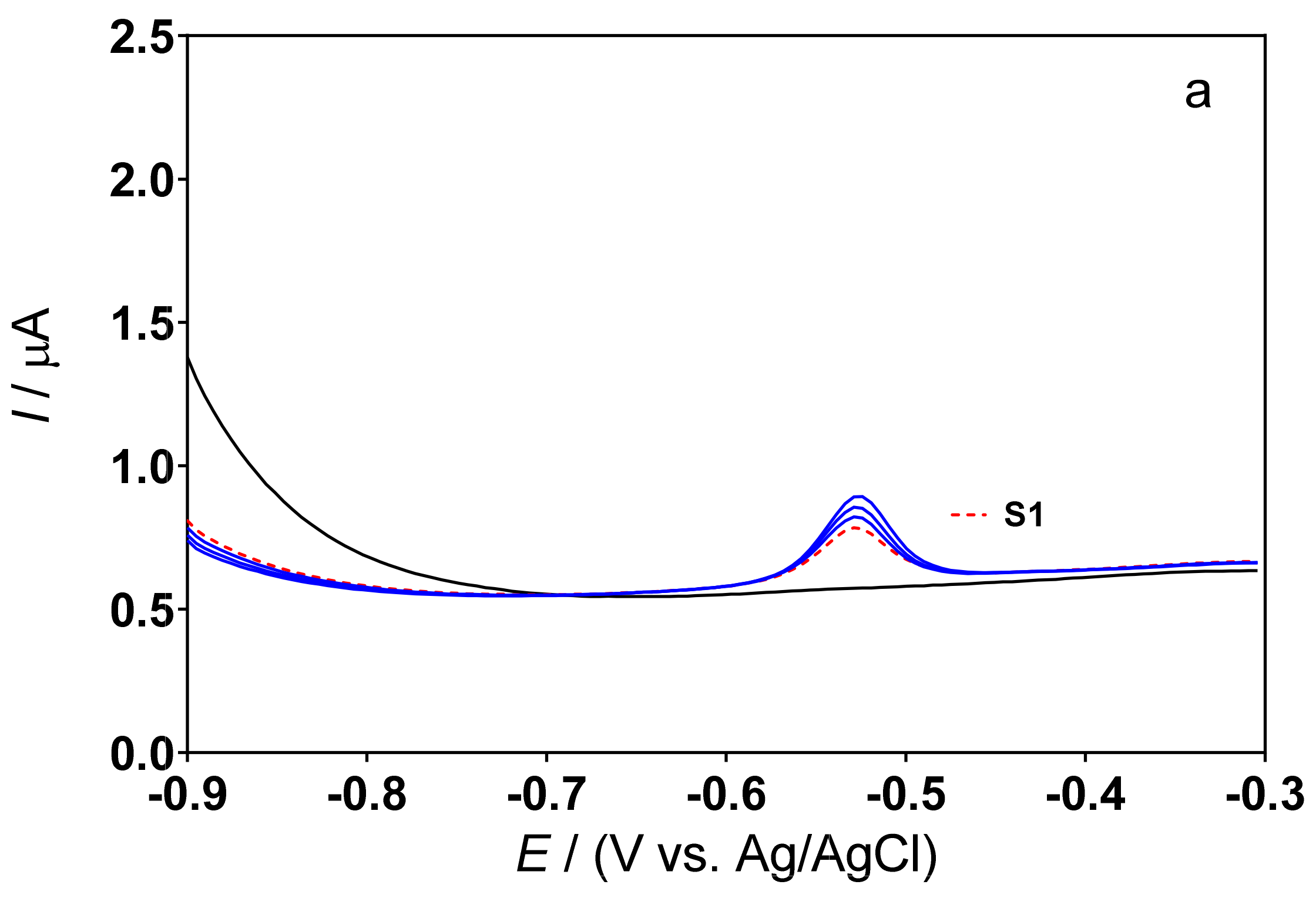
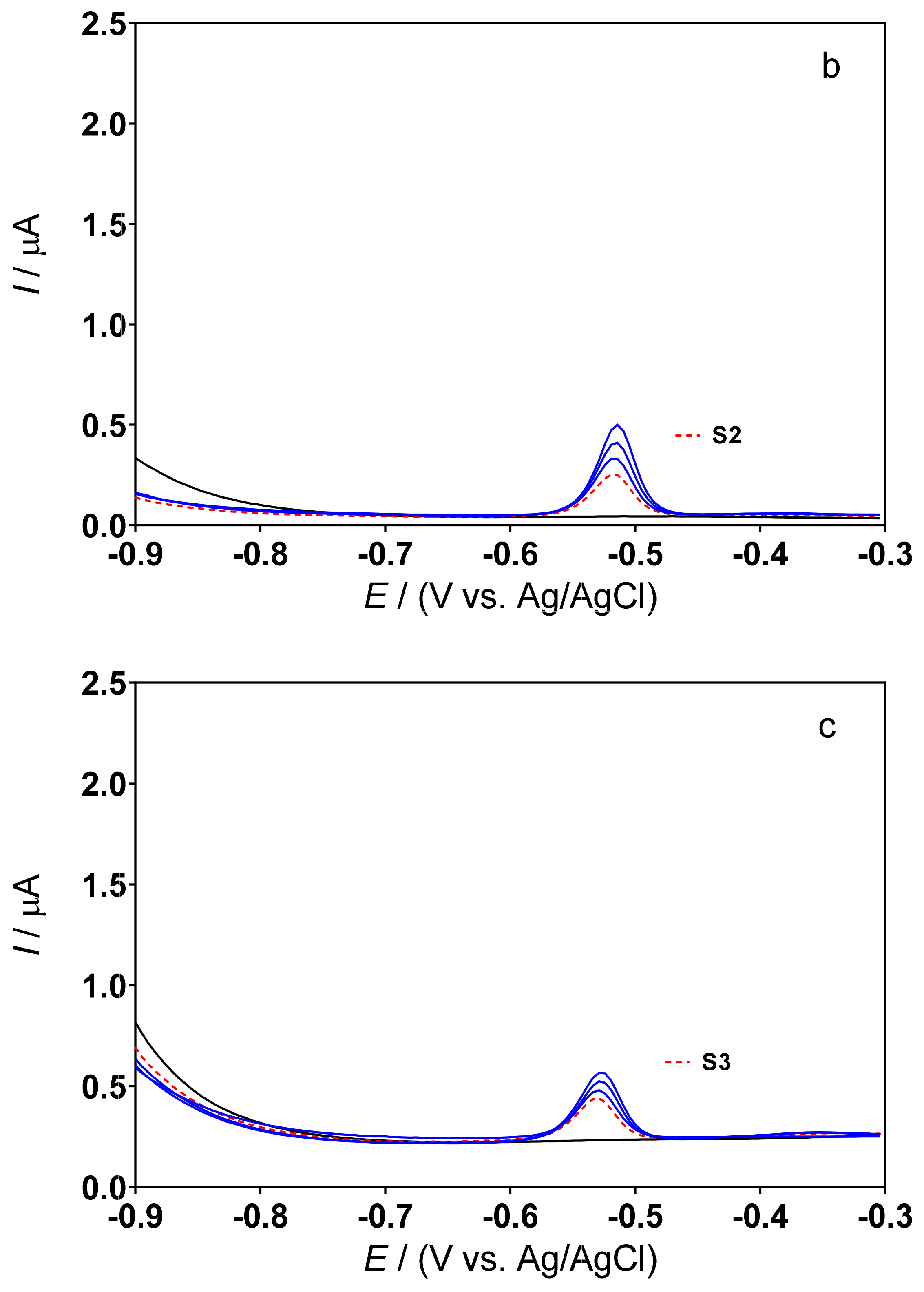
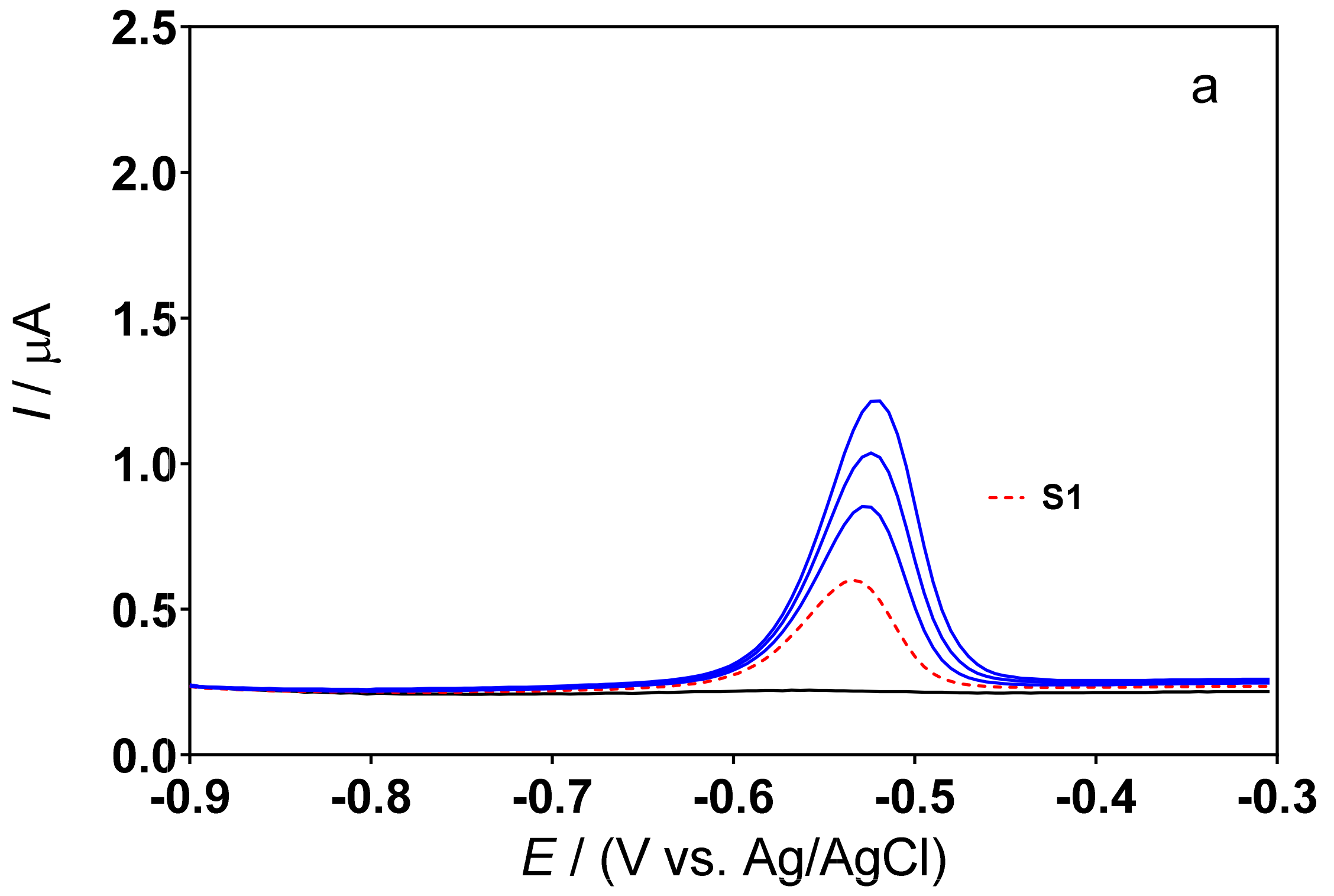
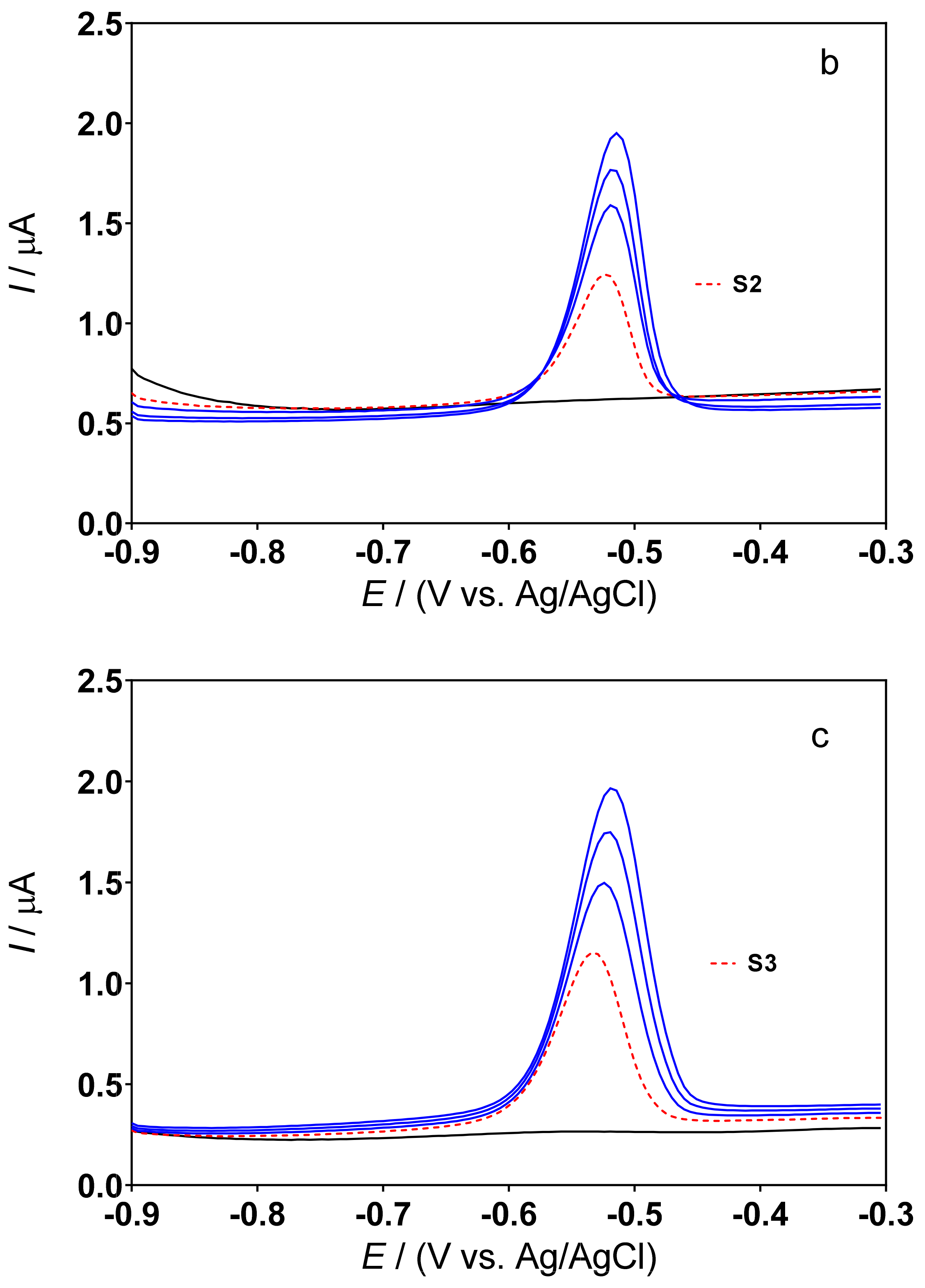
3.3. Determination of Lead Concentration in Hair Dye Samples
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Souza, J.C.; da Silva, B.F.; Morales, D.A.; de Umbuzeiro, G.A.; Zanoni, M.V.B. Assessment of p-aminophenol oxidation by simulating the process of hair dyeing and occurrence in hair salon wastewater and drinking water from treatment plant. J. Hazard. Mater. 2020, 387, 122000. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Tapia, A.; Gonzalez-Guerra, E. Hair cosmetics: Dyes. Actas Dermosifiliogr. 2014, 105, 833. [Google Scholar] [CrossRef] [PubMed]
- Epstein, H. Skin Care Products. In Handbook of Cosmetic Science and Technology; CRC Press: Boca Raton, FL, USA, 2009; ISBN 9781420069686. [Google Scholar]
- Maciel, J.V.; da Silveira, G.D.; Durigon, A.M.M.; Fatibello-Filho, O.; Dias, D. Use of carbon black based electrode as sensor for solid-state electrochemical studies and voltammetric determination of solid residues of lead. Talanta 2022, 236, 122881. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.C. Contaminated and natural lead environments of man. Arch. Environ. Health 1965, 11, 344. [Google Scholar] [CrossRef]
- Pinho, S.; Ladeiro, B. Phytotoxicity by lead as heavy metal focus on oxidative stress. J. Bot. 2012, 2012, 1. [Google Scholar] [CrossRef]
- Agência Nacional de Vigilância Sanitária (Anvisa)—Ministério da Saúde. Resolução da Diretoria Colegiada—RDC No. 15, 26 de Março; Anvisa: Brasilia, Brasil, 2013. [Google Scholar]
- US Food & Drug Administration. FDA to Repeal Color Additive Approval for the Use of Lead Acetate in Hair Dyes. 7 October 2021. Available online: https://www.fda.gov/food/cfsan-constituent-updates/fda-repeal-color-additive-approval-use-lead-acetate-hair-dyes (accessed on 6 January 2022).
- Huseinov, A.; Weese, B.L.; Brewer, B.J.; Alvarez, N.T. Near-electrode pH change for voltammetric detection of insoluble lead carbonate. Anal. Chim. Acta 2021, 1186, 339087. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, B.; He, M.; Hu, B. Switchable solvent based liquid phase microextraction of trace lead and cadmium from environmental and biological samples prior to graphite furnace atomic absorption spectrometry detection. Microchem. J. 2018, 139, 380. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, J.; Tian, Y.; Yao, K.; Xu, M. Detection of heavy metal lead in lettuce leaves based on fluorescence hyperspectral technology combined with deep learning algorithm. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 266, 120460. [Google Scholar] [CrossRef]
- Silva, I.B.; de Araújo, D.M.; Vocciante, M.; Ferro, S.; Martínez-Huitle, C.A.; Dos Santos, E.V. Electrochemical determination of lead using a composite sensor obtained from low-cost green materials: Graphite/cork. Appl. Sci. 2021, 11, 2355. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Doan, T.C.D.; Huynh, T.M.; Nguyen, V.N.P.; Dinh, H.H.; Dang, D.M.T.; Dang, C.M. An electrochemical sensor based on polyvinyl alcohol/chitosan-thermally reduced graphene composite modified glassy carbon electrode for sensitive voltammetric detection of lead. Sens. Actuators B Chem. 2021, 345, 130443. [Google Scholar] [CrossRef]
- Silva, K.N.O.; Paiva, S.S.M.; Souza, F.L.; Silva, D.R.; Martínez-Huitle, C.A.; Santos, E.V. Applicability of electrochemical technologies for removing and monitoring Pb2+ from soil and water. J. Electroanal. Chem. 2018, 816, 171. [Google Scholar] [CrossRef]
- Bilge, S.; Karadurmus, L.; Sınag, A.; Ozkan, S.A. Green synthesis and characterization of carbon-based materials for sensitive detection of heavy metal ions. TrAC Trends Anal. Chem. 2021, 145, 116473. [Google Scholar] [CrossRef]
- Cui, L.; Wu, J.; Ju, H. Electrochemical sensing of heavy metal ions with inorganic, organic and biomaterials. Biosens. Bioelectron. 2015, 63, 276. [Google Scholar] [CrossRef]
- Zhu, G.; Yi, Y.; Chen, J. Recent advances for cyclodextrin-based materials in electrochemical sensing. TrAC Trends Anal. Chem. 2016, 80, 232. [Google Scholar] [CrossRef]
- Zhang, T.; Jin, H.; Fang, Y.; Guan, J.B.; Ma, S.J.; Pan, Y.; Zhang, M.; Zhu, H.; Liu, X.D.; Du, M.L. Detection of trace Cd2+, Pb2+ and Cu2+ ions via porous activated carbon supported palladium nanoparticles modified electrodes using SWASV. Mater. Chem. Phys. 2019, 225, 433. [Google Scholar] [CrossRef]
- Xu, C.; Liu, J.; Bi, Y.; Ma, C.; Bai, J.; Hu, Z.; Zhou, M. Biomass derived worm-like nitrogen-doped-carbon framework for trace determination of toxic heavy metal lead(II). Anal. Chim. Acta 2020, 1116, 16. [Google Scholar] [CrossRef]
- Qin, D.; Gao, S.; Wang, L.; Shen, H.; Yalikun, N.; Sukhrobov, P.; Wagberg, T.; Zhao, Y.; Mamat, X.; Hu, G. Three-dimensional carbon nanofiber derived from bacterial cellulose for use in a Nafion matrix on a glassy carbon electrode for simultaneous voltammetric determination of trace levels of Cd(II) and Pb(II). Microchim. Acta 2017, 184, 2759. [Google Scholar] [CrossRef]
- Pinar Gumus, Z.; Soylak, M. Metal organic frameworks as nanomaterials for analysis of toxic metals in food and environmental applications. TrAC Trends Anal. Chem. 2021, 143, 116417. [Google Scholar] [CrossRef]
- Waheed, A.; Mansha, M.; Ullah, N. Nanomaterials-based electrochemical detection of heavy metals in water: Current status, challenges and future direction. TrAC Trends Anal. Chem. 2018, 105, 37. [Google Scholar] [CrossRef]
- Radhakrishnan, K.; Panneerselvam, P.; Marieeswaran, M. A green synthetic route for the surface-passivation of carbon dots as an effective multifunctional fluorescent sensor for the recognition and detection of toxic metal ions from aqueous solution. Anal. Methods 2019, 11, 490. [Google Scholar] [CrossRef]
- Ghorbani-Kalhor, E. A metal-organic framework nanocomposite made from functionalized magnetite nanoparticles and HKUST-1 (MOF-199) for preconcentration of Cd(II), Pb(II), and Ni(II). Microchim. Acta 2016, 183, 2639. [Google Scholar] [CrossRef]
- Hassanpour, A.; Hosseinzadeh-Khanmiri, R.; Babazadeh, M.; Abolhasani, J.; Ghorbani-Kalhor, E. Determination of heavy metal ions in vegetable samples using a magnetic metal–organic framework nanocomposite sorbent. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 725. [Google Scholar] [CrossRef]
- Monteiro, M.K.S.; Paiva, S.S.M.; da Silva, D.R.; Vilar, V.J.P.; Martínez-Huitle, C.A.; dos Santos, E.V. Novel cork-graphite electrochemical sensor for voltammetric determination of caffeine. J. Electroanal. Chem. 2019, 839, 283. [Google Scholar] [CrossRef]
- Monteiro, M.K.S.; Santos, E.C.M.M.; Silva, D.R.; Martínez-Huitle, C.A.; dos Santos, E.V. Simultaneous determination of paracetamol and caffeine in pharmaceutical formulations and synthetic urine using cork-modified graphite electrodes. J. Solid State Electrochem. 2020, 18, 1789–1800. [Google Scholar] [CrossRef]
- De Araújo, D.M.; Paiva, S.d.S.S.M.; Henrique, J.M.M.; Martínez-Huitle, C.A.; dos Santos, E.V. Green composite sensor for monitoring hydroxychloroquine in different water matrix. Materials 2021, 14, 4990. [Google Scholar] [CrossRef]
- Silva, S.P.; Sabino, M.A.; Fernandas, E.M.; Correlo, V.M.; Boesel, L.F.; Reis, R.L. Cork: Properties, capabilities and applications. Int. Mater. Rev. 2005, 50, 345. [Google Scholar] [CrossRef] [Green Version]
- Desimoni, E.; Brunetti, B. About estimating the limit of detection of heteroscedastic analytical systems. Anal. Chim. Acta 2009, 655, 30. [Google Scholar] [CrossRef]
- Miller, J.N. Basic statistical methods for analytical chemistry. Part 2. Calibration and regression methods. A review. Analyst 1991, 116, 3. [Google Scholar] [CrossRef]
- Mocak, J.; Bond, A.M.; Mitchell, S.; Scollary, G.; Bond, A.M. A statistical overview of standard (IUPAC and ACS) and new procedures for determining the limits of detection and quantification: Application to voltammetric and stripping techniques. Pure Appl. Chem. 1997, 69, 297. [Google Scholar] [CrossRef]
- Honeychurch, K. Trace voltammetric determination of lead at a recycled battery carbon rod electrode. Sensors 2019, 19, 770. [Google Scholar] [CrossRef] [Green Version]
- Visscher, W. Cyclic voltammetry on lead electrodes in sulphuric acid solution. J. Power Sources 1976, 1, 257. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, M.K.S.; da Silva, D.R.; Quiroz, M.A.; Vilar, V.J.P.; Martínez-Huitle, C.A.; dos Santos, E.V. Applicability of cork as novel modifiers to develop electrochemical sensor for caffeine determination. Materials 2021, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Gil, L. New cork-based materials and applications. Materials 2015, 8, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A practical beginner’s guide to cyclic voltammetry. J. Chem. Educ. 2018, 95, 197. [Google Scholar] [CrossRef]
- Brunetti, B.; Desimoni, E. Determination of theophylline at a cysteic acid modified glassy carbon electrode. Electroanalysis 2009, 21, 772. [Google Scholar] [CrossRef]
- Brunetti, B.; Desimoni, E. Voltammetric determination of vitamin B6 in food samples and dietary supplements. J. Food Compos. Anal. 2014, 33, 155. [Google Scholar] [CrossRef]
- Pintor, A.M.A.; Vieira, B.R.C.; Brandaõ, C.C.; Boaventura, R.A.R.; Botelho, C.M.S. Complexation mechanisms in arsenic and phosphorus adsorption onto iron-coated cork granulates. J. Environ. Chem. Eng. 2020, 8, 104184. [Google Scholar] [CrossRef]
- Pintor, A.M.A.; Silvestre-Albero, A.M.; Ferreira, C.I.A.; Pereira, J.P.C.; Vilar, V.J.P.; Botelho, C.M.S.; Rodríguez-Reinoso, F.; Boaventura, R.A.R. Textural and surface characterization of cork-based sorbents for the removal of oil from water. Ind. Eng. Chem. Res. 2013, 52, 16427. [Google Scholar] [CrossRef]
- Danzer, K.; Currie, L.A. Guideline for calibration in analytical chemistry—Part 1: Fundamentals and single component calibration. Pure Appl. Chem. 1998, 70, 993. [Google Scholar] [CrossRef]
- Currie, L.A. Nomenclature in evaluation of analytical methods including detection and quantification capabilities. Pure Appl. Chem. 1995, 67, 1699. [Google Scholar] [CrossRef]
- Oliveira, G.A.; Gevaerd, A.; Mangrich, A.S.; Marcolino-Junior, L.H.; Bergamini, M.F. Biochar obtained from spent coffee grounds: Evaluation of adsorption properties and its application in a voltammetric sensor for lead(II) ions. Microchem. J. 2021, 165, 106114. [Google Scholar] [CrossRef]
- Wang, W.; Bao, N.; Yuan, W.; Si, N.; Bai, H.; Li, H.; Zhang, Q. Simultaneous determination of lead, arsenic, and mercury in cosmetics using a plastic based disposable electrochemical sensor. Microchem. J. 2019, 148, 240. [Google Scholar] [CrossRef]
- Palisoc, S.; Causing, A.M.; Natividad, M. Gold nanoparticle/hexaammineruthenium/Nafion® modified glassy carbon electrodes for trace heavy metal detection in commercial hair dyes. Anal. Methods 2017, 9, 4240. [Google Scholar] [CrossRef]
- Swetha, P.; Chen, J.; Kumar, A.S.; Feng, S.P. High index facets-Ag nanoflower enabled efficient electrochemical detection of lead in blood serum and cosmetics. J. Electroanal. Chem. 2020, 878, 114657. [Google Scholar] [CrossRef]
- Shih, Y.; Zen, J.M.; Kumar, A.S.; Lee, Y.C.; Huang, H.R. Determination of the toxic lead level in cosmetic-hair dye formulations using a screen-printed silver electrode. Bull. Chem. Soc. Jpn. 2004, 77, 311. [Google Scholar] [CrossRef]
- Fardiyah, Q.; Rumhayati, B.; Rosemiyani, I. Determination of lead in cosmetic sampels using coated wire lead(II) ion selective electrode based on phyropillite. UNEJ e-Proceeding 2017, 270–272. Available online: https://jurnal.unej.ac.id/index.php/prosiding/article/view/4239 (accessed on 5 January 2022).
- De Furtado, L.A.; de Lucena, Í.O.; de Fernandes, J.O.; Lepri, F.G.; de Martins, D.L.; Semaan, F.S. New strategies for the simultaneous voltammetric quantification of Pb and Zn in hair cosmetics samples employing chemically modified composite electrodes. Meas. J. Int. Meas. Confed. 2018, 125, 651. [Google Scholar] [CrossRef]
- Currie, L.A. Nomenclature in evaluation of analytical methods including detection and quantification capabilities (IUPAC Recommendations 1995). Anal. Chim. Acta 1999, 391, 105. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrodes | Samples | Method | Electrolyte | Linear Range (µM) | Limit of Detection (LOD, µM) | Ref. |
|---|---|---|---|---|---|---|
| 1 CPME-ACfB300 | Hair dyes and gunshot residues | 2 DPAdSV | 0.1M acetate buffer (pH = 7.0) | 0.13–2.44 | 0.0045 | [44] |
| 3 CB-CP | Hair dyes | 8 DPV | 0.2M phosphate buffer (pH = 5.0) | 0.048–0.48 | – | [4] |
| Disposable plastic electrode | Cosmetic samples | DPSV | 0.8M HNO3—0.1M KCl | 0.24–0.72 | 0.096 | [45] |
| 4 GCE | Hair dyes | 9 ASV | 0.1M NaCl | 7.0–17.5 | 0.021 | [46] |
| 5 AgNF-Pb@GCE | Cosmetic samples | 10 SWV | Acetate buffer (pH 4.5) | 0.4–3.3 | 0.035 | [47] |
| 6 AgSPE | Cosmetic hair dye | SWV | 0.05M Na2SO4 | 0.06–0.79 | 0.031 | [48] |
| 7 CW-ISE | Cosmetic samples | Potentio-metric method | Acetate buffer (pH 5.0) | 10–100 | 8.0 | [49] |
| Graphite/epoxy composite electrodes | Hair cosmetic | 11 SWASV | 0.1M acetate buffer (pH 6.0) | 0.2–1.7 | 0.07 | [50] |
| Cork-graphite | Hair dyes | DPSV | 0.5M H2SO4 | 3.19–54.26 | 1.06 | This work |
| Cork-graphite | Hair dyes | DPSV | 0.1M acetate buffer (pH 4.5) | 3.19–54.26 | 1.26 | This work |
| Sample | Method | [Pb2+]/mg L−1 | Pb Content (%) | Difference (%) *** |
|---|---|---|---|---|
| S1 | ICP–OES | 4.14 ± 1.02 | 0.25 | |
| DPSV * | 4.00 ± 1.65 | 0.20 | 3.4 | |
| DPSV ** | 3.89 ± 1.90 | 0.18 | 6.0 | |
| S2 | ICP–OES | 2.42 ± 0.58 | 0.30 | |
| DPSV * | 2.33 ± 0.23 | 0.26 | 3.7 | |
| DPSV ** | 2.47 ± 0.41 | 0.32 | 2.1 | |
| S3 | ICP–OES | 3.50 ± 0.39 | 0.43 | |
| DPSV * | 2.85 ± 0.25 | 0.34 | 18.5 | |
| DPSV ** | 2.65 ± 0.48 | 0.30 | 24.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barros, T.M.; Medeiros de Araújo, D.; Lemos de Melo, A.T.; Martínez-Huitle, C.A.; Vocciante, M.; Ferro, S.; Vieira dos Santos, E. An Electroanalytical Solution for the Determination of Pb2+ in Progressive Hair Dyes Using the Cork–Graphite Sensor. Sensors 2022, 22, 1466. https://doi.org/10.3390/s22041466
Barros TM, Medeiros de Araújo D, Lemos de Melo AT, Martínez-Huitle CA, Vocciante M, Ferro S, Vieira dos Santos E. An Electroanalytical Solution for the Determination of Pb2+ in Progressive Hair Dyes Using the Cork–Graphite Sensor. Sensors. 2022; 22(4):1466. https://doi.org/10.3390/s22041466
Chicago/Turabian StyleBarros, Thalita Medeiros, Danyelle Medeiros de Araújo, Alana Tamires Lemos de Melo, Carlos Alberto Martínez-Huitle, Marco Vocciante, Sergio Ferro, and Elisama Vieira dos Santos. 2022. "An Electroanalytical Solution for the Determination of Pb2+ in Progressive Hair Dyes Using the Cork–Graphite Sensor" Sensors 22, no. 4: 1466. https://doi.org/10.3390/s22041466